
Synthesis and Characterization of Novel Methyl (3)5-(N-Boc-piperidinyl)-1H-pyrazole-4-carboxylates

 , ,
, ,
Abstract
:
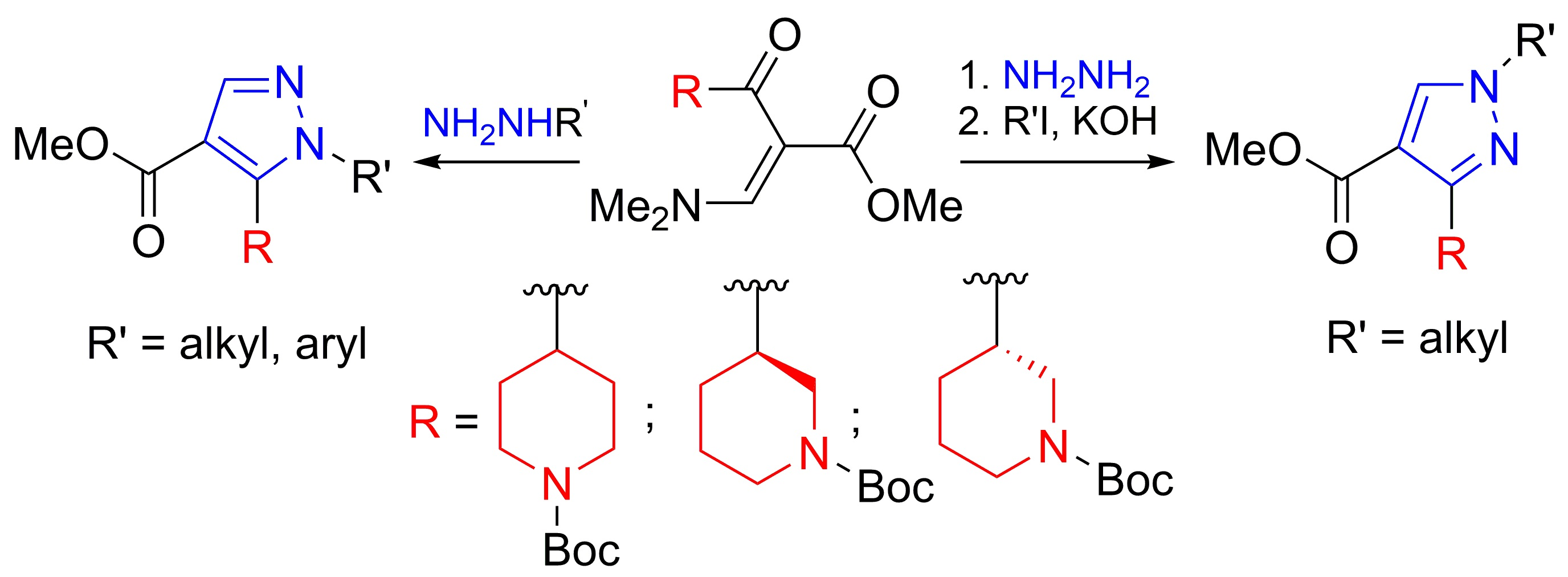
1. Introduction
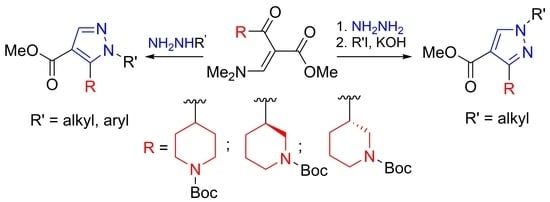
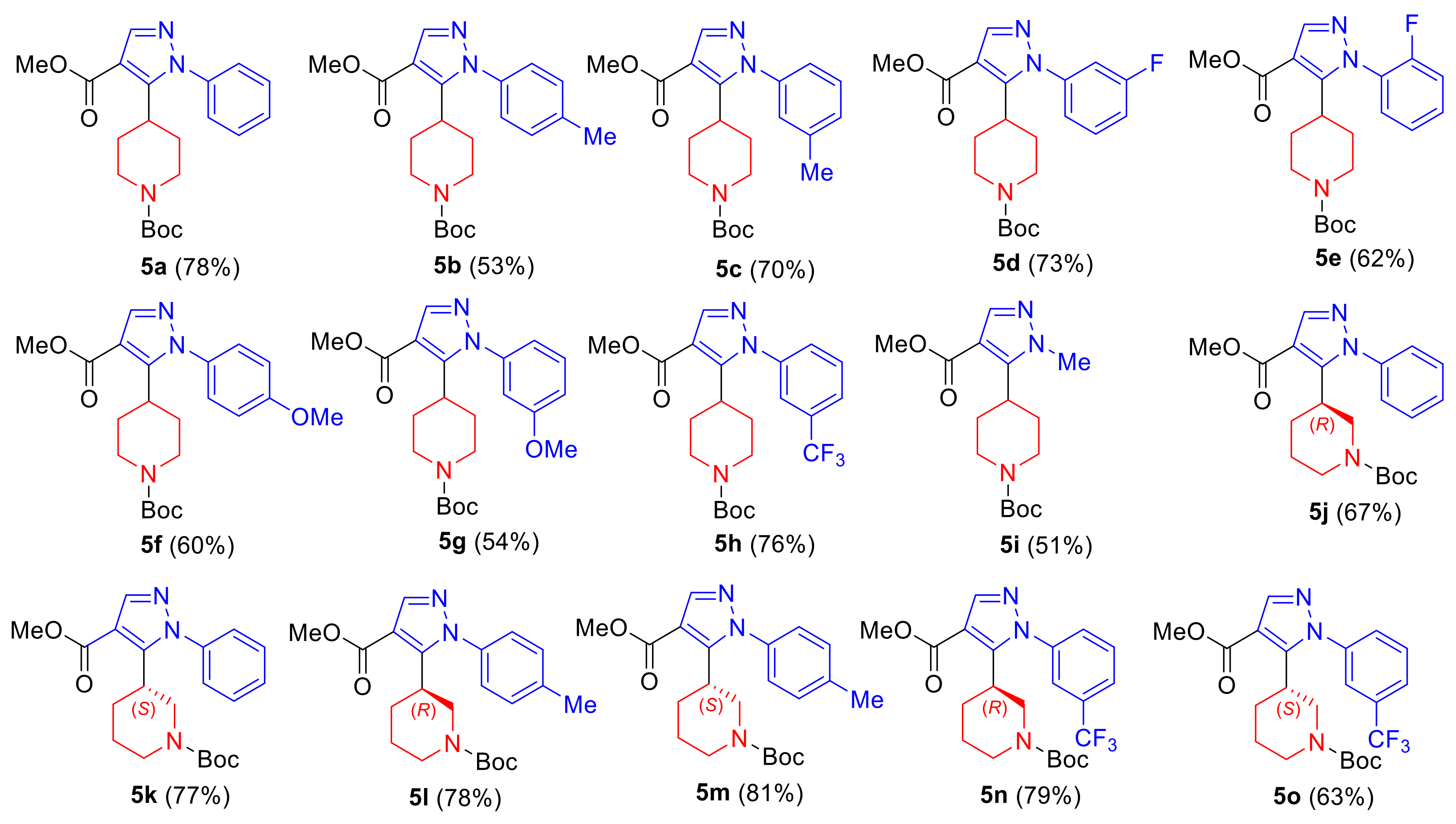
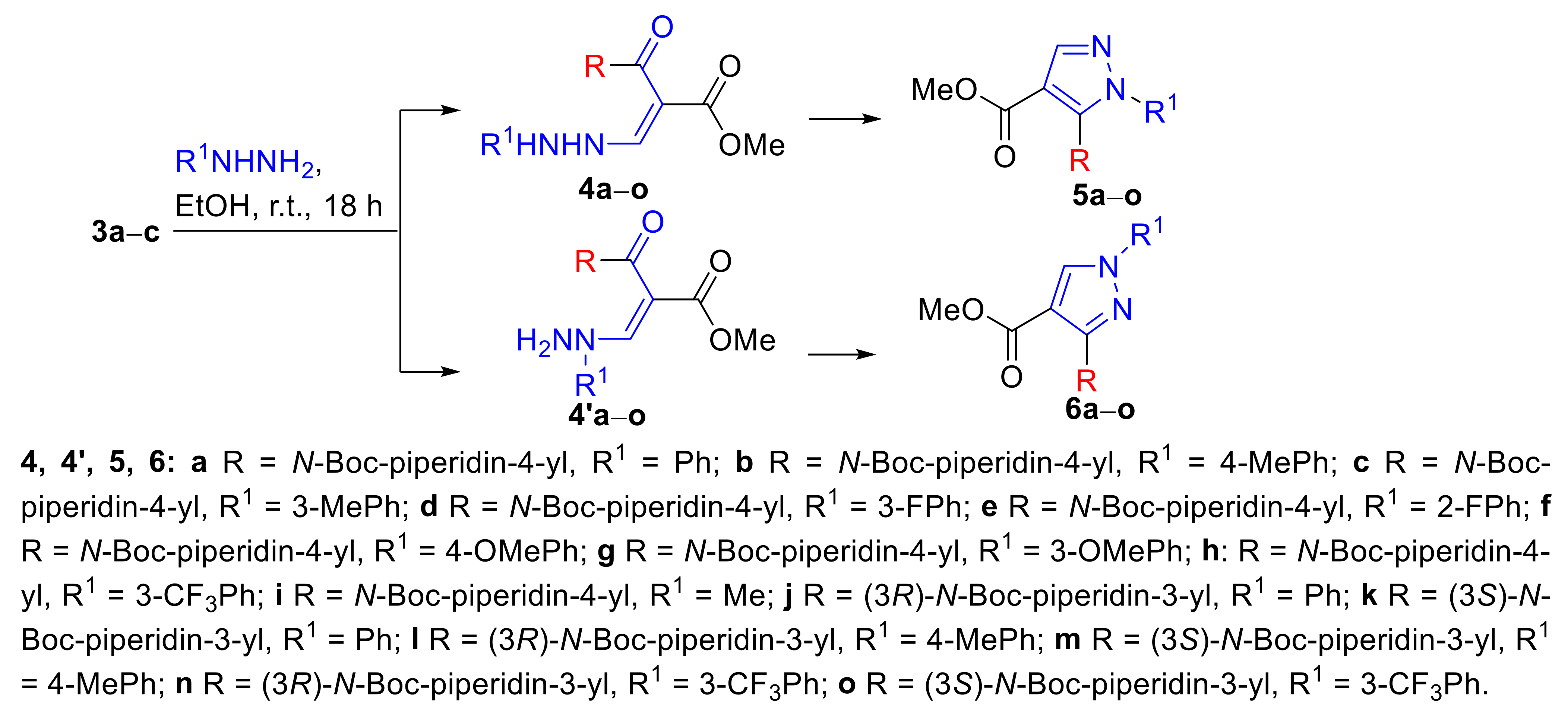
2. Results and Discussion
3. Materials and Methods
3.1. General Information
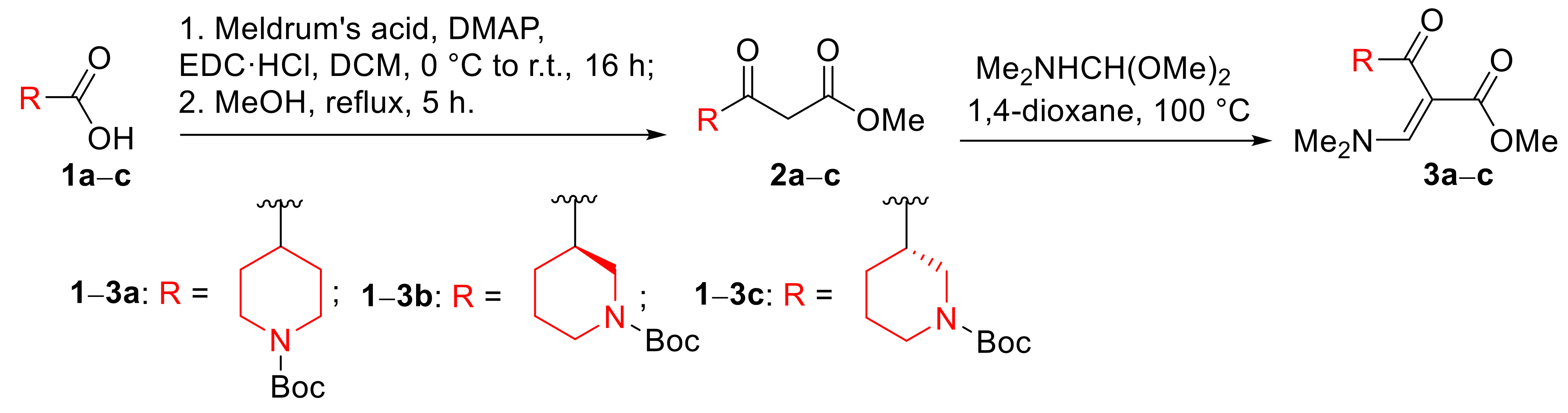
3.2. Synthesis of tert-Butyl 3- and 4-[(2)-3-(Dimethylamino)-2-(methoxycarbonyl)prop-2-enoyl]piperidine-1-carboxylates (3a–c)
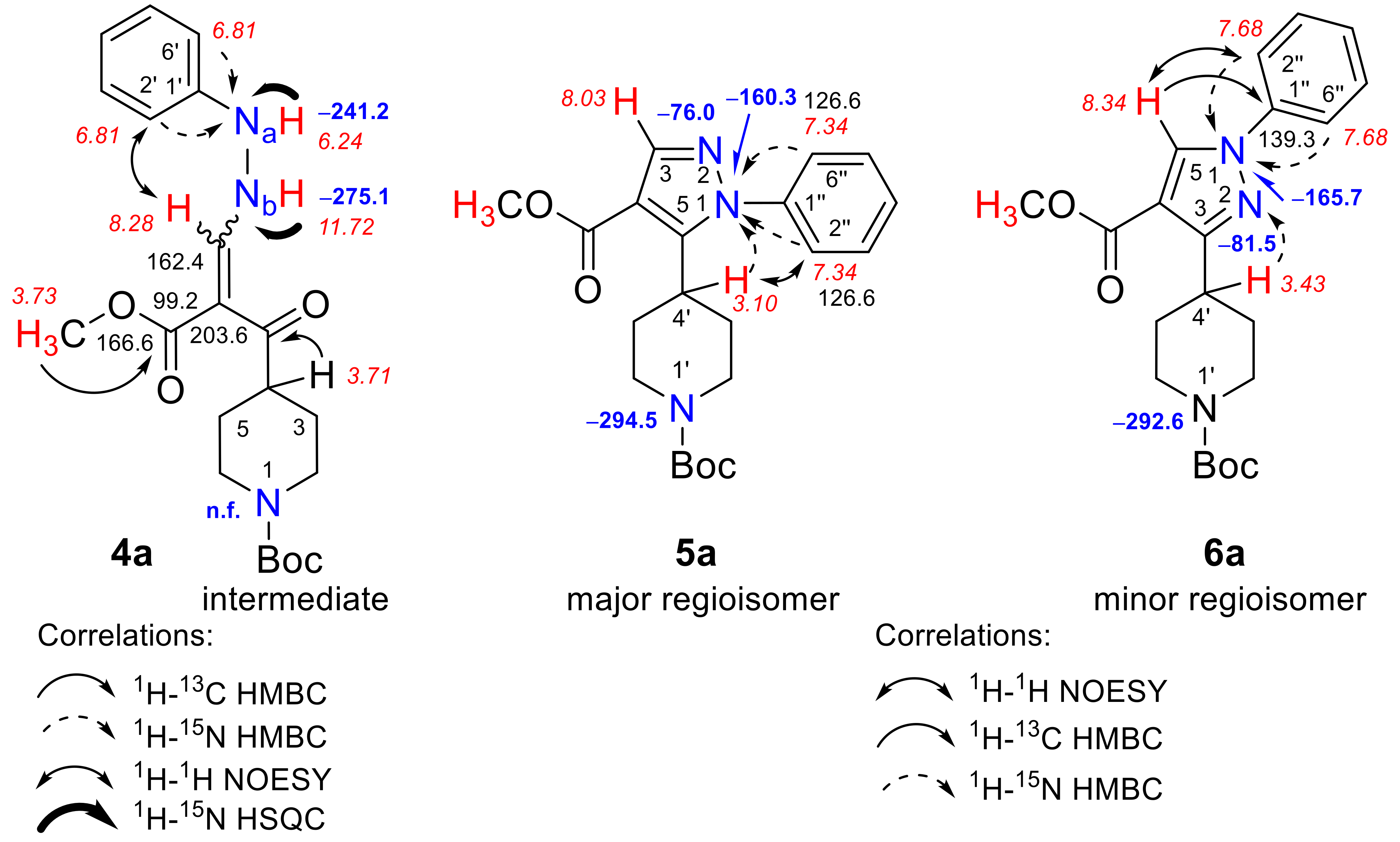
3.3. Synthesis Procedure for the Preparation of Compounds 4a, 5a, and 6a
3.3.1. tert-Butyl 4-[(2E(Z))-2-(methoxycarbonyl)-3-(2-phenylhydrazinyl)prop-2-enoyl]piperidine-1-carboxylate (4a)
3.3.2. tert-Butyl 4-[4-(methoxycarbonyl)-1-phenyl-1H-pyrazol-5-yl]piperidine-1-carboxylate (5a)
3.3.3. tert-Butyl 4-[4-(methoxycarbonyl)-1-phenyl-1H-pyrazol-3-yl]piperidine-1-carboxylate (6a)
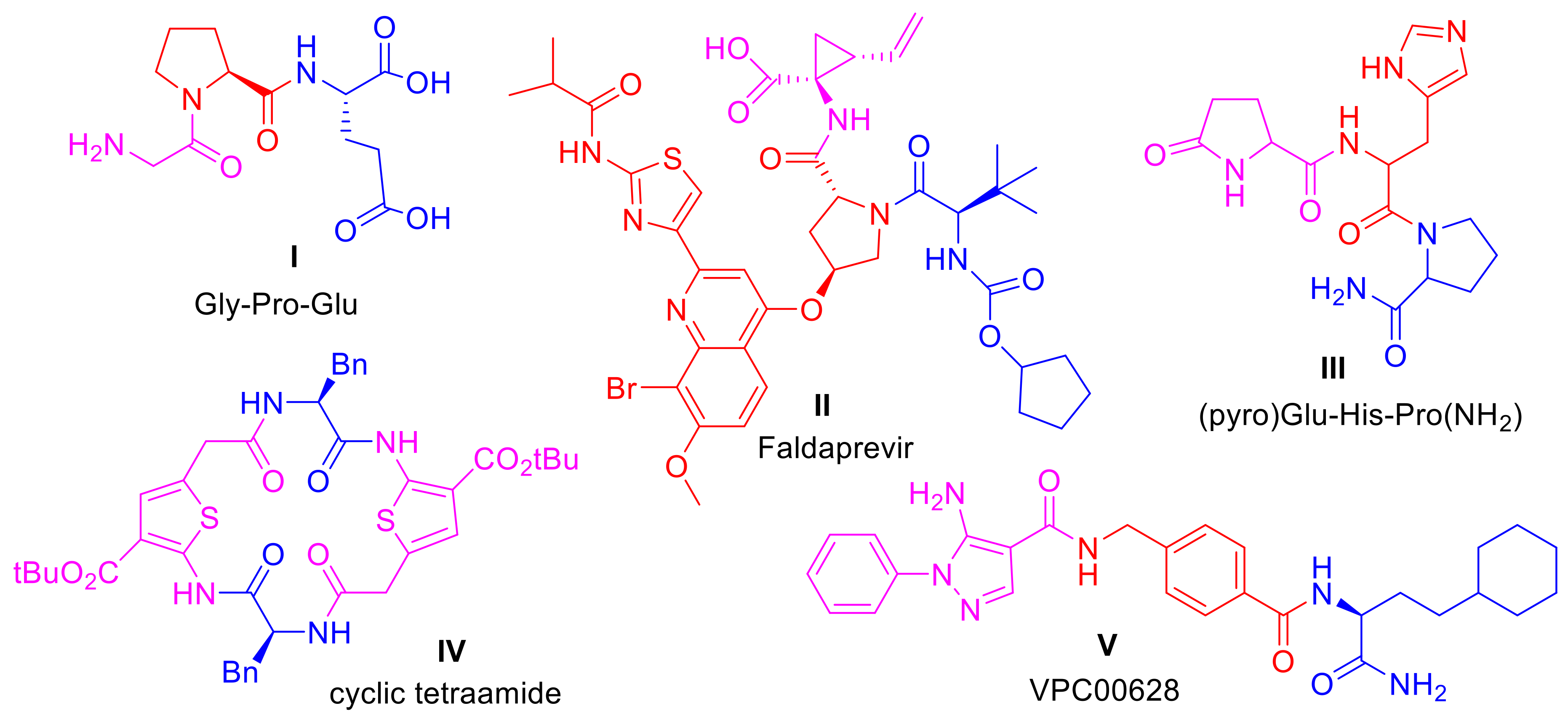
3.4. Synthesis of tert-Butyl 4-[4-(methoxycarbonyl)-1H-pyrazol-5-yl]piperidine-1-carboxylates (5b–o)
3.4.1. tert-Butyl 4-[4-(methoxycarbonyl)-1-(4-methylphenyl)-1H-pyrazol-5-yl]piperidine-1-carboxylate (5b)
3.4.2. tert-Butyl 4-[4-(methoxycarbonyl)-1-(3-methylphenyl)-1H-pyrazol-5-yl]piperidine-1-carboxylate (5c)
3.4.3. tert-Butyl 4-[1-(3-fluorophenyl)-4-(methoxycarbonyl)-1H-pyrazol-5-yl]piperidine-1-carboxylate (5d)
3.4.4. tert-Butyl 4-[1-(2-fluorophenyl)-4-(methoxycarbonyl)-1H-pyrazol-5-yl]piperidine-1-carboxylate (5e)
3.4.5. tert-Butyl 4-[4-(methoxycarbonyl)-1-(4-methoxyphenyl)-1H-pyrazol-5-yl]piperidine-1-carboxylate (5f)
3.4.6. tert-Butyl 4-[4-(methoxycarbonyl)-1-(3-methoxyphenyl)-1H-pyrazol-5-yl]piperidine-1-carboxylate (5g)
3.4.7. tert-Butyl 4-{4-(methoxycarbonyl)-1-[3-(trifluoromethyl)phenyl]-1H-pyrazol-5-yl}piperidine-1-carboxylate (5h)
3.4.8. tert-Butyl 4-[4-(methoxycarbonyl)-1-methyl-1H-pyrazol-5-yl]piperidine-1-carboxylate (5i)
3.4.9. tert-Butyl (3R)-3-[4-(methoxycarbonyl)-1-phenyl-1H-pyrazol-5-yl]piperidine-1-carboxylate (5j)
3.4.10. tert-Butyl (3S)-3-[4-(methoxycarbonyl)-1-phenyl-1H-pyrazol-5-yl]piperidine-1-carboxylate (5k)
3.4.11. tert-Butyl (3R)-3-[4-(methoxycarbonyl)-1-(4-methylphenyl)-1H-pyrazol-5-yl]piperidine-1-carboxylate (5l)
3.4.12. tert-Butyl (3S)-3-[4-(methoxycarbonyl)-1-(4-methylphenyl)-1H-pyrazol-5-yl]piperidine-1-carboxylate (5m)
3.4.13. tert-Butyl (3R)-3-{4-(methoxycarbonyl)-1-[3-(trifluoromethyl)phenyl]-1H-pyrazol-5-yl}piperidine-1-carboxylate (5n)
