
New Nitrogen, Sulfur-, and Selenium-Donating Ligands Derived from Chiral Pyridine Amino Alcohols. Synthesis and Catalytic Activity in Asymmetric Allylic Alkylation



Abstract
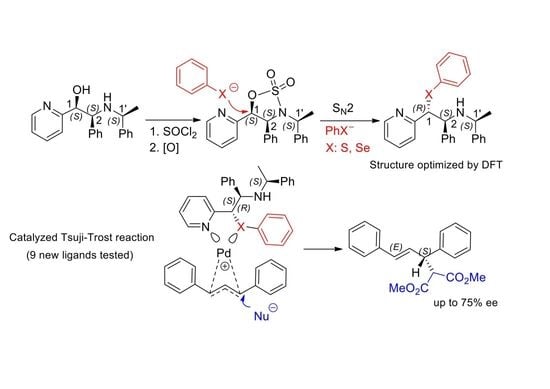
:
1. Introduction
2. Results and Discussion
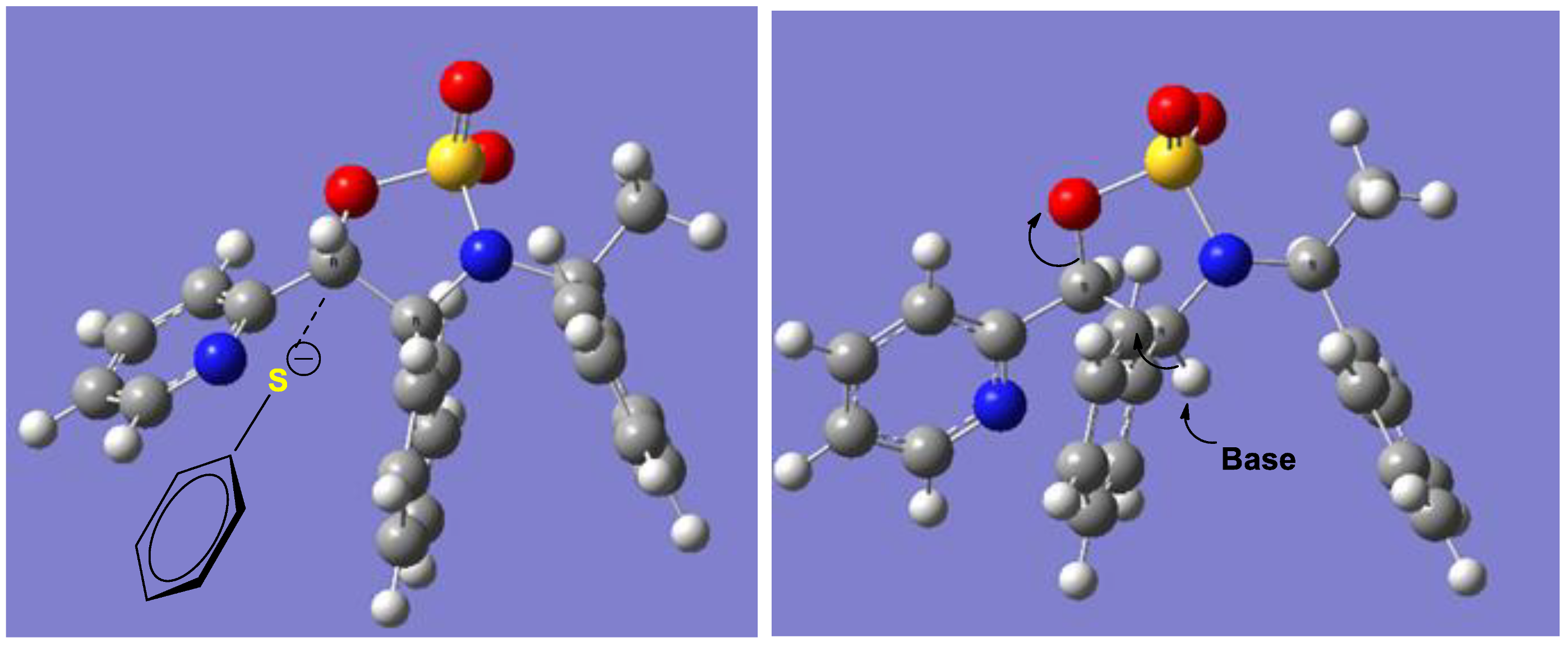
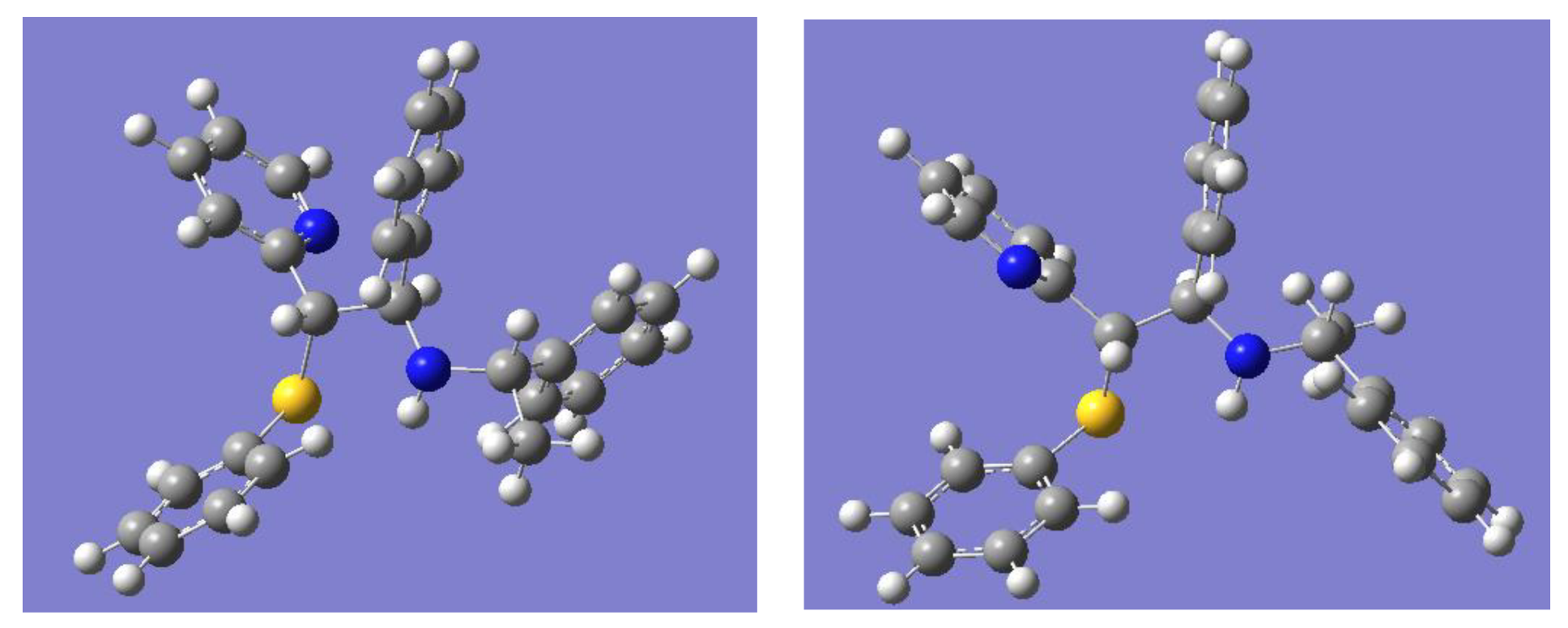
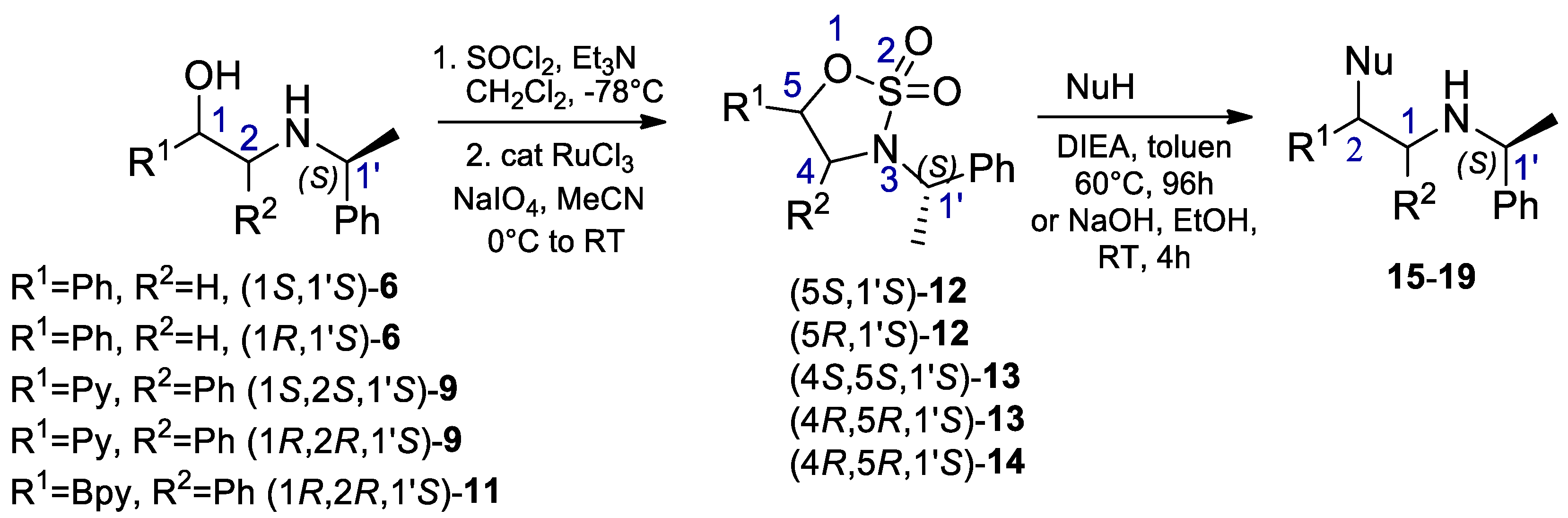
2.1. Synthesis of Sulfur and Selenium Derivatives
2.2. Synthesis of Nitrogen Derivatives (Chiral Schiff Base)
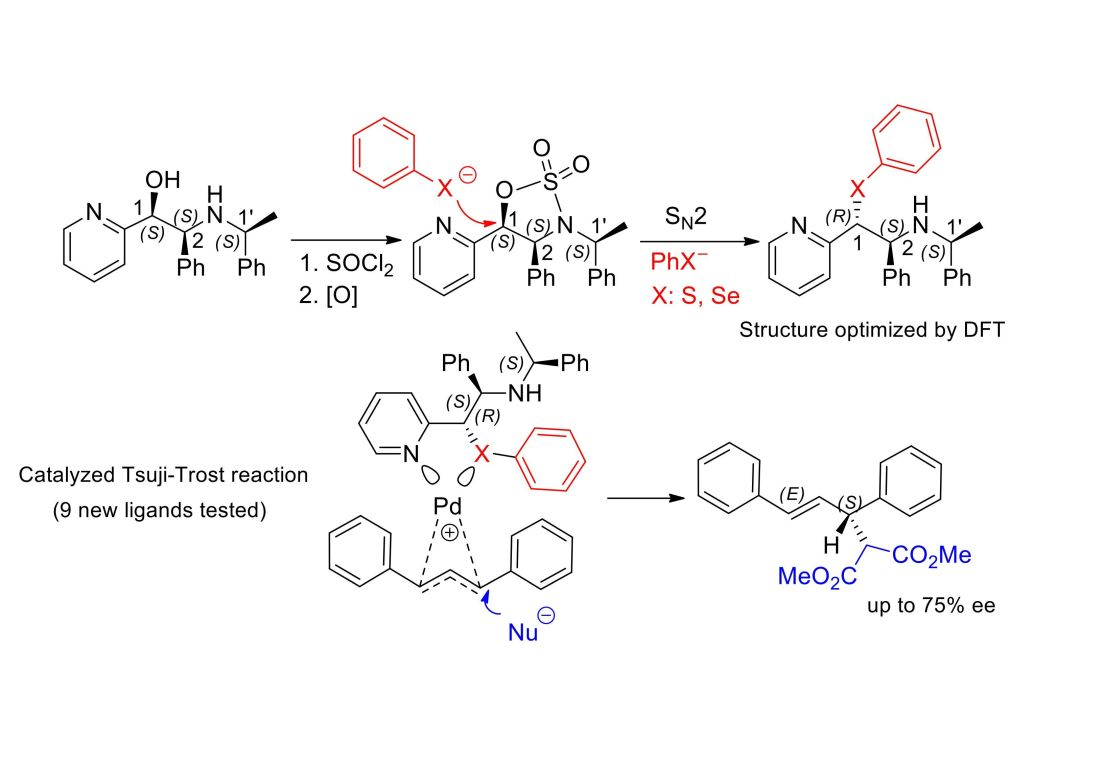
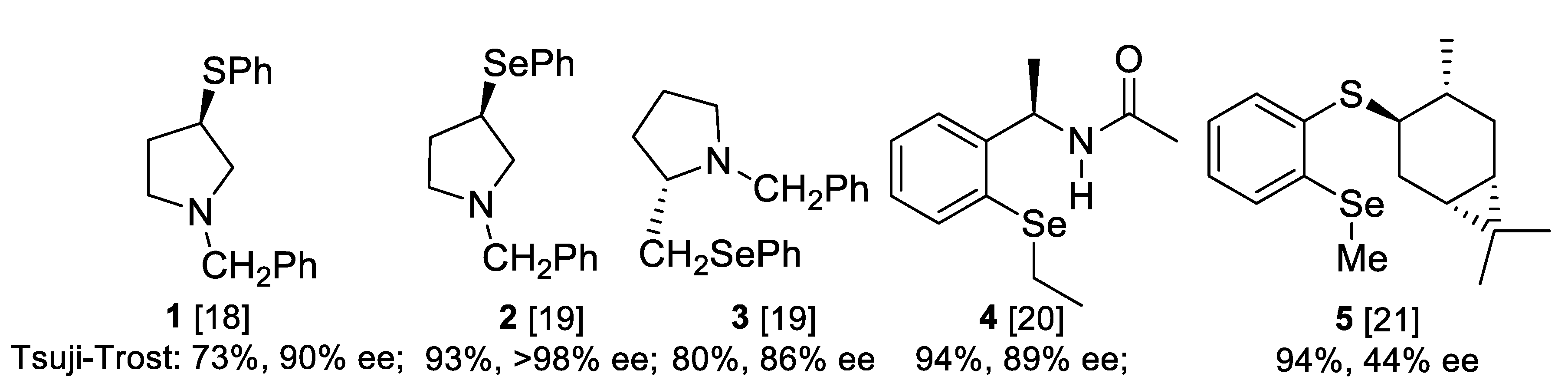
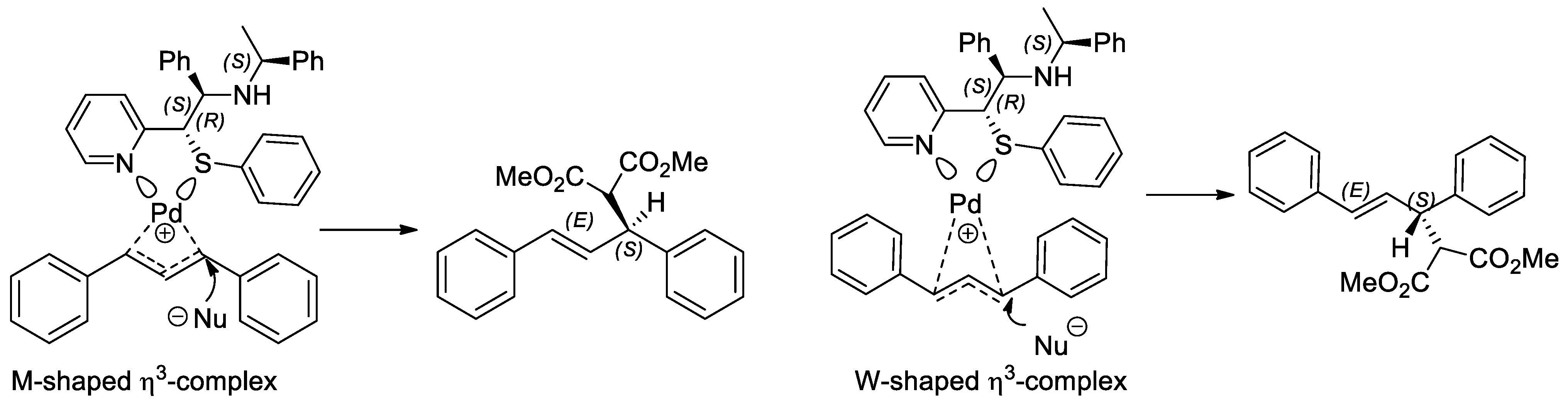
2.3. Application of Chiral S, Se, and N Derivatives in the Tsuji-Trost Reaction
3. Materials and Methods
3.1. General
3.2. General Procedure for the Synthesis of Cyclic Sulfonamidates
3.3. General Procedure for the Synthesis of S and Se Derivatives
3.4. General Procedure for the Synthesis of Mono-Aldimine (Chiral Schiff Base)
3.5. Catalytic Reaction Procedure (Tsuji-Trost)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Ojima, I. Catalysis in Asymmetric Synthesis, 2nd ed.; Wiley-VCH: New York, NY, USA, 2000. [Google Scholar]
- Zhou, Q.-L. Privileged Chiral Ligands and Catalysts, 1st ed.; Wiley-VCH: New York, NY, USA, 2011. [Google Scholar]
- Pellissier, H. Chiral Sulfur Ligands: Asymmetric Catalysis; Royal Society of Chemistry: Cambridge, UK, 2009. [Google Scholar]
