
LP1 and LP2: Dual-Target MOPr/DOPr Ligands as Drug Candidates for Persistent Pain Relief

 ,
,
Abstract
:
1. Introduction
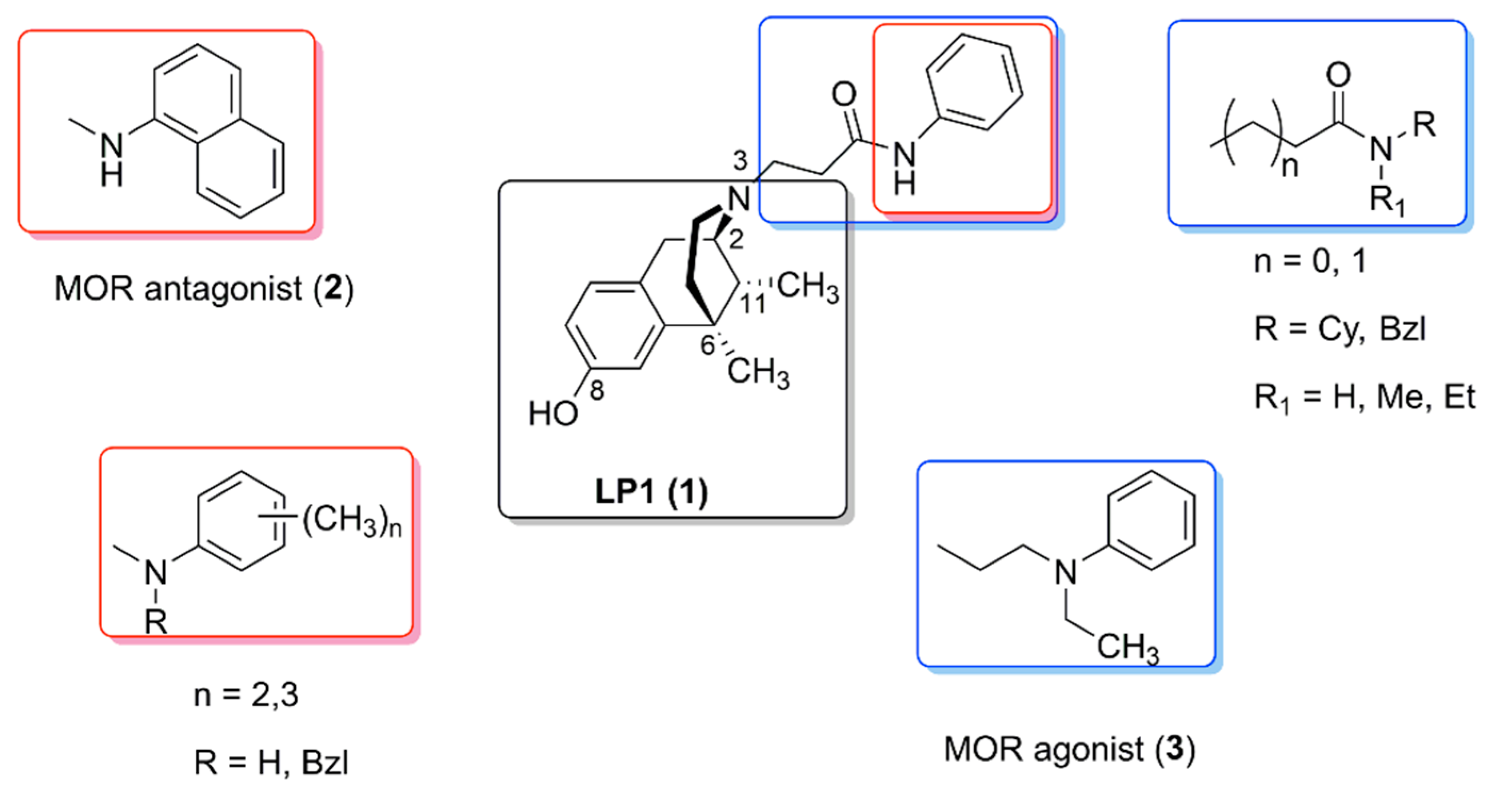
2. Dual-Target Benzomorphan-Based Ligands LP1 and LP2
2.1. Pharmacological LP1 Fingerprint
2.1.1. In Vitro Biological Evaluation
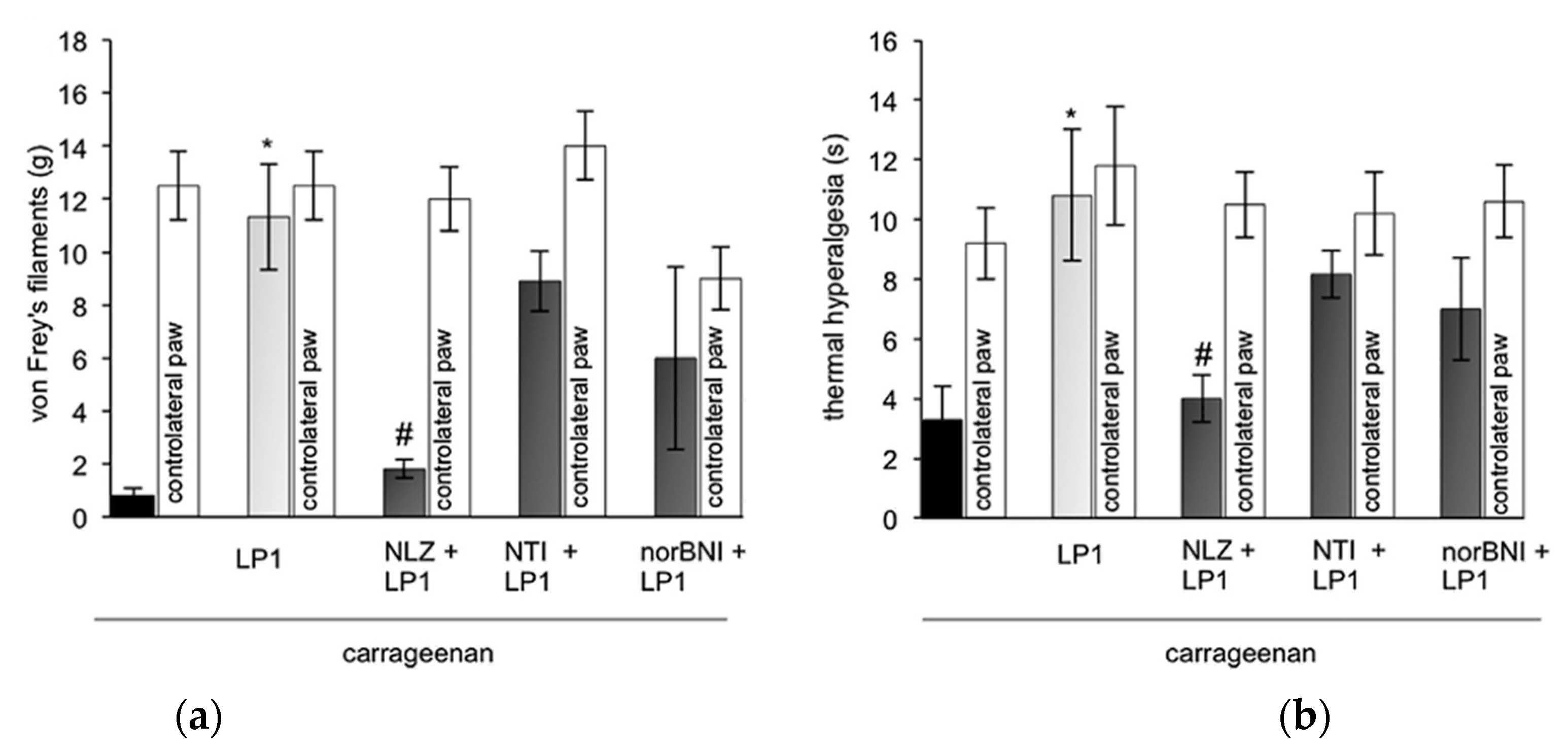
2.1.2. In Vivo Pharmacological Evaluation
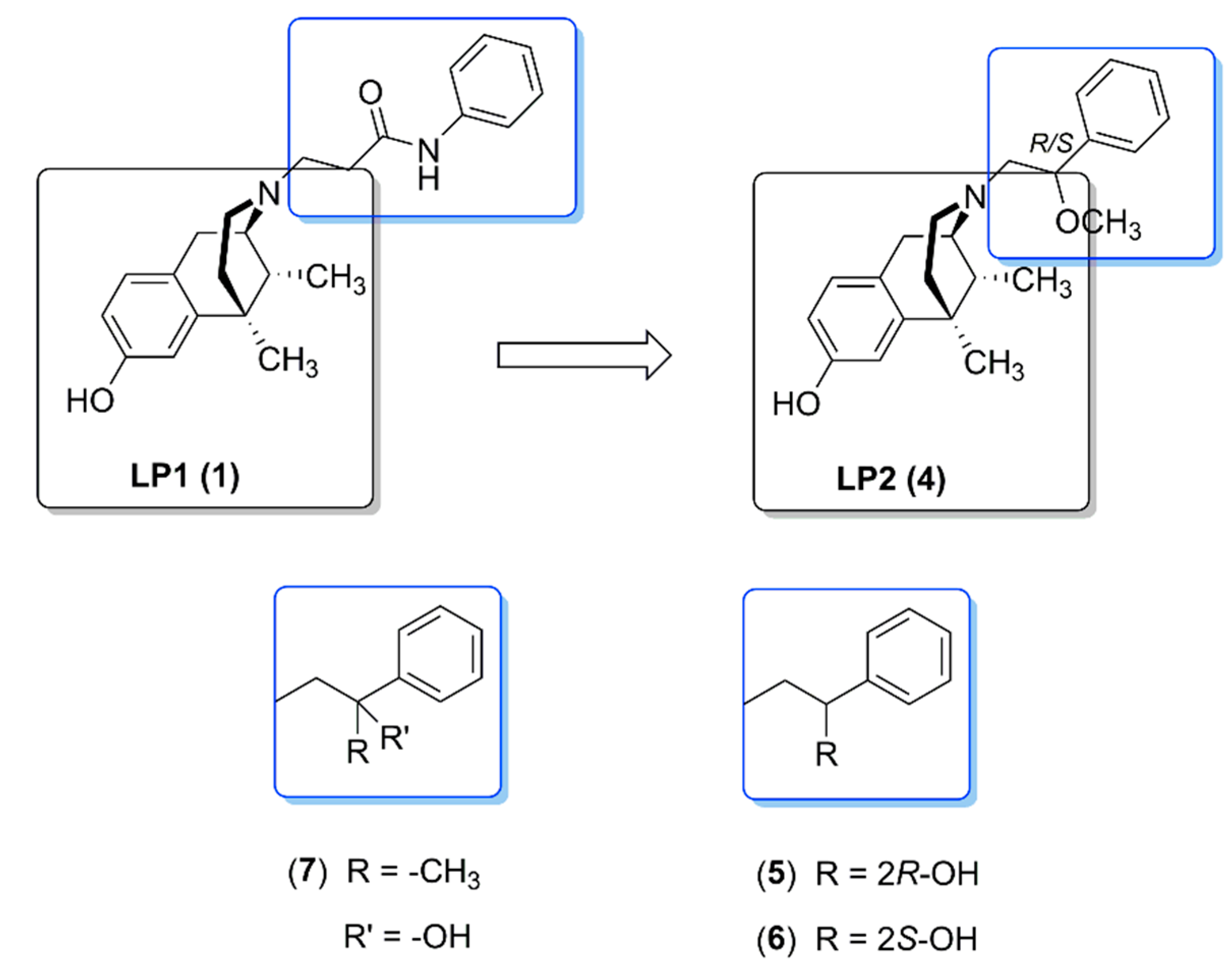
2.2. Pharmacological LP2 Evaluation
2.2.1. In Vitro Biological Evaluation
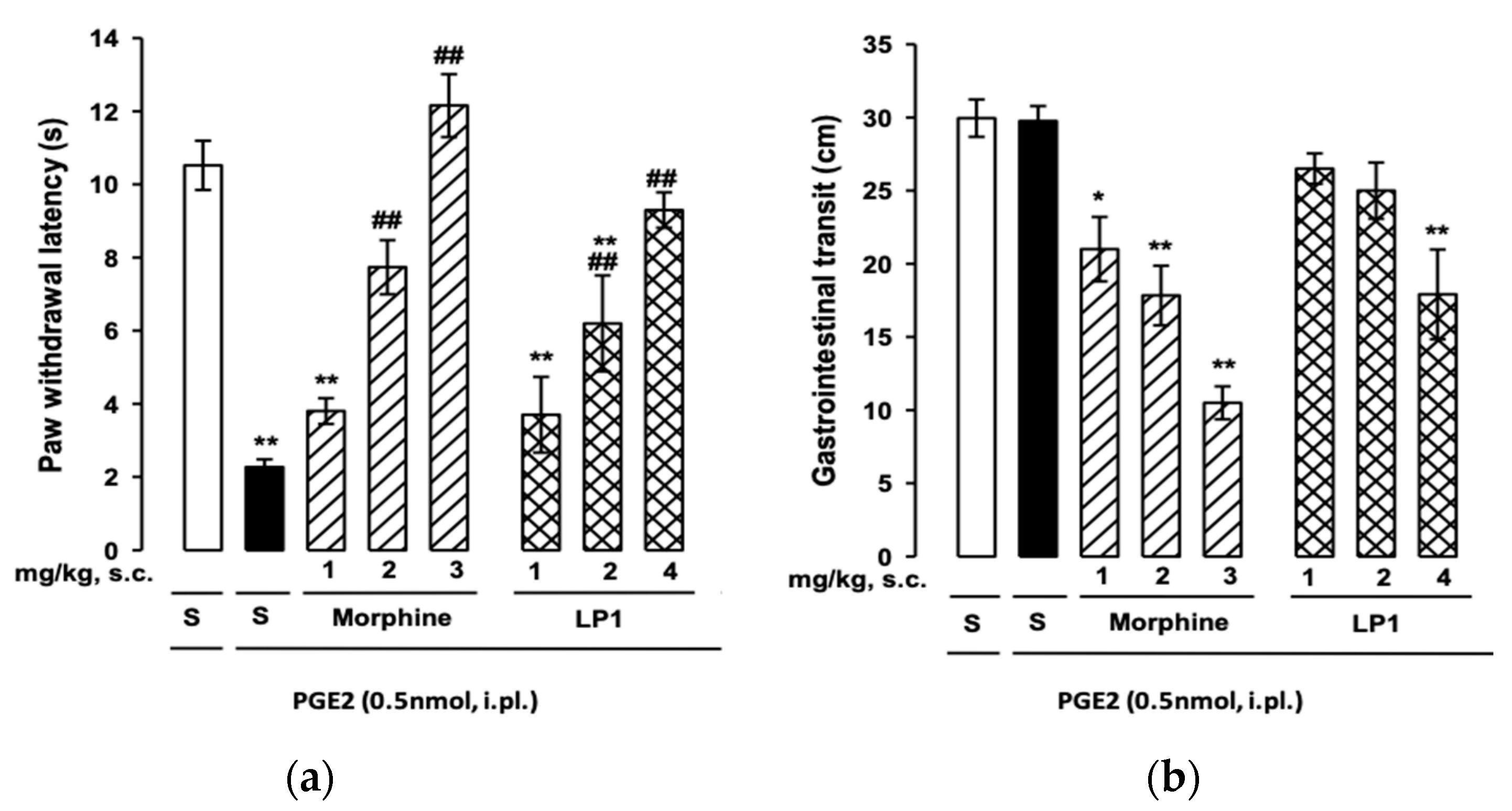
2.2.2. In Vivo Pharmacological Evaluation
3. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Argoff, C. Mechanisms of pain transmission and pharmacologic management. Curr. Med. Res. Opin. 2011, 27, 2019–2031. [Google Scholar] [CrossRef]
- Atchison, J.W.; Herndon, C.M.; Rusie, E. NSAIDs for Musculoskeletal Pain Management: Current Perspectives and Novel Strategies to Improve Safety. J. Manag. Care Pharm. 2013, 19, S3–S19. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Straus, W.L.; Balshaw, R.; Barlas, S.; Vogel, S.; Schnitzer, T.J. A comparison of the efficacy and safety of nonsteroidal antiinflammatory agents versus acetaminophen in the treatment of osteoarthritis: A meta-analysis. Arthritis Rheum. 2004, 51, 746–754. [Google Scholar] [CrossRef]
- Fleming, J.A.; O’Connor, B.D. Use of lidocaine patches for neuropathic pain in a comprehensive cancer centre. Pain Res. Manag. 2009, 14, 381–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, P.S.; Galer, B.S. Review of lidocaine patch 5% studies in the treatment of postherpetic neuralgia. Drugs 2004, 4, 937–947. [Google Scholar] [CrossRef] [PubMed]
- Field, M.J.; Li, Z.; Schwarz, J.B. Ca2+ channel alpha2-delta ligands for the treatment of neuropathic pain. J. Med. Chem. 2007, 50, 2569–2575. [Google Scholar] [CrossRef]
- Beal, B.; Moeller-Bertram, T.; Schilling, J.M.; Wallace, M.S. Gabapentin for once-daily treatment of post-herpetic neuralgia: A review. Clin. Interv. Aging 2012, 7, 249–255. [Google Scholar] [PubMed] [Green Version]
- Levine, J.D.; Gordon, N.C.; Smith, R.; McBryde, R. Desipramine enhances opiate postoperative analgesia. Pain 1986, 27, 45–49. [Google Scholar] [CrossRef]
- McQuay, H.J.; Tramer, M.; Nye, B.A.; Carroll, D.; Wiffen, P.J.; Moore, R.A. A systematic review of antidepressants in neuropathic pain. Pain 1996, 68, 217–227. [Google Scholar] [CrossRef]