3.4.14. tert-Butyl (3S)-3-{4-(methoxycarbonyl)-1-[3-(trifluoromethyl)phenyl]-1H-pyrazol-5-yl}piperidine-1-carboxylate (5o)
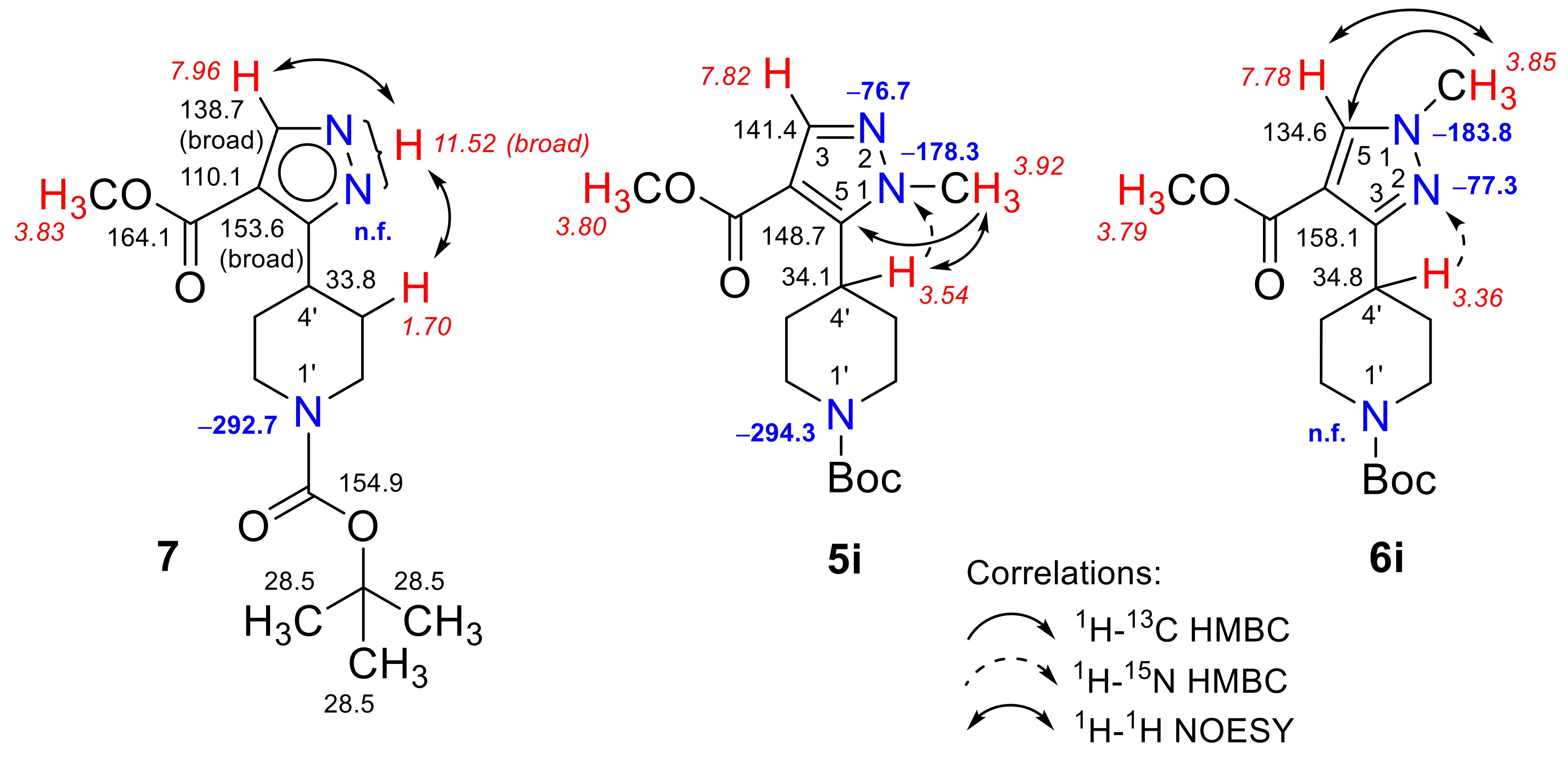
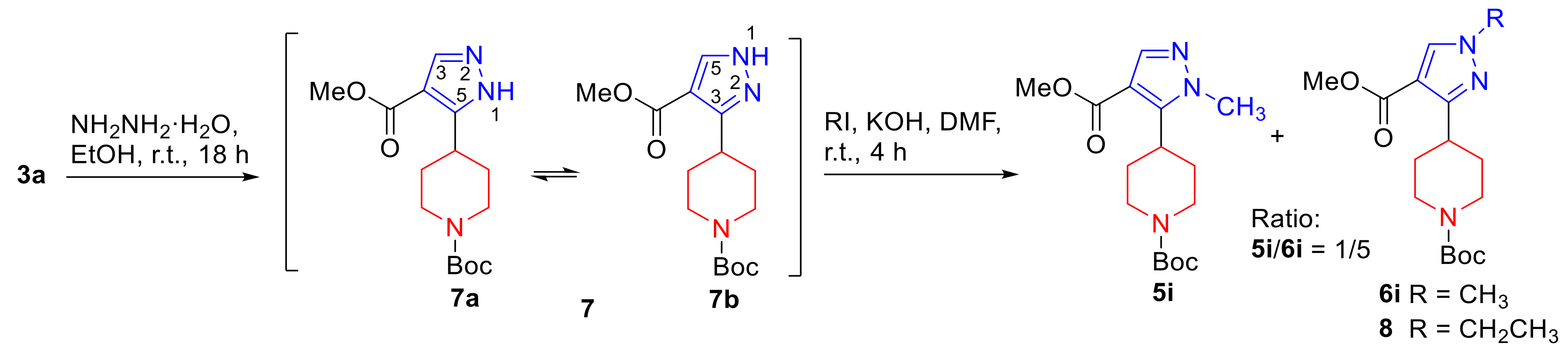
3.5. Synthesis of tert-Butyl 4-[4-(methoxycarbonyl)-1H-pyrazolyl]piperidine-1-carboxylate (7)
3.6. Synthesis of tert-Butyl 4-[4-(methoxycarbonyl)-1H-pyrazolyl]piperidine-1-carboxylates (5i, 6i, 8)
3.6.1. tert-Butyl 4-[4-(methoxycarbonyl)-1-methyl-1H-pyrazol-3-yl]piperidine-1-carboxylate (6i) and tert-Butyl 4-[4-(methoxycarbonyl)-1-methyl-1H-pyrazol-5-yl]piperidine-1-carboxylate (5i)
3.6.2. tert-Butyl 4-[1-ethyl-4-(methoxycarbonyl)-1H-pyrazol-3-yl]piperidine-1-carboxylate (8)
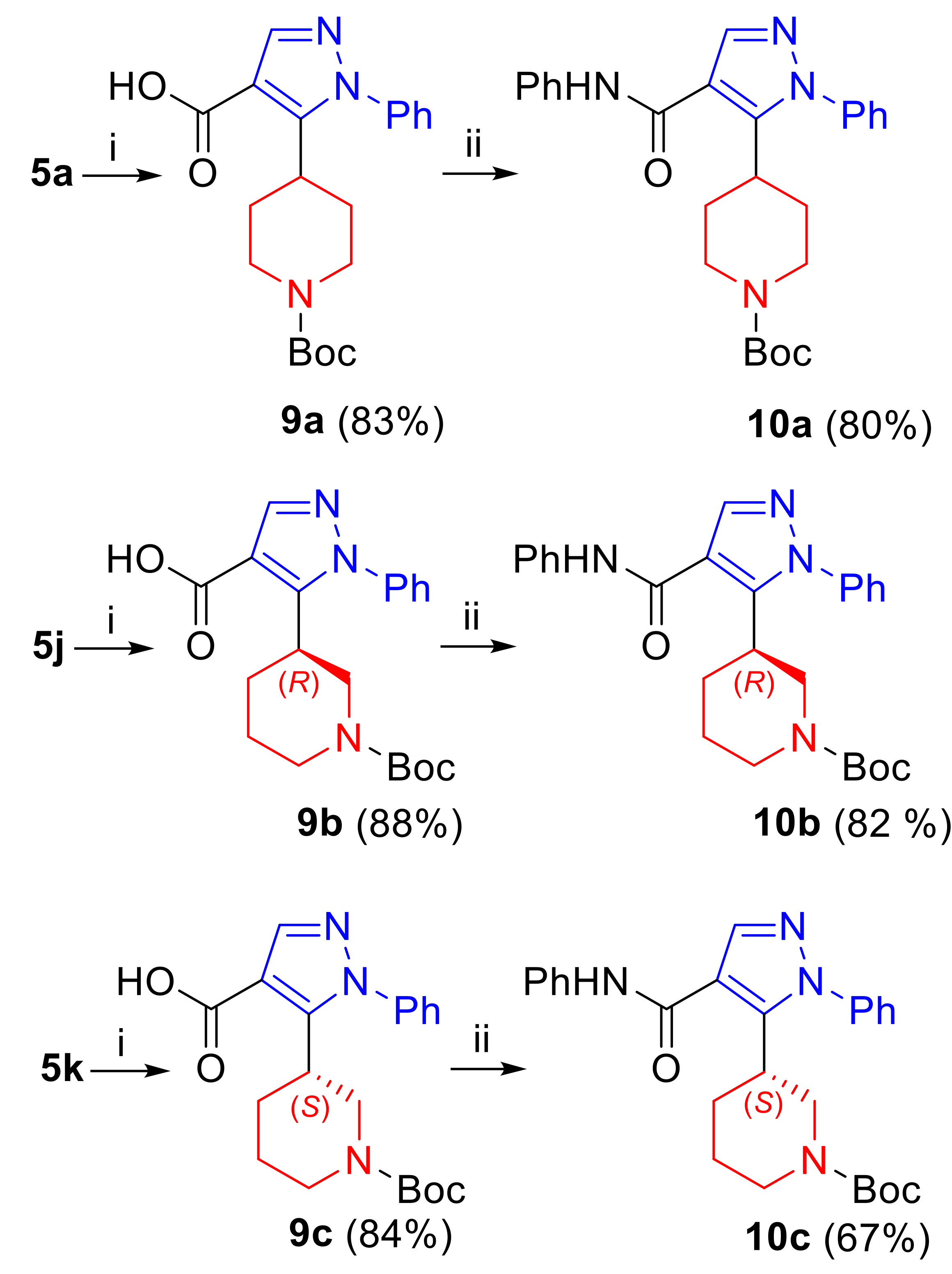
3.7. Synthesis of 5-[1-(tert-Butoxycarbonyl)piperidinyl]-1H-pyrazole-4-carboxylic acids (9a–c)
3.7.1. 5-[1-(tert-Butoxycarbonyl)piperidin-4-yl]-1-phenyl-1H-pyrazole-4-carboxylic Acid (9a)
3.7.2. 5-[(3R)-1-(tert-Butoxycarbonyl)piperidin-3-yl]-1-phenyl-1H-pyrazole-4-carboxylic Acid (9b)
3.7.3. 5-[(3S)-1-(tert-Butoxycarbonyl)piperidin-3-yl]-1-phenyl-1H-pyrazole-4-carboxylic Acid (9c)
3.8. Synthesis of tert-Butyl 3- and 4-[4-(Phenylcarbamoyl)-1H-pyrazol-5-yl]piperidine-1-carboxylates (10a–c)
3.8.1. tert-Butyl 4-[1-phenyl-4-(phenylcarbamoyl)-1H-pyrazol-5-yl]piperidine-1-carboxylate (10a)
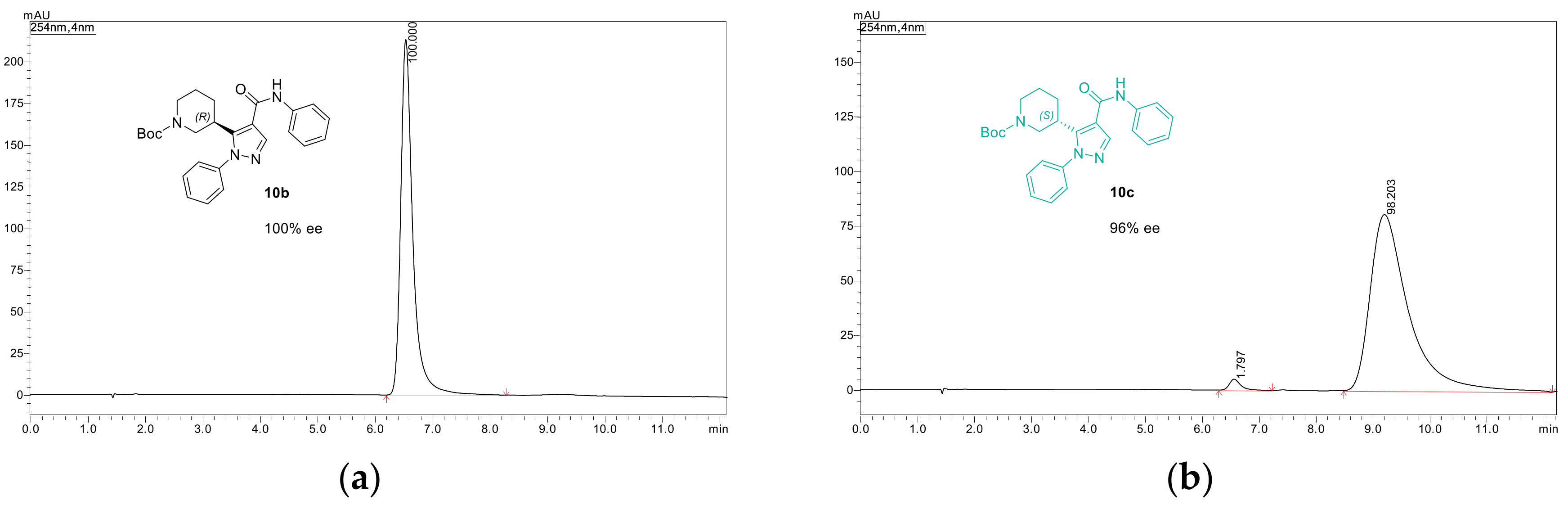
3.8.2. tert-Butyl (3R)-3-[1-phenyl-4-(phenylcarbamoyl)-1H-pyrazol-5-yl]piperidine-1-carboxylate (10b)
3.8.3. tert-Butyl (3S)-3-[1-phenyl-4-(phenylcarbamoyl)-1H-pyrazol-5-yl]piperidine-1-carboxylate (10c)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Singh, P.; Samanta, K.; Das, S.K.; Panda, G. Amino acid chirons: A tool for asymmetric synthesis of heterocycles. Org. Biomol. Chem. 2014, 12, 6297–6339. [Google Scholar] [CrossRef] [PubMed]
- Sardina, F.J.; Rapoport, H. Enantiospecific Synthesis of Heterocycles from α-Amino Acids. Chem. Rev. 1996, 96, 1825–1872. [Google Scholar] [CrossRef] [PubMed]
- Blaskovich, M.A.T. Unusual Amino Acids in Medicinal Chemistry. J. Med. Chem. 2016, 59, 10807–10836. [Google Scholar] [CrossRef] [PubMed]
- Žukauskaitė, A.; Moretto, A.; Peggion, C.; De Zotti, M.; Šačkus, A.; Formaggio, F.; De Kimpe, N.; Mangelinckx, S. Synthesis and Conformational Study of Model Peptides Containing N-Substituted 3-Aminoazetidine-3-carboxylic Acids. Eur. J. Org. Chem. 2014, 2014, 2312–2321. [Google Scholar] [CrossRef]
- Vale, N.; Ferreira, A.; Matos, J.; Fresco, P.; Gouveia, M.J. Amino Acids in the Development of Prodrugs. Molecules 2018, 23, 2318. [Google Scholar] [CrossRef]
