
Cheminformatic Characterization of Natural Antimicrobial Products for the Development of New Lead Compounds


Abstract
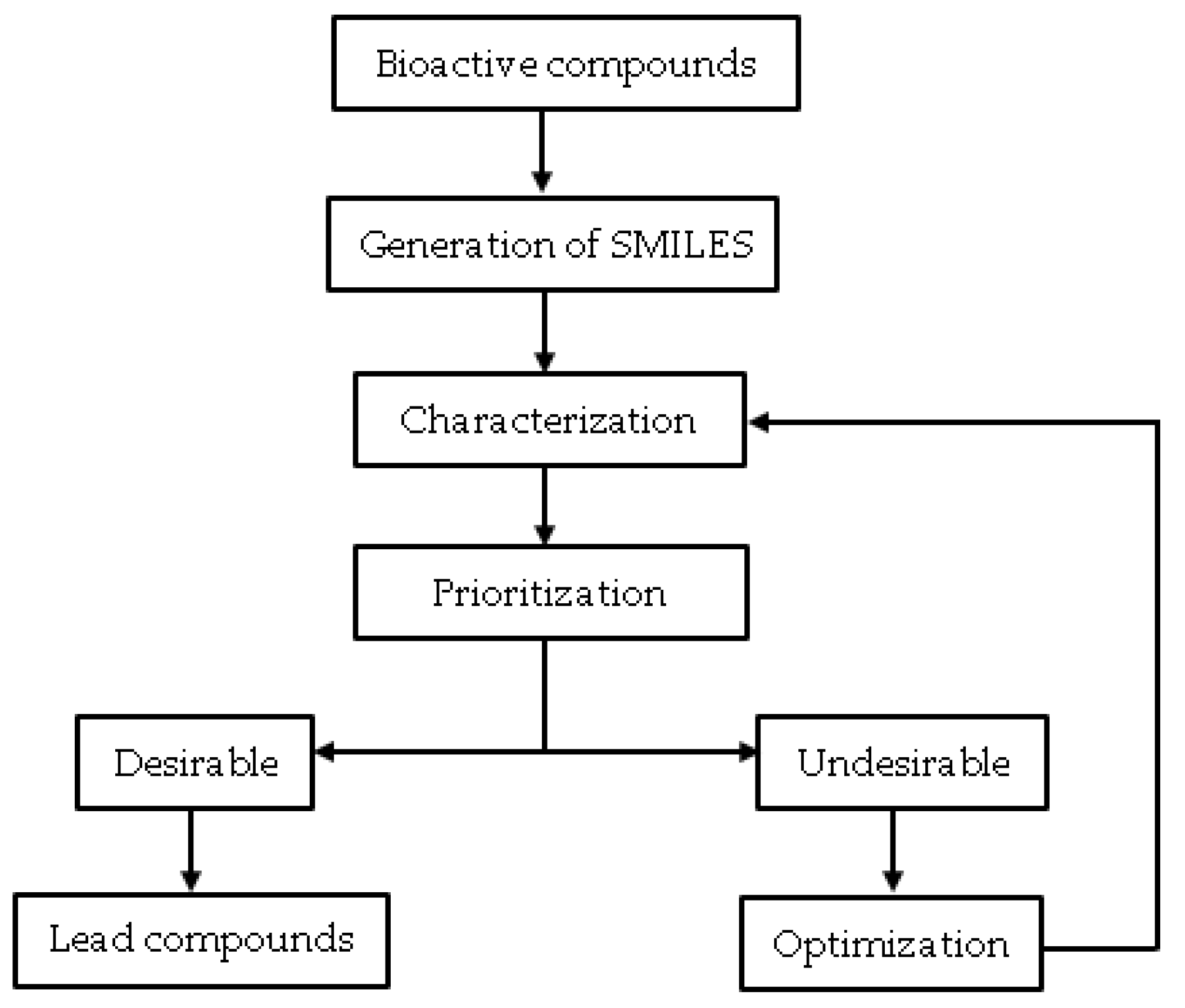
:1. Introduction
2. Natural Products in Antimicrobial Drug Discovery
3. Cheminformatics Techniques in Antimicrobial Drug Discovery and Development
3.1. Overview of Cheminformatics
3.1.1. Hit Profiling: Physicochemical Properties of NPs
Molecular Weight (MW)
Partition Coefficient (logP)
Hydrogen Bonding
3.2. Concept of Drug-Likeness
3.2.1. Lipinski’s Rule of Five (Ro5)
3.2.2. Pharmacokinetics and Toxicity Parameters
3.3. Hit-Prioritization Using the Quantitative Estimate of Drug-Likeness
3.4. Hit Optimization after Hit Profiling
3.5. Cheminformatics Language and Open Access Software Packages for Hit Characterization, Prioritization, and Optimization
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Aslam, B.; Wang, W.; Arshad, M.I.; Khurshid, M.; Muzammil, S.; Rasool, M.H.; Nisar, M.A.; Alvi, R.F.; Aslam, M.A.; Qamar, M.U.; et al. Antibiotic resistance: A rundown of a global crisis. Infect. Drug Resist. 2018, 11, 1645. [Google Scholar] [CrossRef] [PubMed]
- Adedeji, W.A. The treasure called antibiotics. Ann. Ibadan Postgrad. Med. 2016, 14, 56. [Google Scholar]
- Hutchings, M.I.; Truman, A.W.; Wilkinson, B. Antibiotics: Past, present and future. Curr. Opin. Microbiol. 2019, 51, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Silver, L.L. Challenges of antibacterial discovery. Clin. Microbiol. Rev. 2011, 24, 71–109. [Google Scholar] [CrossRef] [PubMed]
- Chokshi, A.; Sifri, Z.; Cennimo, D.; Horng, H. Global contributors to antibiotic resistance. J. Glob. Infect. Dis. 2019, 11, 36. [Google Scholar]
- O’neill, J.M. Antimicrobial Resistance: Tackling a Crisis for the Health and Wealth of Nations; Review on Antimicrobial Resistance: London, UK, 2014. [Google Scholar]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef]
- Seyed, M.A. A comprehensive review on Phyllanthus derived natural products as potential chemotherapeutic and immunomodulators for a wide range of human diseases. Biocatal. Agric. Biotechnol. 2019, 17, 529–537. [Google Scholar] [CrossRef]
- Pereira, F.; Aires-de-Sousa, J. Computational Methodologies in the Exploration of Marine Natural Product Leads. Mar. Drugs 2018, 16, 236. [Google Scholar] [CrossRef]
- Obaid, A.Y.; Voleti, S.; Bora, R.S.; Hajrah, N.H.; Omer, A.M.S.; Sabir, J.S.; Saini, K.S. Cheminformatics studies to analyze the therapeutic potential of phytochemicals from Rhazya stricta. Chem. Cent. J. 2017, 11, 1–21. [Google Scholar] [CrossRef]
- Campillos, M. Computational approaches for the study of the role of small molecules in diseases. Perspect. Sci. 2016, 9, 49–52. [Google Scholar] [CrossRef]
- Shivakumar, R.; Venkatarangaiah, K.; Shastri, S.; Nagaraja, R.B.; Sheshagiri, A. Antibacterial Property and Molecular Docking Studies of Leaf Calli Phytochemicals of Bridelia scandens Wild. Pharmacogn. J. 2018, 10, 1221–1229. [Google Scholar] [CrossRef]
- Abreu, A.C.; Mcbain, J.; Sim, M. Plants as sources of new antimicrobials and resistance-modifying agents. Nat. Prod. Rep. 2012, 29, 1007–1221. [Google Scholar] [CrossRef]
- Dong, L.M.; Huang, L.L.; Dai, H.; Xu, Q.L.; Ouyang, J.K.; Jia, X.C.; Gu, W.X.; Tan, J.W. Anti-MRSA sesquiterpenes from the semi-mangrove plant Myoporum bontioides A. Gray. Mar. Drugs 2018, 16, 438. [Google Scholar] [CrossRef]
- Chen, J.; Li, W.; Yao, H.; Xu, J. Insights into drug discovery from natural products through structural modification. Fitoterapia 2015, 103, 231–241. [Google Scholar] [CrossRef]