- Otocka, S.; Kwiatkowska, M.; Madalińska, L.; Kiełbasiński, P. Chiral Organosulfur Ligands/Catalysts with a Stereogenic Sulfur Atom: Applications in Asymmetric Synthesis. Chem. Rev. 2017, 117, 4147–4181. [Google Scholar] [CrossRef]
- Masdeu-Bulto, A.M.; Dieguez, M.; Martin, E.; Gomez, M. Chiral thioether ligands: Coordination chemistry and asymmetric catalysis. Coord. Chem. Rev. 2003, 242, 159–201. [Google Scholar] [CrossRef]
- Wirth, T. Enantioselective Alkylation of Aldehydes Catalyzed by New Chiral Diselenides. Tetrahedron Lett. 1995, 36, 7849–7852. [Google Scholar] [CrossRef]
- Nishibayashi, Y.; Uemura, S. Selenium compounds as ligands and catalysts in Organoselenium Chemistry: Modern Developments in Organic Synthesis. In Topics in Current Chemistry; Wirth, T., Ed.; Springer: Berlin/Heidelberg, Germany, 2000; Volume 208, pp. 235–255. [Google Scholar]
- Shaw, S.; White, J.D. Asymmetric Catalysis Using Chiral Salen–Metal Complexes: Recent Advances. Chem. Rev. 2019, 119, 9381–9426. [Google Scholar] [CrossRef]
- Trost, B.M.; Van Vrankel, D.L. Asymmetric Transition Metal-Catalyzed Allylic Alkylations. Chem. Rev. 1996, 96, 395–422. [Google Scholar] [CrossRef]
- Tsuji, J. Recollections of organopalladium chemistry. Pure Appl. Chem. 1999, 71, 1539–1547. [Google Scholar] [CrossRef] [Green Version]
- Lubbers, T.; Metz, P. Methods of Organic Chemistry (Houben Weyl), Stereoselective Synthesis; Thieme Verlag: Stuttgart, Germany, 1995; Volume E21c, pp. 2371–2473. [Google Scholar]