- Bonina, F.P.; Arenare, L.; Palagiano, F.; Saija, A.; Nava, F.A.; Trombetta, D.; De Caprariis, P. Synthesis, stability, and pharmacological evaluation of nipecotic acid prodrugs. J. Pharm. Sci. 1999, 88, 561–567. [Google Scholar] [CrossRef]
- Henzi, V.; Reichling, D.B.; Helm, S.W.; MacDermott, A.B. L-proline activates glutamate and glycine receptors in cultured rat dorsal horn neurons. Mol. Pharmacol. 1992, 41, 793–801. [Google Scholar]
- Wang, M.; Rakesh, K.; Leng, J.; Fang, W.-Y.; Ravindar, L.; Gowda, D.C.; Qin, H.-L. Amino acids/peptides conjugated heterocycles: A tool for the recent development of novel therapeutic agents. Bioorg. Chem. 2018, 76, 113–129. [Google Scholar] [CrossRef]
- Walsh, C.T.; O’Brien, R.V.; Khosla, C. Nonproteinogenic Amino Acid Building Blocks for Nonribosomal Peptide and Hybrid Polyketide Scaffolds. Angew. Chem. Int. Ed. 2013, 52, 7098–7124. [Google Scholar] [CrossRef]
- Cerminara, I.; Chiummiento, L.; Funicello, M.; Guarnaccio, A.; Lupattelli, P. Heterocycles in Peptidomimetics and Pseudopeptides: Design and Synthesis. Pharmaceuticals 2012, 5, 297–316. [Google Scholar] [CrossRef]
- Hollis, S.J. Heterocyclic in Peptide Chemistry. Ph. D. Thesis, The Open University, Milton Keynes, UK, 2000. [Google Scholar]
- Kothapalli, Y.; Puthukanoori, R.K.; Alapati, S.R. Enantiospecific first total synthesis of 7a(S)-p-hydroxyphenopyrrozin. Tetrahedron Lett. 2012, 53, 1891–1893. [Google Scholar] [CrossRef]
- Lin, C.-H.; Hong, B.-C.; Lee, G.-H. Asymmetric synthesis of functionalized pyrrolizidines by an organocatalytic and pot-economy strategy. RSC Adv. 2016, 6, 8243–8247. [Google Scholar] [CrossRef]
- Luna-Freire, K.R.; Scaramal, J.P.S.; Resende, J.A.L.C.; Tormena, C.F.; Oliveira, F.L.; Aparicio, R.; Coelho, F. An asymmetric substrate-controlled Morita–Baylis–Hillman reaction as approach for the synthesis of pyrrolizidinones and pyrrolizidines. Tetrahedron 2014, 70, 3319–3326. [Google Scholar] [CrossRef]
- Dockerty, P.; Edens, J.G.; Tol, M.B.; Angeles, D.M.; Domenech, A.; Liu, Y.; Hirsch, A.K.H.; Veening, J.-W.; Scheffers, D.-J.; Witte, M.D. Bicyclic enol cyclocarbamates inhibit penicillin-binding proteins. Org. Biomol. Chem. 2016, 15, 894–910. [Google Scholar] [CrossRef]
- Lu, X.; Shi, L.; Zhang, H.; Jiang, Y.; Ma, D. Assembly of N-substituted pyrrolo[2,1-c][1,4]benzodiazepine-5,11-diones via copper catalyzed aryl amination. Tetrahedron 2010, 66, 5714–5718. [Google Scholar] [CrossRef]