- Özakin, S.; İnce, E. Genome and metabolome mining of marine obligate Salinispora strains to discover new natural products. Turk. J. Biol. 2019, 43, 28–36. [Google Scholar] [CrossRef]
- Wright, G.D. Unlocking the potential of natural products in drug discovery. Microb. Biotechnol. 2019, 12, 55–57. [Google Scholar] [CrossRef] [PubMed]
- Zengin, G.; Uysal, A.; Aktumsek, A.; Mocan, A.; Mollica, A.; Locatelli, M.; Custodio, L.; Neng, N.R.; Nogueira, J.M.F.; Aumeeruddy-elal, Z.; et al. ScienceDirect Euphorbia denticulata Lam.: A promising source of phyto-pharmaceuticals for the development of novel functional formulations. Biomed. Pharmacother. 2017, 87, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Harvey, A.L. Natural products in drug discovery. Drug Discov. Today 2008, 13, 894–901. [Google Scholar] [CrossRef]
- Harvey, A.L.; Edrada-Ebel, R.; Quinn, R.J. The re-emergence of natural products for drug discovery in the genomics era. Nat. Rev. Drug Discov. 2015, 14, 111–129. [Google Scholar] [CrossRef] [PubMed]
- Jaradat, N.A.; Al-Masri, M.; Zaid, A.N.; Hussein, F.; Al-Rimawi, F.; Mokh, A.A.; Mokh, J.A.; Ghonaim, S. Phytochemical, antimicrobial and antioxidant preliminary screening of a traditional Palestinian medicinal plant, Ononis pubescens L. Eur. J. Integr. Med. 2017, 14, 46–51. [Google Scholar] [CrossRef]
- Bueno, J. In vitro antimicrobial activity of natural products using minimum inhibitory concentrations: Looking for new chemical entities or predicting clinical response. Med. Aromat. Plants 2012, 1, 412–2167. [Google Scholar] [CrossRef]
- Gibbons, S. Phytochemicals for bacterial resistance-strengths, weaknesses and opportunities. Planta Med. 2008, 74, 594–602. [Google Scholar] [CrossRef] [PubMed]
- Cos, P.; Vlietinck, A.J.; Berghe, D.V.; Maes, L. Anti-infective potential of natural products: How to develop a stronger in vitro ‘proof-of-concept’. J. Ethnopharmacol. 2006, 106, 290–302. [Google Scholar] [CrossRef] [PubMed]
- Dej-adisai, S.; Parndaeng, K. Determination of Phytochemical Compounds, and Tyrosinase Inhibitory and Antimicrobial Activities of Bioactive Compounds from Streblus ilicifolius ( S Vidal ) Corner. Trop. J. Pharm. Res. 2016, 15, 497–506. [Google Scholar] [CrossRef]
- Yang, S.; Tseng, C.; Wang, P.; Lu, P.; Weng, Y. Pterostilbene, a Methoxylated Resveratrol Derivative, Efficiently Eradicates Planktonic, Biofilm, and Intracellular MRSA by Topical Application. Front. Microbiol. 2017, 8, 1103. [Google Scholar] [CrossRef]
- de Bruijn, W.J.; Araya-Cloutier, C.; Bijlsma, J.; de Swart, A.; Sanders, M.G.; de Waard, P.; Gruppen, H.; Vincken, J.P. Phytochemistry Letters Antibacterial prenylated stilbenoids from peanut (Arachis hypogaea). Phytochem. Lett. 2018, 28, 13–18. [Google Scholar] [CrossRef]
- Saleem, M.; Nazir, M.; Ali, S.; Hussain, H.; Lee, S. Antimicrobial natural products: An update on future antibiotic drug candidates. Nat. Prod. Rep. 2010, 27, 238–254. [Google Scholar] [CrossRef] [PubMed]
- Bonvicini, F.; Antognoni, F.; Mandrone, M.; Protti, M.; Mercolini, L.; Lianza, M.; Gentilomi, G.A.; Poli, F. All Aspects of Plant Biology Phytochemical analysis and antibacterial activity towards methicillin-resistant Staphylococcus aureus of leaf extracts from Argania spinosa (L.) Skeels. Plant Biosyst. Int. J. Deal. Asp. Plant Biol. 2017, 151, 649–656. [Google Scholar]
- Liu, M.; El-hossary, E.M.; Oelschlaeger, T.A.; Donia, M.S.; Quinn, R.J.; Abdelmohsen, U.R. Review Potential of marine natural products against drug-resistant bacterial infections. Lancet Infect. Dis. 2019, 3099, 1–9. [Google Scholar]