- Pfalz, A.; Lautens, M. Comprehensive Asymmetric Catalysis; Jacobsen, E.N., Pfaltz, A., Yamamoto, H., Eds.; Springer: Berlin/Heidelberg, Germany, 1999; Volume 2, pp. 834–884. [Google Scholar]
- Oslob, J.D.; Åkermark, B.; Helquist, P.; Norrby, P.-O. Steric Influences on the Selectivity in Palladium-Catalyzed Allylation. Organometallics 1997, 16, 3015–3021. [Google Scholar] [CrossRef]
- Trost, B.M.; Crawley, M.L. Asymmetric Transition-Metal-Catalyzed Allylic Alkylations: Applications in Total Synthesis. Chem. Rev. 2003, 103, 2921–2943. [Google Scholar] [CrossRef] [PubMed]
- Transition Metal Catalyzed Enantioselective Allylic Substitution in Organic Synthesis; Kazmaier, U. (Ed.) (Topics in Organometallic Chemistry); Springer: Berlin/Heidelberg, Germany, 2012; Volume 38. [Google Scholar]
- Pàmies, O.; Margalef, J.; Cañellas, S.; James, J.; Judge, E.; Guiry, P.J.; Moberg, C.; Bäckvall, J.-E.; Pfaltz, A.; Pericas, M.A.; et al. Recent Advances in Enantioselective Pd-Catalyzed Allylic Substitution: From Design to Applications. Chem. Rev. 2021, 121, 4373–4505. [Google Scholar] [CrossRef]
- Noreen, S.; Zahoor, A.F.; Ahmad, S.; Shahzadi, I.; Irfan, A.; Faiz, S. Novel Chiral Ligands for Palladium-catalyzed Asymmetric Allylic Alkylation/Asymmetric Tsuji-Trost Reaction: A Review. Curr. Org. Chem. 2019, 23, 1168–1213. [Google Scholar] [CrossRef]