- Wang, H.; Jiang, Y.; Gao, K.; Ma, D. Facile synthesis of 1,4-benzodiazepin-3-ones from o-bromobenzylamines and amino acids via a cascade coupling/condensation process. Tetrahedron 2009, 65, 8956–8960. [Google Scholar] [CrossRef]
- Villalgordo, J.M.; Heimgartner, H. Synthesis of a novel heterospirocyclic 3-(N-methyl-N-phenylamino)-2H-azirine and its use as an amino acid equivalent in the preparation of a model tripeptide. Tetrahedron 1993, 49, 7215–7222. [Google Scholar] [CrossRef]
- Heimgartner, H.; Strässler, C.; Linden, A. Synthesis of Tripeptides Containing Heterocyclic α-Amino Acids by Using Heterospirocyclic 3-Amino-2H-azirines. Heterocycles 2018, 97, 333. [Google Scholar] [CrossRef]
- Shimazaki, M.; Hasegawa, J.; Kan, K.; Nomura, K.; Nose, Y.; Kondo, H.; Ohashi, T.; Watanabe, K. Synthesis of captopril starting from an optically active.BETA.-hydroxy acid. Chem. Pharm. Bull. 1982, 30, 3139–3146. [Google Scholar] [CrossRef]
- Luer, M.S.; Rhoney, D.H. Tiagabine: A Novel Antiepileptic Drug. Ann. Pharmacother. 1998, 32, 1173–1180. [Google Scholar] [CrossRef]
- Leach, J.P.; Brodie, M.J. Tiagabine. Lancet 1998, 351, 203–207. [Google Scholar] [CrossRef]
- Chorghade, M.S.; Ellegaard, P.; Lee, E.C.; Petersen, H.; Sørensen, P.O. Synthesis of Desmethyl Tiagabine. Heterocycles 1994, 37, 783. [Google Scholar] [CrossRef]
- Andersen, K.E.; Sørensen, J.L.; Lau, J.; Lundt, B.F.; Petersen, H.; Huusfeldt, P.O.; Suzdak, P.D.; Swedberg, M.D.B. Synthesis of Novel γ-Aminobutyric Acid (GABA) Uptake Inhibitors. 5.1Preparation and Structure−Activity Studies of Tricyclic Analogues of Known GABA Uptake Inhibitors. J. Med. Chem. 2001, 44, 2152–2163. [Google Scholar] [CrossRef] [PubMed]
- Seth, A.; Sharma, P.A.; Tripathi, A.; Choubey, P.K.; Srivastava, P.; Tripathi, P.N.; Shrivastava, S.K. Design, synthesis, evaluation and molecular modeling studies of some novel N-substituted piperidine-3-carboxylic acid derivatives as potential anticonvulsants. Med. Chem. Res. 2018, 27, 1206–1225. [Google Scholar] [CrossRef]
- Schaarschmidt, M.; Höfner, G.; Wanner, K.T. Synthesis and Biological Evaluation of Nipecotic Acid and Guvacine Derived 1,3-Disubstituted Allenes as Inhibitors of Murine GABA Transporter mGAT1. ChemMedChem 2019, 14, 1135–1151. [Google Scholar] [CrossRef]
- Saura, J.; Curatolo, L.; Williams, C.E.; Gatti, S.; Benatti, L.; Peeters, C.; Guan, J.; Dragunow, M.; Post, C.; Faull, R.L.M.; et al. Neuroprotective effects of Gly-Pro-Glu, the N- terminal tripeptide of IGF-1, in the hippocampus in vitro. NeuroReport 1999, 10, 161–164. [Google Scholar] [CrossRef]