- Mico, J.A.; Ardid, D.; Berrocoso, E.; Eschalier, A. Antidepressants and pain. Trends Pharmacol. Sci. 2006, 27, 348–354. [Google Scholar] [CrossRef]
- Golembiewski, J.; Rakic, A.M. Opioids. J. Perianesth. Nurs. 2010, 25, 258–260. [Google Scholar] [CrossRef]
- Bekhit, M.H. Opioid-induced hyperalgesia and tolerance. Am. J. Ther. 2010, 17, 498–510. [Google Scholar] [CrossRef]
- Benyamin, R.; Trescot, A.M.; Datta, S.; Buenaventura, R.; Adlaka, R.; Sehgal, N.; Glaser, S.E.; Vallejo, R. Opioid complications and side effects. Pain Physician 2008, 11, S181–S200. [Google Scholar]
- Dworkin, R.H.; O′Connor, A.B.; Backonja, M.; Farrar, J.T.; Finnerup, N.B.; Jensen, T.S.; Kalso, E.A.; Loeser, J.D.; Miaskowski, C.; Nurmikko, T.J.; et al. Pharmacologic management of neuropathic pain: Evidence-based recommendations. Pain 2007, 132, 237–251. [Google Scholar] [CrossRef]
- Breivik, E.K.; Barkvoll, P.; Skovlund, E. Combining diclofenac with acetaminophen or acetaminophen–codeine after oral surgery: A randomized, double-blind single-dose study. Clin. Pharmacol. Ther. 1999, 66, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Gatti, A.; Sabato, A.F.; Occhioni, R.; Baldeschi, G.C.; Reale, C. Controlled-release oxycodone and pregabalin in the treatment of neuropathic pain: Results of a multicenter Italian study. Eur. Neurol. 2009, 61, 129–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plummer, J.L.; Cmielewski, P.L.; Gourlay, G.K.; Owen, H.; Cousins, M.J. Antinociceptive and motor effects of intrathecal morphine combined with intrathecal clonidine, noradrenaline, carbachol or midazolam in rats. Pain 1992, 49, 145–152. [Google Scholar] [CrossRef]
- Richards, P.; Riff, D.; Kelen, R.; Stern, W. Analgesic and adverse effects of a fixed-dose morphine-oxycodone combination (MoxDuo®) in the treatment of postoperative pain. J. Opioid Manag. 2011, 7, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, B.D. Dual-opioid therapy: Changing the paradigm. Introduction. Pain Med. 2012, 13, S1-3. [Google Scholar] [PubMed] [Green Version]
- Hermanns, K.; Junker, U.; Nolte, T. Prolonged-release oxycodone/naloxone in the treatment of neuropathic pain—Results from a large observational study. Expert Opin. Pharmacother. 2012, 13, 299–311. [Google Scholar] [CrossRef]