- Siedlecka, R.; Wojaczyńska, E.; Skarżewski, J. Chiral pyrrolidine thioethers: Effective nitrogen–sulfur donating ligands in palladium-catalyzed asymmetric allylic alkylations. Tetrahedron Asymmetry 2004, 15, 1437–1444. [Google Scholar] [CrossRef]
- Zielińska-Błajet, M.; Siedlecka, R.; Skarżewski, J. Chiral phenylselenyl derivatives of pyrrolidine and Cinchona alkaloids: Nitrogen-selenium donating ligands in palladium-catalyzed asymmetric allylic alkylation. Tetrahedron Asymmetry 2007, 18, 131–136. [Google Scholar] [CrossRef]
- Andrade, L.H.; Silva, A.V.; Milani, P.; Koszelewski, D.; Kroutil, W. ω-Transaminases as efficient biocatalysts to obtain novel chiral selenium-amine ligands for Pd-catalysis. Org. Biomol. Chem. 2010, 8, 2043–2051. [Google Scholar] [CrossRef]
- Ścianowski, J.; Pacuła, A.J.; Zielińska-Błajet, M.; Wojtczak, A. New diphenyl diselenides o-substituted by an O(S,Se)-caranyl skeleton—Synthesis and application in asymmetric reactions. New J. Chem. 2016, 40, 6697–6705. [Google Scholar] [CrossRef]
- Sehnem, J.A.; Vargas, F.; Milani, P.; Nascimento, V.; Braga, L.A. Modular Synthesis of Chiral N-Protected β-Seleno Amines and Amides via Cleavage of 2-Oxazolidinones and Application in Palladium-Catalyzed Asymmetric Allylic Alkylation. Synthesis 2008, 8, 1262–1268. [Google Scholar] [CrossRef]