- Alves, M.J.; Ferreira, I.C.F.R.; Froufe, H.J.C.; Abreu, R.M.V.; Martins, A.; Pintado, M. Antimicrobial activity of phenolic compounds identified in wild mushrooms, SAR analysis and docking studies. J. Appl. Microbiol. 2013, 115, 346–357. [Google Scholar] [CrossRef]
- Tóth, B.; Liktor-Busa, E.; Urbán, E.; Csorba, A.; Jakab, G.; Hohmann, J.; Vasas, A. Fitoterapia Antibacterial screening of Juncaceae species native to the Carpathian Basin against resistant strains and LC-MS investigation of phenanthrenes responsible for the effect. Fitoterapia 2016, 115, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Al-ani, I.; Zimmermann, S.; Reichling, J.; Wink, M. Phytomedicine Pharmacological synergism of bee venom and melittin with antibiotics and plant secondary metabolites against multi-drug resistant microbial pathogens. Phytomedicine 2015, 22, 245–255. [Google Scholar] [CrossRef]
- Obiang-Obounou, B.W.; Kang, O.H.; Choi, J.G.; Keum, J.H.; Kim, S.B.; Mun, S.H.; Shin, D.W.; Kim, K.W.; Park, C.B.; Kim, Y.G.; et al. The mechanism of action of sanguinarine against methicillin-resistant Staphylococcus aureus. J. Toxicol. Sci. 2011, 36, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Eom, S.; Kim, Y.; Kim, S. Marine bacteria: Potential sources for compounds to overcome antibiotic resistance. Appl. Microbiol. Biotechnol. 2013, 97, 4763–4773. [Google Scholar] [CrossRef]
- Hatano, T.; Eerdunbayaer, C.Y.; Kuroda, T.; Shimozu, Y. We are IntechOpen the world’s leading publisher of Open Access books Built by scientists, for scientists TOP 1%. In Licorice as a Resource for Pharmacologically Active Phenolic Substances: Antioxidant and Antimicrobial Effects. Biological Activities and Action Mechanisms of Licorice Ingredients; InTech: Rijeka, Croatia, 2017; pp. 59–75. [Google Scholar]
- Cha, J.; Lee, J.; Choi, K.M.; Choi, S.; Park, J.H. Synergistic Effect between Cryptotanshinone and Antibiotics against Clinic Methicillin and Vancomycin-Resistant Staphylococcus aureus. Evid. Based Complement. Altern. Med. 2014, 2014, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Prada-Gracia, D.; Huerta-Yépez, S.; Moreno-Vargas, L.M. Application of computational methods for anticancer drug discovery, design, and optimization. Boletín Médico Del Hosp. Infant. México 2016, 73, 411–423. [Google Scholar]
- Gillet, V.J. Applications of Chemoinformatics in Drug Discovery 2. 2 Computer Representation of Chemical Structures; Wiley: Hoboken, NJ, USA, 2019. [Google Scholar]
- Egieyeh, S.; Malan, S.F.; Christoffels, A. Cheminformatics techniques in antimalarial drug discovery and development from natural products 1: Basic concepts. Phys. Sci. Rev. 2019, 4, 1–11. [Google Scholar] [CrossRef]
- Xu, J.; Hagler, A. Chemoinformatics and drug discovery. Molecules 2002, 7, 566–600. [Google Scholar] [CrossRef]
- Jónsdóttir, S.Ó.; Jørgensen, F.S.; Brunak, S. Structural bioinformatics Prediction methods and databases within chemoinformatics: Emphasis on drugs and drug candidates. Bioinformatics 2005, 21, 2145–2160. [Google Scholar] [CrossRef]
- Medina-Franco, J.L. Advances in computational approaches for drug discovery based on natural products. Rev. Latinoam. Química 2013, 41, 95–110. [Google Scholar]
- Chen, Y.; Kirchmair, J. Cheminformatics in Natural Product-based Drug Discovery. Mol. Inform. 2020, 39, 2000171. [Google Scholar] [CrossRef] [PubMed]
- Wetzel, S.; Schuffenhauer, A.; Roggo, S.; Ertl, P.; Waldmann, H. Cheminformatic analysis of natural products and their chemical space. Chim. Int. J. Chem. 2007, 61, 355–360. [Google Scholar] [CrossRef]