- Leppert, W.; Zajaczkowska, R.; Wordliczek, J. The role of oxycodone/naloxone in the management of patients with pain and opioid-induced constipation. Expert Opin. Pharmacother. 2019, 20, 511–522. [Google Scholar] [CrossRef]
- Armstrong, S.C.; Cozza, K.L. Pharmacokinetic drug interactions of morphine, codeine, and their derivatives: Theory and clinical reality, Part II. Psychosomatics 2003, 44, 515–520. [Google Scholar] [CrossRef] [Green Version]
- Anighoro, A.; Bajorath, J.; Rastelli, G. Polypharmacology: Challenges and opportunities in drug discovery. J. Med. Chem. 2014, 57, 7874–7887. [Google Scholar] [CrossRef] [PubMed]
- Morphy, R.; Rankovic, R. Designed multiple ligands. An emerging drug discovery paradigm. J. Med. Chem. 2005, 48, 6523–6543. [Google Scholar] [CrossRef]
- Turnaturi, R.; Arico, G.; Ronsisvalle, G.; Pasquinucci, L.; Parenti, C. Multitarget Opioid/Non-opioid Ligands: A Potential Approach in Pain Management. Curr. Med. Chem. 2016, 23, 4506–4528. [Google Scholar] [CrossRef]
- Burgess, G.; Williams, D. The discovery and development of analgesics: New mechanisms, new modalities. J. Clin. Investig. 2010, 120, 3753–3759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turnaturi, R.; Aricò, G.; Ronsisvalle, G.; Parenti, C.; Pasquinucci, L. Multitarget opioid ligands in pain relief: New players in an old game. Eur. J. Med. Chem. 2016, 108, 211–228. [Google Scholar] [CrossRef]
- Stevenson, G.W.; Folk, J.E.; Linsenmayer, D.C.; Rice, K.C.; Negus, S.S. Opioid interactions in rhesus monkeys: Effects of delta + mu and delta + kappa agonists on schedule-controlled responding and thermal nociception. J. Pharmacol. Exp. Ther. 2003, 307, 1054–1064. [Google Scholar] [CrossRef] [PubMed]
- Negus, S.S.; Bear, A.E.; Folk, J.E.; Rice, K.C. Role of delta opioid efficacy as a determinant of mu/delta opioid interactions in rhesus monkeys. Eur. J. Pharmacol. 2009, 602, 92–100. [Google Scholar] [CrossRef] [Green Version]
- Sykes, K.T.; White, S.R.; Hurley, R.W.; Mizoguchi, H.; Tseng, L.F.; Hammond, D.L. Mechanisms responsible for the enhanced antinociceptive effects of micro- opioid receptor agonists in the rostral ventromedial medulla of male rats with persistent inflammatory pain. J. Pharmacol. Exp. Ther. 2007, 322, 813–821. [Google Scholar] [CrossRef]