- Margalef, J.; Borras, C.; Alegre, S.; Pamies, O.; Diéguez, M.A. readily accessible and modular carbohydrate-derived thioether/selenoether-phosphite ligand library for Pd-catalyzed asymmetric allylic substitutions. Dalton Trans. 2019, 48, 12632–12643. [Google Scholar] [CrossRef] [PubMed]
- Vargas, F.; Sehnem, J.A.; Galetto, F.Z.; Braga, A.L. Modular chiral β-selenium-, sulfur-, and tellurium amides: Synthesis and application in the palladium-catalyzed asymmetric allylic alkylation. Tetrahedron 2008, 64, 392–398. [Google Scholar] [CrossRef]
- Wosińska-Hyrydczuk, M.; Skarżewski, J. 2-Oxiranyl-pyridines: Synthesis and Regioselective Epoxide Ring Openings with Chiral Amines as a Route to Chiral Ligands. Heteroat. Chem. 2019. [Google Scholar] [CrossRef]
- Wosińska-Hrydczuk, M.; Boratyński, P.J.; Skarżewski, J. Regioselective and Stereodivergent Synthesis of Enantiomerically Pure Vic-Diamines from Chiral β-Amino Alcohols with 2-Pyridyl and 6-(2,2′-Bipyridyl) Moieties. Molecules 2020, 25, 727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hata, T.; Sekine, M. The synthesis of S-phenyl nucleoside phosphorothioates. Chem. Lett. 1974, 15, 837–838. [Google Scholar] [CrossRef]
- Valentine, D.H.; Hillhouse, J.H. Alkyl Phosphines as Reagents and Catalysts in Organic Synthesis. Synthesis 2003, 3, 317–344. [Google Scholar] [CrossRef]
- Grieco, P.A.; Grilman, S.; Nishizawa, M. Organoselenium Chemistry. A Facile One-Step Synthesis of Alkyl Aryl Selenides from Alcohols. J. Org. Chem. 1976, 41, 1485–1486. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Lodewyk, M.W.; Siebert, M.R.; Tantillo, D.J. Computational prediction of 1H and 13C chemical shifts: A useful tool for natural Product, Mechanistic, and Synthetic Organic Chemistry. Chem. Rev. 2012, 112, 1839–1862. [Google Scholar] [CrossRef]
- Liu, G.L.; Chen, W.; Jiang, Q.-Y.; Bai, X.-F.; Li, Z.; Xu, Z.; Xu, L.-W. A D-Camphor-Based Schiff Base as a Highly Efficient N, P Ligand for Enantioselective Palladium-Catalyzed Allylic Substitutions. ChemCatChem 2016, 8, 1495–1499. [Google Scholar] [CrossRef]
- Szabo, K.J. Effects of the Ancillary Ligands on Palladium−Carbon Bonding in (η3−Allyl)palladium Complexes. Implications for Nucleophilic Attack at the Allylic Carbons. Organometallics 1996, 15, 1128–1133. [Google Scholar] [CrossRef]
- Pfaltz, A. From Corrin Chemistry to Asymmetric Catalysis—A Personal Account. Synlett 1999, 835–842. [Google Scholar] [CrossRef]
- Laveson, W.; Reid, G. For the coordination of transition metals to chalcogen ethers. In Comprehensive Coordination Chemistry II; Lever, A.B.P., Ed.; Elsevier: Oxford, UK, 2004; Volume 1, pp. 395–398. [Google Scholar]
- Niu, J.-L.; Wang, M.-C.; Kong, P.-P.; Chen, Q.-T.; Zhu, Y.; Song, M.-P. Origin of enantioselectivity with heterobidentate sulfide-tertiary amine (sp3) ligands in palladium-catalyzed allylic substitution. Tetrahedron 2009, 65, 8869–8878. [Google Scholar] [CrossRef]