- Willett, P. From chemical documentation to chemoinformatics: 50 years of chemical information science. J. Inf. Sci. 2008, 34, 477–499. [Google Scholar] [CrossRef]
- Medina-Franco, J.L.; Saldívar-González, F.I. Cheminformatics to characterize pharmacologically active natural products. Biomolecules 2020, 10, 1566. [Google Scholar] [CrossRef] [PubMed]
- Wenlock, M.C.; Barton, P. In silico physicochemical parameter predictions. Mol. Pharm. 2013, 10, 1224–1235. [Google Scholar] [CrossRef]
- Chandrasekaran, B.; Abed, S.N.; Al-Attraqchi, O.; Kuche, K.; Tekade, R.K. Computer-aided prediction of pharmacokinetic (ADMET) properties. In Dosage Form Design Parameters; Elsevier: Amsterdam, The Netherlands, 2018; pp. 731–755. [Google Scholar]
- Gleeson, M.P.; Leeson, D.; Waterbeemd, H.A.N.V.A.N.D.E. Physicochemical Properties and Compound Quality. In The Handbook of Medicinal Chemistr; The Royal Society of Chemistry: London, UK, 2015; pp. 1–31. ISBN 9781782621836. [Google Scholar]
- Schneider, G. Prediction of drug-like properties. In Madame Curie Bioscience Database [Internet]; Landes Bioscience: Austin, TX, USA, 2013. [Google Scholar]
- Vallianatou, T.; Giaginis, C.; Tsantili-Kakoulidou, A. The impact of physicochemical and molecular properties in drug design: Navigation in the “drug-like” chemical space. In GeNeDis 2014; Springer: Berlin/Heidelberg, Germany, 2015; pp. 187–194. [Google Scholar]
- Lobell, M.; Hendrix, M.; Hinzen, B.; Keldenich, J.; Meier, H.; Schmeck, C.; Schohe-Loop, R.; Wunberg, T.; Hillisch, A. In silico ADMET traffic lights as a tool for the prioritization of HTS hits. ChemMedChem 2006, 1, 1229–1236. [Google Scholar] [CrossRef] [PubMed]
- Meanwell, N.A. Improving drug candidates by design: A focus on physicochemical properties as a means of improving compound disposition and safety. Chem. Res. Toxicol. 2011, 24, 1420–1456. [Google Scholar] [CrossRef] [PubMed]
- Lagorce, D.; Douguet, D.; Miteva, M.A.; Villoutreix, B.O. Computational analysis of calculated physicochemical and ADMET properties of protein-protein interaction inhibitors. Sci. Rep. 2017, 7, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C. Physicochemical properties and the discovery of orally active drugs: Technical and people issues. In Proceedings of the In Molecular Informatics: Confronting Complexity, Beilstein-Institute Workshop, Bozen, Italy, 13–16 May 2002; pp. 59–78. [Google Scholar]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef]
- Ebejer, J.P.; Charlton, M.H.; Finn, P.W. Are the physicochemical properties of antibacterial compounds really different from other drugs ? J. Cheminform. 2016, 8, 1–9. [Google Scholar] [CrossRef]
- O’Shea, R.O.; Moser, H.E. Physicochemical Properties of Antibacterial Compounds: Implications for Drug Discovery. J. Med. Chem. 2008, 51, 2871–2878. [Google Scholar] [CrossRef]
- Konaklieva, M.I. Addressing Antimicrobial Resistance through New Medicinal and Synthetic Chemistry Strategies. SLAS Discov. Adv. Life Sci. R&D 2019, 24, 419–439. [Google Scholar]
- Bhal, S.K. LogP—Making Sense of the Value; Advanced Chemistry Development, Inc.: Toronto, ON, Canada, 2007; pp. 1–4. [Google Scholar]
- Oselusi, S.O.; Egieyeh, S.A.; Christoffels, A. Cheminformatic Profiling and Hit Prioritization of Natural Products with Activities against Methicillin-Resistant Staphylococcus aureus, (Forthcoming). 2021.
- Ropponen, H.K.; Richter, R.; Hirsch, A.K.; Lehr, C.M. Mastering the Gram-Negative Bacterial Barrier–Chemical Approaches to Increase Bacterial Bioavailability of Antibiotics. Adv. Drug Deliv. Rev. 2021, 172, 339–360. [Google Scholar] [CrossRef]