- Gengo, P.J.; Pettit, H.O.; O’Neill, S.J.; Wei, K.; McNutt, R.; Bishop, M.J.; Chang, K.J. DPI-3290 [(+)-3-((alpha-R)-alpha-((2S,5R)-4-allyl-2,5-dimethyl-1-piperazinyl)-3-hydroxybenzyl)-N-(3- fluorophenyl)-N-methylbenzamide]. I. A mixed opioid agonist with potent antinociceptive activity. J. Pharmacol. Exp. Ther. 2003, 307, 1221–1226. [Google Scholar] [CrossRef]
- Healy, J.R.; Bezawada, P.; Shim, J.; Jones, J.W.; Kane, M.A.; MacKerell, A.D., Jr.; Coop, A.; Matsumoto, R.R. Synthesis, modeling, and pharmacological evaluation of UMB 425, a mixed μ agonist/δ antagonist opioid analgesic with reduced tolerance liabilities. ACS Chem. Neurosci. 2013, 4, 1256–1266. [Google Scholar] [CrossRef] [Green Version]
- Harland, A.A.; Yeomans, L.; Griggs, N.W.; Anand, J.P.; Pogozheva, I.D.; Jutkiewicz, E.M.; Traynor, J.R.; Mosberg, H.I. Further Optimization and Evaluation of Bioavailable, Mixed-Efficacy μ-Opioid Receptor (MOR) Agonists/δ-Opioid Receptor (DOR) Antagonists: Balancing MOR and DOR Affinities. J. Med. Chem. 2015, 58, 8952–8969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turnaturi, R.; Marrazzo, A.; Parenti, C.; Pasquinucci, L. Benzomorphan scaffold for opioid analgesics and pharmacological tools development: A comprehensive review. Eur. J. Med. Chem. 2018, 148, 410–422. [Google Scholar] [CrossRef] [PubMed]
- Turnaturi, R.; Montenegro, L.; Marrazzo, A.; Parenti, R.; Pasquinucci, L.; Parenti, C. Benzomorphan skeleton, a versatile scaffold for different targets: A comprehensive review. Eur. J. Med. Chem. 2018, 155, 492–502. [Google Scholar] [CrossRef]
- Turnaturi, R.; Pasquinucci, L.; Chiechio, S.; Grasso, M.; Marrazzo, A.; Amata, E.; Dichiara, M.; Prezzavento, O.; Parenti, C. Exploiting the Power of Stereochemistry in Drug Action: 3-[(2S,6S,11S)-8-Hydroxy-6,11-dimethyl-1,4,5,6-tetrahydro-2,6-methano-3-benzazocin-3(2>H)-yl]-N-phenylpropanamide as Potent Sigma-1 Receptor Antagonist. ACS Chem. Neurosci. 2020, 11, 999–1005. [Google Scholar] [CrossRef] [PubMed]
- Bidlack, J.M.; Cohen, D.J.; McLaughlin, J.P.; Lou, R.; Ye, Y.; Wentland, M.P. 8-Carboxamidocyclazocine: A long-acting, novel benzomorphan. J. Pharmacol. Exp. Ther. 2002, 302, 374–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wentland, M.P.; VanAlstine, M.; Kucejko, R.; Lou, R.; Cohen, D.J.; Parkhill, A.L.; Bidlack, J.M. Redefining the structure-activity relationships of 2,6-methano-3-benzazocines. 4. Opioid receptor binding properties of 8-[N-(4′-phenyl)-phenethyl)carboxamido] analogues of cyclazocine and ethylketocycalzocine. J. Med. Chem. 2006, 49, 5635–5639. [Google Scholar] [CrossRef] [PubMed]