- Van Dort, M.E.; Jung, Y.-W.; Sherman, P.S.; Kilbourn, M.R.; Wieland, D.M. Fluorine for Hydroxy Substitution in Biogenic Amines: Asymmetric Synthesis and Biological Evaluation of Fluorine-18-Labeled -Fluorophenylalkylamines as Model Systems. J. Med. Chem. 1995, 38, 810–815. [Google Scholar] [CrossRef]
- Silva, D.R.C.; Maria, E.J.; Suárez Ordóñeza, R.M.; Thierry, J.; Cariou, K.; Dodd, R.H. Synthesis of Orthogonally N-Protected, C-4 Functionalized Cyclic Guanidines from L-Serine. Synlett 2017, 28, 815–818. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sulfonamidates | R1 | R2 | NuH | Yield [%] a | Product 15–19 |
|---|---|---|---|---|---|
| (5S,1′S)-12 | Ph | H | PhSH | 49 | (2R,1′S)-15 |
| (5R,1′S)-12 | Ph | H | PhSH | 21 a | (2S,1′S)-15 |
| (4R,5R,1′S)-13 | Py | Ph | PhSH | 30, 58 a | (1R,2S,1′S)-16 |
| (4S,5S,1′S)-13 | Py | Ph | PhSH | 5 b | (1S,2R,1′S)-16 |
| (4R,5R,1′S)-13 | Py | Ph | PhCH2SH | 47 a | (1R,2S,1′S)-17 |
| (4R,5R,1′S)-14 | Bpy | Ph | PhSH | 56 a | (1R,2S,1′S)-18 |
| (4R,5R,1′S)-13 | Py | Ph | PhSeH | 10 a | (1R,2S,1′S)-19 |

| Chiral Diamine | R1 | R2 | R3 | Yield [%] | Product |
|---|---|---|---|---|---|
| (1S,2R,1′S)-20 | Py | Ph | Me | 70 | (1S,2R,1′S)-22 |
| (2R,1′S)-21 | Ph | H | tBu | 60 | (2R,1′S)-23 |
| (2R,1′S)-21 | Ph | H | Me | 68 | (2R,1′S)-24 |

| Chiral Ligand | Conversion [%] a | Ee [%] Configuration |
|---|---|---|
| (2R,1′S)-15 | 15 | Rac |
| (2S,1′S)-15 | 30 | 31 (R) |
| (1R,2S,1′S)-16 | 100 | 58 (S) |
| (1R,2S,1′S)-17 | 98 | 26 (S) |