- Schwöbel, J.A.H.; Ebert, R.; Kühne, R.; Schüürmann, G. Prediction models for the Abraham hydrogen bond donor strength: Comparison of semi-empirical, ab initio, and DFT methods. J. Phys. Org. Chem. 2011, 24, 1072–1080. [Google Scholar] [CrossRef]
- Yunta, M.J. It is important to compute intramolecular hydrogen bonding in drug design. Am. J. Model. Optim. 2017, 5, 24–57. [Google Scholar] [CrossRef]
- Wade, R.C.; Goodford, P.J. The role of hydrogen-bonds in drug binding. Prog. Clin. Biol. Res. 1989, 289, 433–444. [Google Scholar] [PubMed]
- Zhao, H.; Huang, D. Hydrogen bonding penalty upon ligand binding. PLoS ONE 2011, 6, e19923. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Oezguen, N.; Urvil, P.; Ferguson, C.; Dann, S.M.; Savidge, T.C. Regulation of protein-ligand binding affinity by hydrogen bond pairing. Sci. Adv. 2016, 2, e1501240. [Google Scholar] [CrossRef]
- Lacret, R.; Oves-Costales, D.; Gómez, C.; Díaz, C.; De la Cruz, M.; Pérez-Victoria, I.; Vicente, F.; Genilloud, O.; Reyes, F. New ikarugamycin derivatives with antifungal and antibacterial properties from Streptomyces zhaozhouensis. Mar. Drugs 2015, 13, 128–140. [Google Scholar] [CrossRef]
- Rankovic, Z. CNS drug design: Balancing physicochemical properties for optimal brain exposure. J. Med. Chem. 2015, 58, 2584–2608. [Google Scholar] [CrossRef] [PubMed]
- Bickerton, G.R.; Paolini, G.V.; Besnard, J.; Muresan, S.; Hopkins, A.L. Quantifying the chemical beauty of drugs. Nat. Chem. 2012, 4, 90–98. [Google Scholar] [CrossRef]
- Ntie-Kang, F.; Nyongbela, K.D.; Ayimele, G.A.; Shekfeh, S. “Drug-likeness” properties of natural compounds. In Fundamental Concepts; De Gruyter: Berlin, Germany, 2020; pp. 81–102. ISBN 3110579359. [Google Scholar]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Mignani, S.; Rodrigues, J.; Tomas, H.; Jalal, R.; Singh, P.P.; Majoral, J.P.; Vishwakarma, R.A. Present drug-likeness filters in medicinal chemistry during the hit and lead optimization process: How far can they be simplified ? Drug Discov. Today 2018, 23, 605–615. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Tian, S.; Wang, J.; Li, Y.; Li, D.; Xu, L.; Hou, T. The application of in silico drug-likeness predictions in pharmaceutical research. Adv. Drug Deliv. Rev. 2015, 86, 2–10. [Google Scholar] [CrossRef]
- Krämer, S.D.; Aschmann, H.E.; Hatibovic, M.; Hermann, K.F.; Neuhaus, C.S.; Brunner, C.; Belli, S. When barriers ignore the “rule-of-five”. Adv. Drug Deliv. Rev. 2016, 101, 62–74. [Google Scholar] [CrossRef]
- Boufridi, A.; Quinn, R.J. Harnessing the properties of natural products. Annu. Rev. Pharmacol. Toxicol. 2018, 58, 451–470. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Paul, J.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2012, 64, 4–17. [Google Scholar] [CrossRef]
- Zhang, M.-Q.; Wilkinson, B. Drug discovery beyond the ‘rule-of-five’. Curr. Opin. Biotechnol. 2007, 18, 478–488. [Google Scholar] [CrossRef] [PubMed]
- Abad-Zapatero, C. A Sorcerer’s apprentice and the rule of five: From rule-of-thumb to commandment and beyond. Drug Discov. Today 2007, 12, 995–997. [Google Scholar] [CrossRef] [PubMed]
- Benet, L.Z.; Hosey, C.M.; Ursu, O.; Oprea, T.I. BDDCS, the rule of 5 and drugability. Adv. Drug Deliv. Rev. 2016, 101, 89–98. [Google Scholar] [CrossRef]
- Segall, M. Advances in multiparameter optimization methods for de novo drug design. Expert Opin. Drug Discov. 2014, 9, 803–817. [Google Scholar] [CrossRef]