- Minelli, A.; Conte, C.; Cacciatore, I.; Cornacchia, C.; Pinnen, F. Molecular mechanism underlying the cerebral effect of Gly-Pro-Glu tripeptide bound to l-dopa in a Parkinson’s animal model. Amino Acids 2012, 43, 1359–1367. [Google Scholar] [CrossRef]
- Kanda, T.; Yokosuka, O.; Omata, M. Faldaprevir for the Treatment of Hepatitis C. Int. J. Mol. Sci. 2015, 16, 4985–4996. [Google Scholar] [CrossRef]
- White, P.W.; Llinàs-Brunet, M.; Amad, M.; Bethell, R.C.; Bolger, G.; Cordingley, M.G.; Duan, J.; Garneau, M.; Lagacé, L.; Thibeault, D.; et al. Preclinical Characterization of BI 201335, a C-Terminal Carboxylic Acid Inhibitor of the Hepatitis C Virus NS3-NS4A Protease. Antimicrob. Agents Chemother. 2010, 54, 4611–4618. [Google Scholar] [CrossRef]
- Bilodeau, F.; Bailey, M.D.; Bhardwaj, P.K.; Bordeleau, J.; Forgione, P.; Garneau, M.; Ghiro, E.; Gorys, V.; Halmos, T.; Jolicoeur, E.S.; et al. Synthesis and optimization of a novel series of HCV NS3 protease inhibitors: 4-Arylproline analogs. Bioorg. Med. Chem. Lett. 2013, 23, 4267–4271. [Google Scholar] [CrossRef]
- Folkers, K.; Enzmann, F.H. Tripeptidine Having the Activity of the Thyrotropin Releasing Hormone. U.S. Patent 3,746,697, 17 July 1973. [Google Scholar]
- Ngo, H.T.; Berndt, H.; Wilsdorf, M.; Lentz, D.; Reissig, H.-U. Linear and Cyclic Hybrids of Alternating Thiophene-Amino Acid Units: Synthesis and Effects of Chirality on Conformation and Molecular Packing. Chem. A Eur. J. 2013, 19, 15155–15165. [Google Scholar] [CrossRef]
- Raja, R.; Hemaiswarya, S.; Ganesan, V.; de Carvalho, I.S. Recent developments in therapeutic applications of Cyanobacteria. Crit. Rev. Microbiol. 2015, 42, 1–12. [Google Scholar] [CrossRef]
- Shi, B.; Zhou, Y.; Huang, Y.; Zhang, J.; Li, X. Recent advances on the encoding and selection methods of DNA-encoded chemical library. Bioorg. Med. Chem. Lett. 2017, 27, 361–369. [Google Scholar] [CrossRef]
- Usanov, D.L.; Chan, A.I.; Maianti, J.P.; Liu, D.R. Second-generation DNA-templated macrocycle libraries for the discovery of bioactive small molecules. Nat. Chem. 2018, 10, 704–714. [Google Scholar] [CrossRef]
- Madsen, D.; Azevedo, C.; Micco, I.; Petersen, L.K.; Vest Hansen, N.J. An overview of DNA-encoded libraries: A versatile tool for drug discovery. Prog. Med. Chem. 2020, 59, 181–249. [Google Scholar] [CrossRef]
- Blakskjaer, P.; Heitner, T.; Vest Hansen, N.J. Fidelity by design: Yoctoreactor and binder trap enrichment for small-molecule DNA-encoded libraries and drug discovery. Curr. Opin. Chem. Biol. 2015, 26, 62–71. [Google Scholar] [CrossRef]