| (1R,2S,1′S)-18 | 89 | 13 (R) |
| (1R,2S,1′S)-19 | 70 | 50 (S) |
| (1S,2R,1′S)-22 | 98 b | 49 (S) |
| (2R,1′S)-23 | 66 b | 38 (R) |
| (2R,1′S)-24 | 69 b | 38 (R) |
| Chiral Ligand | Temperature [°C] | Conversion [%] | Ee [%] Configuration | Time [h] |
|---|---|---|---|---|
| (1R,2S,1′S)-16 | 0–4 | 97 | 70 (S) | 48 |
| (1R,2S,1′S)-16 | −18 | 75 | 75 (S) | 96 |
| (1R,2S,1′S)-16 | −30 | 5 | 73 (S) | 10 |
| (1S,2R,1′S)-22 | −18 | 96 | 71 (S) | 96 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wosińska-Hrydczuk, M.; Skarżewski, J. New Nitrogen, Sulfur-, and Selenium-Donating Ligands Derived from Chiral Pyridine Amino Alcohols. Synthesis and Catalytic Activity in Asymmetric Allylic Alkylation. Molecules 2021, 26, 3493. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26123493
Wosińska-Hrydczuk M, Skarżewski J. New Nitrogen, Sulfur-, and Selenium-Donating Ligands Derived from Chiral Pyridine Amino Alcohols. Synthesis and Catalytic Activity in Asymmetric Allylic Alkylation. Molecules. 2021; 26(12):3493. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26123493
Chicago/Turabian StyleWosińska-Hrydczuk, Marzena, and Jacek Skarżewski. 2021. "New Nitrogen, Sulfur-, and Selenium-Donating Ligands Derived from Chiral Pyridine Amino Alcohols. Synthesis and Catalytic Activity in Asymmetric Allylic Alkylation" Molecules 26, no. 12: 3493. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26123493