- Wager, T.T.; Hou, X.; Verhoest, P.R.; Villalobos, A. Moving beyond rules: The development of a central nervous system multiparameter optimization (CNS MPO) approach to enable alignment of druglike properties. ACS Chem. Neurosci. 2010, 1, 435–449. [Google Scholar] [CrossRef]
- Vrbanac, J.; Slauter, R. ADME in drug discovery. In A Comprehensive Guide to Toxicology in Nonclinical Drug Development; Elsevier: Amsterdam, The Netherlands, 2017; pp. 39–67. [Google Scholar]
- Sharma, A.; Saini, R. Pharmacokinetic profiling of anticancer phytocompounds using computational approach. Phytochem. Anal. 2018, 29, 559–568. [Google Scholar] [CrossRef]
- Wang, J.; Urban, L. The impact of early ADME profiling on drug discovery and development strategy. DDW Drug Discov. World 2004, 5, 73–86. [Google Scholar]
- Skariyachan, S.; Krishnan, R.S.; Siddapa, S.B.; Salian, C.; Bora, P.; Sebastian, D. Computer aided screening and evaluation of herbal therapeutics against MRSA infections. Bioinformation 2011, 7, 222. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, S.I.; Chaudhari, H.K. Design, synthesis, in-silico studies and biological screening of quinazolinone analogues as potential antibacterial agents against MRSA. Bioorg. Med. Chem. 2019, 27, 2676–2688. [Google Scholar] [CrossRef] [PubMed]
- Ekins, S.; Mestres, J.; Testa, B. In silico pharmacology for drug discovery: Methods for virtual ligand screening and profiling. Br. J. Pharmacol. 2007, 152, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Yusof, I.; Segall, M.D. Considering the impact drug-like properties have on the chance of success. Drug Discov. Today 2013, 18, 659–666. [Google Scholar] [CrossRef] [PubMed]
- Kirkpatrick, P. Shades of chemical beauty. Nat. Rev. Drug Discov. 2012, 11, 107. [Google Scholar] [CrossRef]
- Egieyeh, S.A.; Syce, J.; Malan, S.F.; Christoffels, A. Prioritization of anti–malarial hits from nature: Chemo–informatic profiling of natural products with in vitro antiplasmodial activities and currently registered anti–malarial drugs. Malar. J. 2016, 15, 1–23. [Google Scholar] [CrossRef]
- Kim, S.-K.; Lee, S. Drug-likeness and Oral bioavailability for Chemical Compounds of Medicinal Materials Constituting Oryeong-san. Korea J. Herbol. 2018, 33, 19–37. [Google Scholar]
- Guo, Z. The modification of natural products for medical use. Acta Pharm. Sin. B 2017, 7, 119–136. [Google Scholar] [CrossRef]
- Chen, Y.; Zhao, J.; Qiu, Y.; Yuan, H.; Khan, S.I.; Hussain, N.; Choudhary, M.I.; Zeng, F.; Guo, D.; Khan, I.A.; et al. Fitoterapia Prenylated fl avonoids from the stems and roots of Tripterygium wilfordii. Fitoterapia 2017, 119, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.; Morris-Natschke, S.L.; Lee, K.H. Strategies for the Optimization of Natural Leads to Anticancer Drugs or Drug Candidates. Med. Res. Rev. 2016, 36, 32–91. [Google Scholar] [CrossRef]
- Harrold, M.W.; Zavod, R.M.; Harrold, M.W.; Zavod, R.M. Functional group characteristics and roles. In Basic Concepts in Medical Chemistry; Harrold, M.W., Zavod, R.M., Eds.; American Society of Health-System Pharmacists: Bethesda, MD, USA, 2013; pp. 15–50. [Google Scholar]
- Wunberg, T.; Hendrix, M.; Hillisch, A.; Lobell, M.; Meier, H.; Schmeck, C.; Wild, H.; Hinzen, B. Improving the hit-to-lead process: Data-driven assessment of drug-like and lead-like screening hits. Drug Discov. Today 2006, 11, 175–180. [Google Scholar] [CrossRef]
- Sharifi-Rad, J.; Ozleyen, A.; Boyunegmez Tumer, T.; Oluwaseun Adetunji, C.; El Omari, N.; Balahbib, A.; Taheri, Y.; Bouyahya, A.; Martorell, M.; Martins, N. Natural products and synthetic analogs as a source of antitumor drugs. Biomolecules 2019, 9, 679. [Google Scholar] [CrossRef]