- Hansen, M.H.; Blakskjær, P.; Petersen, L.K.; Hansen, T.H.; Hojfeldt, J.; Gothelf, K.V.; Vest Hansen, N.J. A Yoctoliter-Scale DNA Reactor for Small-Molecule Evolution. J. Am. Chem. Soc. 2009, 131, 1322–1327. [Google Scholar] [CrossRef]
- Galloway, W.R.; Isidro-Llobet, A.; Spring, D.R. Diversity-oriented synthesis as a tool for the discovery of novel biologically active small molecules. Nat. Commun. 2010, 1, 80. [Google Scholar] [CrossRef]
- Petersen, L.K.; Blakskjær, P.; Chaikuad, A.; Christensen, A.; Dietvorst, J.; Holmkvist, J.; Knapp, S.; Korinek, M.; Larsen, L.K.; Pedersen, A.; et al. Novel p38α MAP kinase inhibitors identified from yoctoReactor DNA-encoded small molecule library. MedChemComm 2016, 7, 1332–1339. [Google Scholar] [CrossRef]
- Iškauskienė, M.; Ragaitė, G.; Sløk, F.A.; Šačkus, A. Facile synthesis of novel amino acid-like building blocks by N-alkylation of heterocyclic carboxylates with N-Boc-3-iodoazetidine. Mol. Divers. 2020, 24, 1235–1251. [Google Scholar] [CrossRef]
- Malinauskienė, V.; Kveselytė, A.; Dzedulionytė, K.; Bieliauskas, A.; Burinskas, S.; Sløk, F.A.; Šačkus, A. L-Proline and related chiral heterocyclic amino acids as scaffolds for the synthesis of functionalized 2-amino-1,3-selenazole-5-carboxylates. Chem. Heterocycl. Compd. 2018, 54, 469–473. [Google Scholar] [CrossRef]
- Sackus, A.; Kveselyte, A.; Malinauskiene, V.; Dzedulionyte, K.; Bieliauskas, A.; Burinskas, S.; Krikstolaityte, S.; Slok, F.A. Heterocyclic amino acids as scaffolds for the synthesis of functionalized chiral 1,3-thiazole and 1,3-selenazole derivatives. In Proceedings of the 256th National Meeting and Exposition of the American-Chemical-Society (ACS), Boston, MA, USA, 19–23 August 2018; p. 586. [Google Scholar]
- Fustero, S.; Sánchez-Roselló, M.; Barrio, P.; Fuentes, A.S. From 2000 to Mid-2010: A Fruitful Decade for the Synthesis of Pyrazoles. Chem. Rev. 2011, 111, 6984–7034. [Google Scholar] [CrossRef] [PubMed]
- Karrouchi, K.; Radi, S.; Ramli, Y.; Taoufik, J.; Mabkhot, Y.N.; Al-Aizari, F.A.; Ansar, M. Synthesis and Pharmacological Activities of Pyrazole Derivatives: A Review. Molecules 2018, 23, 134. [Google Scholar] [CrossRef] [PubMed]
- Ansari, A.; Ali, A.; Asif, M.; Shamsuzzaman, S. Review: Biologically active pyrazole derivatives. New J. Chem. 2016, 41, 16–41. [Google Scholar] [CrossRef]