- May, E.L.; Aceto, M.D.; Bowman, E.R.; Bentley, C.; Martin, B.R.; Harris, L.S.; Medzihradsky, F.; Mattson, M.V.; Jacobson, A.E. Antipodal alpha-N-(methyl through decyl)-N-normetazocines (5,9 alpha-dimethyl-2′-hydroxy-6,7- benzomorphans): In vitro and in vivo properties. J. Med. Chem. 1994, 37, 3408–3418. [Google Scholar] [CrossRef]
- Casy, A.F.; Parfitt, R.T. Opioid Analgesics: Chemistry and Receptors; Plenum Press: New York, NY, USA, 1986. [Google Scholar]
- Pasquinucci, L.; Prezzavento, O.; Marrazzo, A.; Amata, E.; Ronsisvalle, S.; Georgoussi, Z.; Fourla, D.D.; Scoto, G.M.; Parenti, C.; Aricò, G.; et al. Evaluation of N-substitution in 6,7-benzomorphan compounds. Bioorg. Med. Chem. 2010, 18, 4975–4982. [Google Scholar] [CrossRef]
- Pasquinucci, L.; Parenti, C.; Turnaturi, R.; Aricò, G.; Marrazzo, A.; Prezzavento, O.; Ronsisvalle, S.; Georgoussi, Z.; Fourla, D.D.; Scoto, G.M.; et al. The benzomorphan-based LP1 ligand is a suitable MOR/DOR agonist for chronic pain treatment. Life Sci. 2012, 90, 66–70. [Google Scholar] [CrossRef]
- Pasquinucci, L.; Turnaturi, R.; Aricò, G.; Parenti, C.; Pallaki, P.; Georgoussi, Z.; Ronsisvalle, S. Evaluation of N-substituent structural variations in opioid receptor profile of LP1. Bioorg. Med. Chem. 2016, 24, 2832–2842. [Google Scholar] [CrossRef] [PubMed]
- Pasquinucci, L.; Parenti, C.; Amata, E.; Georgoussi, Z.; Pallaki, P.; Camarda, V.; Calò, G.; Arena, E.; Montenegro, L.; Turnaturi, R. Synthesis and Structure-Activity Relationships of (-)-cis-N-Normetazocine-Based LP1 Derivatives. Pharmaceuticals 2018, 11, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turnaturi, R.; Parenti, C.; Prezzavento, O.; Marrazzo, A.; Pallaki, P.; Georgoussi, Z.; Amata, E.; Pasquinucci, L. Synthesis and Structure-Activity Relationships of LP1 Derivatives: N-Methyl-N-phenylethylamino Analogues as Novel MOR Agonists. Molecules 2018, 23, 677. [Google Scholar] [CrossRef] [Green Version]
- Pasquinucci, L.; Parenti, C.; Ruiz-Cantero, M.C.; Georgoussi, Z.; Pallaki, P.; Cobos, E.J.; Amata, E.; Marrazzo, A.; Prezzavento, O.; Arena, E.; et al. Novel N-Substituted Benzomorphan-Based Compounds: From MOR-Agonist/DOR-Antagonist to Biased/Unbiased MOR Agonists. ACS Med. Chem. Lett. 2020, 11, 678–685. [Google Scholar] [CrossRef] [PubMed]