- David, L.; Thakkar, A.; Mercado, R.; Engkvist, O. Molecular representations in AI-driven drug discovery: A review and practical guide. J. Cheminform. 2020, 12, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W. Computational Methods in Drug Discovery. Pharmacol. Rev. 2014, 66, 334–395. [Google Scholar] [CrossRef]
- Dong, J.; Wang, N.-N.; Yao, Z.-J.; Zhang, L.; Cheng, Y.; Ouyang, D.; Lu, A.-P.; Cao, D.-S. ADMETlab: A platform for systematic ADMET evaluation based on a comprehensively collected ADMET database. J. Cheminform. 2018, 10, 1–11. [Google Scholar] [CrossRef]
- Fereidoonnezhad, M.; Mostoufi, A.; Eskandari, M.; Zali, S.; Aliyan, F. Multitarget drug design, molecular docking and PLIF studies of novel Tacrine− Coumarin hybrids for the treatment of Alzheimer’s disease. Iran. J. Pharm. Res. IJPR 2018, 17, 1217. [Google Scholar]
- Kar, S.; Leszczynski, J. Open access in silico tools to predict the ADMET profiling of drug candidates. Expert Opin. Drug Discov. 2020, 15, 1473–1487. [Google Scholar] [CrossRef]
- Dhanda, S.K.; Singla, D.; Mondal, A.K.; Raghava, G.P.S. DrugMint: A webserver for predicting and designing of drug-like molecules. Biol. Direct 2013, 8, 1–12. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug- likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Wirth, M.; Zoete, V.; Michielin, O.; Sauer, W.H.B. SwissBioisostere: A database of molecular replacements for ligand design. Nucleic Acids Res. 2013, 41, D1137–D1143. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef] [PubMed]
- Sander, T.; Freyss, J.; von Kor, M.; Rufener, C. DataWarrior: An Open-Source Program For Chemistry Aware Data Visualization And Analysis. J. Chem. Inf. Model. 2015, 55, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Feunang, Y.D.; Fiamoncini, J.; Gil, A.; Fuente, D.; Greiner, R. BioTransformer: A comprehensive computational tool for small molecule metabolism prediction and metabolite identification. J. Cheminform. 2019, 11, 1–25. [Google Scholar]
- Falcón-Cano, G.; Molina, C.; Cabrera-Pérez, M.Á. ADME Prediction with KNIME: Development and Validation of a Publicly Available Workflow for the Prediction of Human Oral Bioavailability. J. Chem. Inf. Model. 2020, 60, 2660–2667. [Google Scholar] [CrossRef]
- Guha, R.; Willighagen, E.; Zdrazil, B.; Jeliazkova, N. What is the role of cheminformatics in a pandemic? J. Cheminform. 2021, 13, 1–3. [Google Scholar] [CrossRef]


{kind=link}
| SN | Natural Compound | Structure | Source of Compounds | Pathogen | Average Reported MIC (μg/mL) Value | No of the Tested Strains | Reference |
|---|---|---|---|---|---|---|---|
| 1 | Resveratrol |  | Fruits such as grapes, peanuts, and cranberries | Methicillin-resistant Staphylococcus aureus (MRSA) | 1.25 | 3 | [25,26] |
| 2 | Pterostilbene |  | Majorly found in fruits such as blueberries | MRSA | 0.078 | 2 | [26,27] |
| 3 | 7-amino-4-methylcoumarin |  | Endophytic Xylaria | Shigellaflexneri | 6.3 | 1 | [28] |
| 4 | Quercetin |  | Plants (Guss extracts) | MRSA | 31.2–125 | 29 | [29] |
| 5 | Anthracimycin |  | Marine actinobacteria | Bacillus anthracis | 0.031 | 1 | [30] |
| 6 | Protocatechuic acid |  | Plants and mushroom | Escherichia coli | 1 | 1 | [31] |
| 7 | Juncuenin D |  | Plants (Juncaceae family) | MRSA | 12.5 | 1 | [32] |
| 8 | Sanguinarine |  | Plants (Papaveraceae families) | MRSA | 3.12–6.25 | 15 | [33,34] |
| 9 | Vanillic acid |  | Mushroom | Escherichia coli | 0.5 | 1 | [31] |
| 10 | Abyssomicin C |  | Marine bacterium (Verrucosispora AB-18-032) | MRSA | 4 | 1 | [35] |