- Souza, T.F.; Silva, M.J.V.; Silva, R.G.M.; Gonçalves, D.S.; Simon, P.A.; Jacomini, A.P.; Basso, E.A.; Moura, S.; Martins, M.A.P.; Back, D.F.; et al. Regiochemical Control of Pyrazoles by Solvent and β-Enamino Diketone Structure: Regioselective Synthesis of 4,5-Disubstituted N -Phenylpyrazoles. Asian J. Org. Chem. 2017, 6, 627–633. [Google Scholar] [CrossRef]
- Gao, W.; Lau, T.; Pan, S.; Phillips, D.P.; Wang, X. Compounds and Compositions as TGR5 Agonists. WO Patent WO2012/082947A1, 14 December 2011. [Google Scholar]
- Brooks, D.W.; Lu, L.D.-L.; Masamune, S. C-Acylation under Virtually Neutral Conditions. Angew. Chem. Int. Ed. 1979, 18, 72–74. [Google Scholar] [CrossRef]
- Thomassigny, C.; Le Bouc, G.; Greck, C. Asymmetric syntheses of functionalized pyrrolizidin-3-ones. Arkivoc 2012, 2012, 231. [Google Scholar] [CrossRef]
- Semenov, V.; Samultsev, D.O.; Krivdin, L.B. Substitution effects in the 15N NMR chemical shifts of heterocyclic azines evaluated at the GIAO-DFT level. Magn. Reson. Chem. 2018, 56, 767–774. [Google Scholar] [CrossRef]
- Chernyshev, K.A.; Semenov, V.A.; Krivdin, L.B. Quantum-chemical calculations of NMR chemical shifts of organic molecules: VIII. Solvation effects on 15N NMR chemical shifts of nitrogen-containing heterocycles. Russ. J. Org. Chem. 2013, 49, 379–383. [Google Scholar] [CrossRef]
- Claramunt, R.M.; López, C.; Maria, M.D.S.; Sanz, D.; Elguero, J. The Use of NMR Spectroscopy to Study Tautomerism. ChemInform 2007, 38, 169–206. [Google Scholar] [CrossRef]
- Secrieru, A.; O’Neill, P.M.; Cristiano, M.L.S. Revisiting the Structure and Chemistry of 3(5)-Substituted Pyrazoles. Molecules 2019, 25, 42. [Google Scholar] [CrossRef]
- Arbačiauskienė, E.; Krikštolaitytė, S.; Mitrulevičienė, A.; Bieliauskas, A.; Martynaitis, V.; Bechmann, M.; Roller, A.; Šačkus, A.; Holzer, W. On the Tautomerism of N-Substituted Pyrazolones: 1,2-Dihydro-3H-pyrazol-3-ones versus 1H-Pyrazol-3-ols. Molecules 2018, 23, 129. [Google Scholar] [CrossRef]
- Aguilar-Parrilla, F.; Cativiela, C.; Diaz-De-Villegas, M.D.; Limbach, H.-H.; Smith, J.A.S.; Toiron, C. The tautomerism of 3(5)-phenylpyrazoles: An experimental (1H, 13C, 15N NMR and X-ray crystallography) study. J. Chem. Soc. Perkin Trans. 2 1992, 2, 1737–1742. [Google Scholar] [CrossRef]
- Tarabová, D.; Šoralová, S.; Breza, M.; Fronc, M.; Holzer, W.; Milata, V. Use of activated enol ethers in the synthesis of pyrazoles: Reactions with hydrazine and a study of pyrazole tautomerism. Beilstein J. Org. Chem. 2014, 10, 752–760. [Google Scholar] [CrossRef]
- Huang, A.; Wo, K.; Lee, S.Y.C.; Kneitschel, N.; Chang, J.; Zhu, K.; Mello, T.; Bancroft, L.; Norman, N.J.; Zheng, S.-L. Regioselective Synthesis, NMR, and Crystallographic Analysis of N1-Substituted Pyrazoles. J. Org. Chem. 2017, 82, 8864–8872. [Google Scholar] [CrossRef]