- Pasquinucci, L.; Turnaturi, R.; Prezzavento, O.; Arena, E.; Aricò, G.; Georgoussi, Z.; Parenti, R.; Cantarella, G.; Parenti, C. Development of novel LP1-based analogues with enhanced delta opioid receptor profile. Bioorg. Med. Chem. 2017, 25, 4745–4752. [Google Scholar] [CrossRef]
- Pasquinucci, L.; Turnaturi, R.; Calò, G.; Pappalardo, F.; Ferrari, F.; Russo, G.; Arena, E.; Montenegro, L.; Chiechio, S.; Prezzavento, O.; et al. (2S)-N-2-methoxy-2-phenylethyl-6,7-benzomorphan compound (2S-LP2): Discovery of a biased mu/delta opioid receptor agonist. Eur. J. Med. Chem. 2019, 168, 189–198. [Google Scholar] [CrossRef]
- Parenti, C.; Turnaturi, R.; Aricò, G.; Marrazzo, A.; Prezzavento, O.; Ronsisvalle, S.; Scoto, G.M.; Ronsisvalle, G.; Pasquinucci, L. Antinociceptive profile of LP1, a non-peptide multitarget opioid ligand. Life Sci. 2012, 90, 957–961. [Google Scholar] [CrossRef] [PubMed]
- Parenti, C.; Turnaturi, R.; Aricò, G.; Gramowski-Voss, A.; Schroeder, O.H.; Marrazzo, A.; Prezzavento, O.; Ronsisvalle, S.; Scoto, G.M.; Ronsisvalle, G.; et al. The multitarget opioid ligand LP1′s effects in persistent pain and in primary cell neuronal cultures. Neuropharmacology 2013, 71, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Accolla, M.L.; Turnaturi, R.; Sarpietro, M.G.; Ronsisvalle, S.; Castelli, F.; Pasquinucci, L. Differential scanning calorimetry approach to investigate the transfer of the multitarget opioid analgesic LP1 to biomembrane model. Eur. J. Med. Chem. 2014, 77, 84–90. [Google Scholar] [CrossRef]
- Mizoguchi, H.; Watanabe, H.; Hayashi, T.; Sakurada, W.; Sawai, T.; Fujimura, T.; Sakurada, T.; Sakurada, S. Possible involvement of Dynorphin A-(1–17) release via μ1-opioid receptors in spinal antinociception by endomorphin-2. J. Pharmacol. Exp. Ther. 2006, 317, 362–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakurada, S.; Sawai, T.; Mizoguchi, H.; Watanabe, H.; Watanabe, C.; Yonezawa, A.; Morimoto, M.; Sato, T.; Komatsu, T.; Sakurada, T. Possible involvement of dynorphin A release via mu1-opioid receptor on supraspinal antinociception of endomorphin-2. Peptides 2008, 29, 1554–1560. [Google Scholar] [CrossRef]
- Roy, S.; Liu, H.C.; Loh, H.H. mu-Opioid receptor-knockout mice: The role of mu-opioid receptor in gastrointestinal transit. Brain Res. Mol. Brain Res. 1998, 56, 281–283. [Google Scholar]