| 11 | MC21-A(Bromophene) |  | Marine bacterium (Pseudoalteromonas phenolica) | MRSA | 1–2 | 10 | [35] |
| 12 | Canthin-6-one |  | Plant (Allium neapolitanum) | Mycobacterium smegmatis 14468, M. phlei ATCC 11758, Staphylococcus aureus 1199B and S. aureus XU212 | 8 | 4 | [28] |
| 13 | Psoracorylifol A |  | The seeds of Psoralea corylifolia | Helicobacter pylori | 12.5–25 | 2 | [28] |
| 14 | Erycristagallin |  | The stem of a plant (Erythrina subumbrans) | MRSA | 0.78–1.56 | 4 | [28] |
| 15 | Hardwickiic acid |  | The stem bark of the plant (Irvingia gabonensis) | MRSA | 19.53 | 1 | [28] |
| 16 | Mangostanin |  | Fruit (Garcinia cowa) | MRSA | 4.0 | 1 | [28] |
| 17 | Mutactimycin C |  | Saccharothrix sp. | Microccocus leteus and Klebsiella pneumoniae | 5 | 2 | [28] |
| 18 | Protocatechuic acid |  | Plants and mushroom | MRSA | 1 | 1 | [31] |
| 19 | Gancaonin G |  | Plants (Glycyrrhiza uralensis) | MRSA | 16 | 4 | [36] |
| 20 | 3′-(γ,γ-dimethylallyl)-kievitone |  | Plants (Glycyrrh uralensis) | MRSA | 8 | 4 | [36] |
| 21 | Licoisoflavone B |  | Plants (Glycyrrhiza uralensis) | MRSA | 32 | 2 | [36] |
| 22 | Cryptotanshinon |  | Root of Salvia miltiorrhiza | MRSA | 0.5–8 | 16 | [37] |
| Tool Name | Function | Algorithm | Identifier | Reference |
|---|---|---|---|---|
| ADMETlab | Drug-likeness evaluation, profiling of ADMET, and subsequent prioritization of chemical entities | Random Forests (RF), Support Vector Machine (SVM), etc. | http://admet.scbdd.com/ | [103] |
| DruLiTo | Physicochemical properties, Drug-likeness rules, QED score | SVM, QSAR | http://www.niper.gov.in/pi_dev_tools/DruLiToWeb/DruLiTo_index.html | [104,105] |
| Drugmint | Predicting the drug-likeness, QED score, and optimization | SVM | http://crdd.osdd.net/oscadd/drugmint/ | [106] |
| SwissADME | Physicochemical properties, ADME, Rule-based drug-likeness, and Optimization | SVM and Bayesian techniques | http://www.swissadme.ch/ | [107] |
| SwissBioisostere | Optimization | Hussain-Rea algorithm | http://www.swissbioisostere.ch/ | [108] |
| pkCSM | Physicochemical properties, Rule-based drug-likeness, ADMET parameters | Graph-based structural signatures | http://structure.bioc.cam.ac.uk/pkcsm | [109] |
| DataWarrior | Physicochemical properties, Rule-based drug-likeness, Toxicity prediction, prioritization, and optimization (through the generation of Structure−Activity Landscape Index) | Stereo-enhanced Morgan-algorithm | https://openmolecules.org/datawarrior/ | [110] |
| Galaxy | Physicochemical properties, QED score | Structural similarity | https://usegalaxy.eu/ | [71] |
| BioTransformer | Prediction f drug metabolism | Machine learning algorithms | www.biotransformer.ca | [111] |
| Knime | Molecular descriptors and ADME | Machine learning | https://www.knime.com/ | [112] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oselusi, S.O.; Christoffels, A.; Egieyeh, S.A. Cheminformatic Characterization of Natural Antimicrobial Products for the Development of New Lead Compounds. Molecules 2021, 26, 3970. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26133970
Oselusi SO, Christoffels A, Egieyeh SA. Cheminformatic Characterization of Natural Antimicrobial Products for the Development of New Lead Compounds. Molecules. 2021; 26(13):3970. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26133970
Chicago/Turabian StyleOselusi, Samson Olaitan, Alan Christoffels, and Samuel Ayodele Egieyeh. 2021. "Cheminformatic Characterization of Natural Antimicrobial Products for the Development of New Lead Compounds" Molecules 26, no. 13: 3970. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26133970