- Liu, X.; Qiao, L.; Zhai, Z.; Cai, P.; Cantrell, C.L.; Tan, C.; Weng, J.; Han, L.; Wu, H. Novel 4-pyrazole carboxamide derivatives containing flexible chain motif: Design, synthesis and antifungal activity. Pest. Manag. Sci. 2019, 75, 2892–2900. [Google Scholar] [CrossRef]
- Gewehr, M.; Dietz, J.; Grote, T.; Blettner, C.; Grammenos, W.; Hünger, U.; Müller, B.; Schieweck, F.; Schwögler, A.; Lohmann, J.K.; et al. Pyrazole Carboxylic Acid Anilides, Method for the Production Thereof and Agents Containing Them for Controlling Pathogenic Fungi. WO Patent WO2006087343A1, 15 February 2006. [Google Scholar]
- Braun, S.; Kalinowski, H.O.; Berger, S. 150 and more Basic NMR Experiments: A Practical Course, 2nd ed.; ACS: New York, NY, USA, 2000. [Google Scholar]
- Buevich, A.V.; Williamson, R.T.; Martin, G.E. NMR Structure Elucidation of Small Organic Molecules and Natural Products: Choosing ADEQUATE vs HMBC. J. Nat. Prod. 2014, 77, 1942–1947. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Solvent * | t (h) | 4a/5a/6a ** | t (h) | 4a/5a/6a ** | 5a, Yield (%) *** |
|---|---|---|---|---|---|---|
| 1 | EtOH | 1 | 52.4/46.8/0.8 | 18 | 0/99.5/0.5 | 78 |
| 2 | ACN | 1 | 27.6/68.2/4.2 | 18 | 0/95.2/4.8 | 75 |
| 3 | CCl4 | 1 | 58.3/27.5/14.2 | 18 | 18.3/67.9/13.8 | 54 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matulevičiūtė, G.; Arbačiauskienė, E.; Kleizienė, N.; Kederienė, V.; Ragaitė, G.; Dagilienė, M.; Bieliauskas, A.; Milišiūnaitė, V.; Sløk, F.A.; Šačkus, A. Synthesis and Characterization of Novel Methyl (3)5-(N-Boc-piperidinyl)-1H-pyrazole-4-carboxylates. Molecules 2021, 26, 3808. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26133808
Matulevičiūtė G, Arbačiauskienė E, Kleizienė N, Kederienė V, Ragaitė G, Dagilienė M, Bieliauskas A, Milišiūnaitė V, Sløk FA, Šačkus A. Synthesis and Characterization of Novel Methyl (3)5-(N-Boc-piperidinyl)-1H-pyrazole-4-carboxylates. Molecules. 2021; 26(13):3808. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26133808
Chicago/Turabian StyleMatulevičiūtė, Gita, Eglė Arbačiauskienė, Neringa Kleizienė, Vilija Kederienė, Greta Ragaitė, Miglė Dagilienė, Aurimas Bieliauskas, Vaida Milišiūnaitė, Frank A. Sløk, and Algirdas Šačkus. 2021. "Synthesis and Characterization of Novel Methyl (3)5-(N-Boc-piperidinyl)-1H-pyrazole-4-carboxylates" Molecules 26, no. 13: 3808. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26133808