- Pradhan, A.A.; Smith, M.L.; Kieffer, B.L.; Evans, C.J. Ligand-directed signalling within the opioid receptor family. Br. J. Pharmacol. 2012, 167, 960–969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu Sin Chung, P.; Boehrer, A.; Stephan, A.; Matifas, A.; Scherrer, G.; Darcq, E.; Befort, K.; Kieffer, B.L. Delta opioid receptors expressed in forebrain GABAergic neurons are responsible for SNC80-induced seizures. Behav. Brain Res. 2015, 278, 429–434. [Google Scholar] [CrossRef] [Green Version]
- Gendron, L.; Cahill, C.M.; von Zastrow, M.; Schiller, P.W.; Pineyro, G. Molecular pharmacology of d-opioid receptors. Pharmacol. Rev. 2016, 68, 631–700. [Google Scholar] [CrossRef] [Green Version]
- Vicente-Sanchez, A.; Segura, L.; Pradhan, A.A. The delta opioid receptor tool box. Neuroscience 2016, 338, 145–159. [Google Scholar] [CrossRef] [Green Version]
- Turnaturi, R.; Chiechio, S.; Salerno, L.; Rescifina, A.; Pittalà, V.; Cantarella, G.; Tomarchio, E.; Parenti, C.; Pasquinucci, L. Progress in the development of more effective and safer analgesics for pain management. Eur. J. Med. Chem. 2019, 183, 111701. [Google Scholar] [CrossRef]
- Conibear, A.E.; Asghar, J.; Hill, R.; Henderson, G.; Borbely, E.; Tekus, V.; Helyes, Z.; Palandri, J.; Bailey, C.; Starke, I.; et al. A Novel G Protein-Biased Agonist at the δ Opioid Receptor with Analgesic Efficacy in Models of Chronic Pain. J. Pharmacol. Exp. Ther. 2020, 372, 224–236. [Google Scholar] [CrossRef] [Green Version]
- Pasquinucci, L.; Turnaturi, R.; Montenegro, L.; Caraci, F.; Chiechio, S.; Parenti, C. Simultaneous targeting of MOR/DOR: A useful strategy for inflammatory pain modulation. Eur. J. Pharmacol. 2019, 847, 97–102. [Google Scholar] [CrossRef]
- Vicario, N.; Pasquinucci, L.; Spitale, F.M.; Chiechio, S.; Turnaturi, R.; Caraci, F.; Tibullo, D.; Avola, R.; Gulino, R.; Parenti, R.; et al. Simultaneous Activation of Mu and Delta Opioid Receptors Reduces Allodynia and Astrocytic Connexin 43 in an Animal Model of Neuropathic Pain. Mol. Neurobiol. 2019, 56, 7338–7354. [Google Scholar] [CrossRef] [PubMed]
- Vicario, N.; Turnaturi, R.; Spitale, F.M.; Torrisi, F.; Zappalà, A.; Gulino, R.; Pasquinucci, L.; Chiechio, S.; Parenti, C.; Parenti, R. Intercellular communication and ion channels in neuropathic pain chronicization. Inflamm. Res. 2020, 69, 841–850. [Google Scholar] [CrossRef] [PubMed]
- Polo, S.; Díaz, A.F.; Gallardo, N.; Leánez, S.; Balboni, G.; Pol, O. Treatment with the Delta Opioid Agonist UFP-512 Alleviates Chronic Inflammatory and Neuropathic Pain: Mechanisms Implicated. Front. Pharmacol. 2019, 10, 283. [Google Scholar]
- Grasso, M.; Caruso, G.; Musso, N.; Turnaturi, R.; Pasquinucci, L.; Vicario, N.; Spitale, F.M.; Parenti, R.; Chiechio, S.; Parenti, C.; et al. The Multimodal MOR/DOR Agonist LP2 Exerts Analgesic Effects in an Animal Model. In Proceedings of the 40° Congresso Nazionale SIF—Società Italiana di Farmacologia, Digital Edition. 9–13 March 2021. [Google Scholar]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compd | Ki (nM) ± S.D. | IC50 (nM) ± S.D. | ED50 mg/kg | |||
|---|---|---|---|---|---|---|
| GPI e | MVD e | Tail Flick Test | ||||
| MOPr | DOPr | KOPr | MOPr | DOPr | ||
| LP1(1) a | 0.83 ± 0.05 | 29.1 ± 1 | 110 ± 6 | 6.8 ± 0.03 b | pA2 of 7.2 | 2.03 |
| (2) b | 38 ± 4 | 210 ± 30 | 800 ± 160 | pA2 of 8.6 | - | AD50 2.0 |
| IC50 (nM) ± S.D. | ||||||
| cAMP inhibition | ||||||
| (3) c | 6.1 ± 0.5 | 147 ± 5.7 | 31 ± 1.3 | 11.5 ± 2.5 | - | 4.33 |
| IC50 (nM) ± S.D. | ||||||
| GPI e | MVD e | |||||
| LP2 (4) d | 1.08 ± 0.10 | 6.60 ± 0.60 | 15.22 ± 0.80 | 21.5 ± 1.3 | 4.4 ± 0.7 | 0.9 |
| (5) d | 2.47 ± 0.3 | 9.6 ± 0.5 | 7.30 ± 0.2 | 49.2 ± 2 | 10.8 ± 1.3 | 1.3 |
| (6) d | 0.5 ± 0.2 | 0.8 ± 0.2 | 2.22 ± 0.2 | 9.9 ± 0.9 | 11.8 ± 1 | 1.0 |
| (7) d | 60 ± 1.3 | 160 ± 2.8 | 10.51 ± 0.9 | 194.4 ± 33 | 793.5 ± 43 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pasquinucci, L.; Parenti, C.; Georgoussi, Z.; Reina, L.; Tomarchio, E.; Turnaturi, R. LP1 and LP2: Dual-Target MOPr/DOPr Ligands as Drug Candidates for Persistent Pain Relief. Molecules 2021, 26, 4168. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26144168
Pasquinucci L, Parenti C, Georgoussi Z, Reina L, Tomarchio E, Turnaturi R. LP1 and LP2: Dual-Target MOPr/DOPr Ligands as Drug Candidates for Persistent Pain Relief. Molecules. 2021; 26(14):4168. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26144168
Chicago/Turabian StylePasquinucci, Lorella, Carmela Parenti, Zafiroula Georgoussi, Lorena Reina, Emilia Tomarchio, and Rita Turnaturi. 2021. "LP1 and LP2: Dual-Target MOPr/DOPr Ligands as Drug Candidates for Persistent Pain Relief" Molecules 26, no. 14: 4168. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26144168