
Chiral Imidazolium Prolinate Salts as Efficient Synzymatic Organocatalysts for the Asymmetric Aldol Reaction

 ,
, 
Abstract
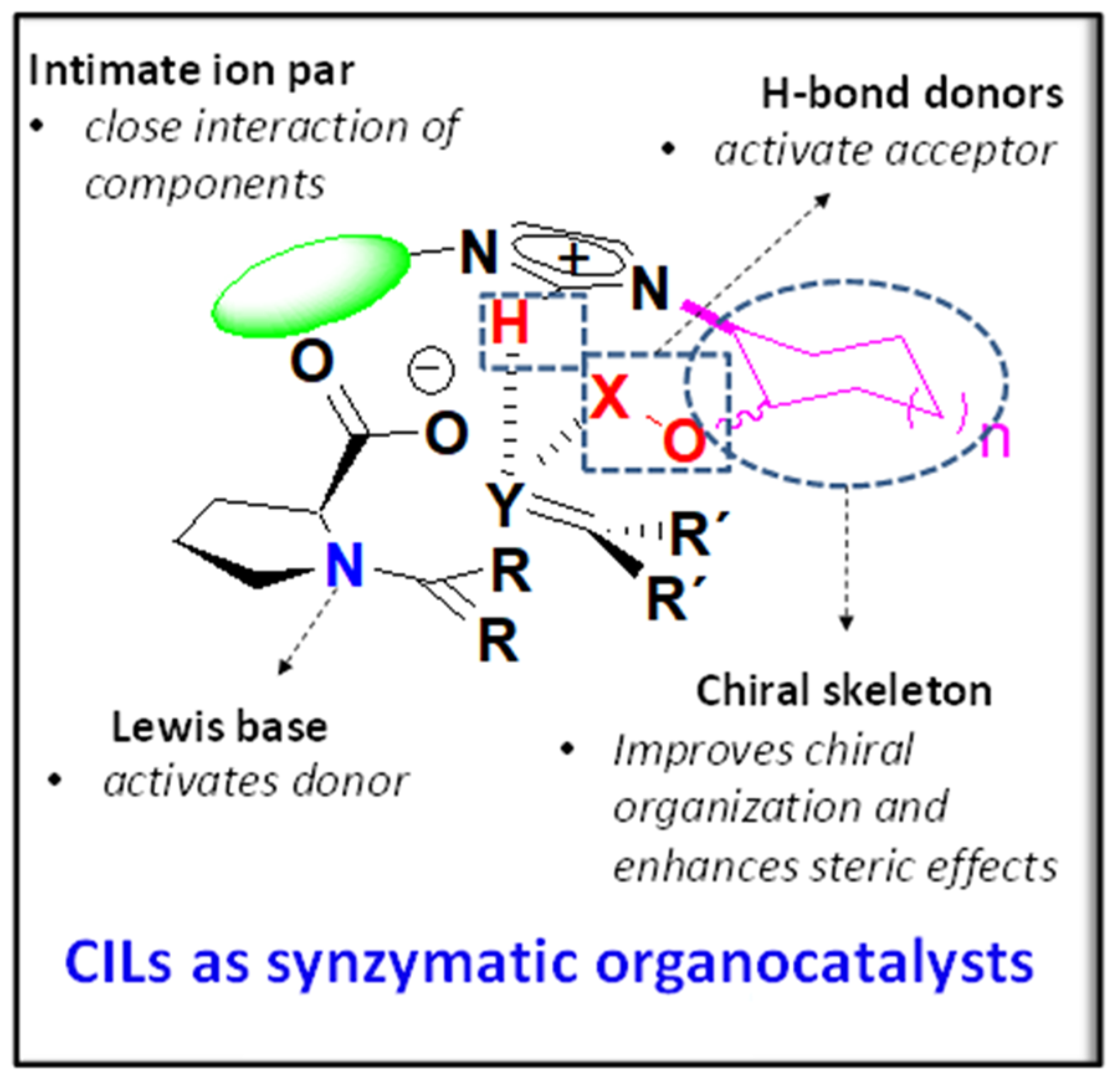
:1. Introduction
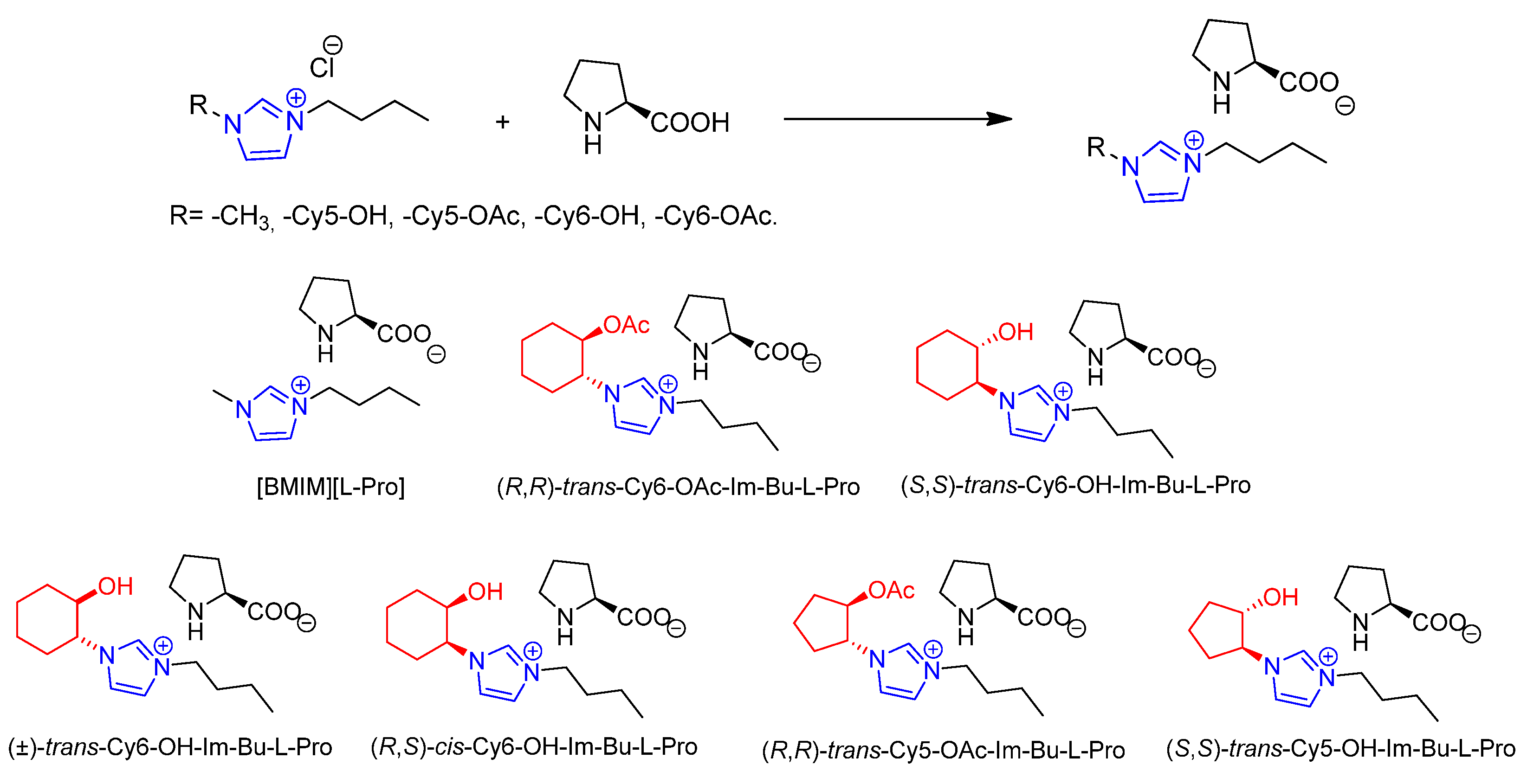
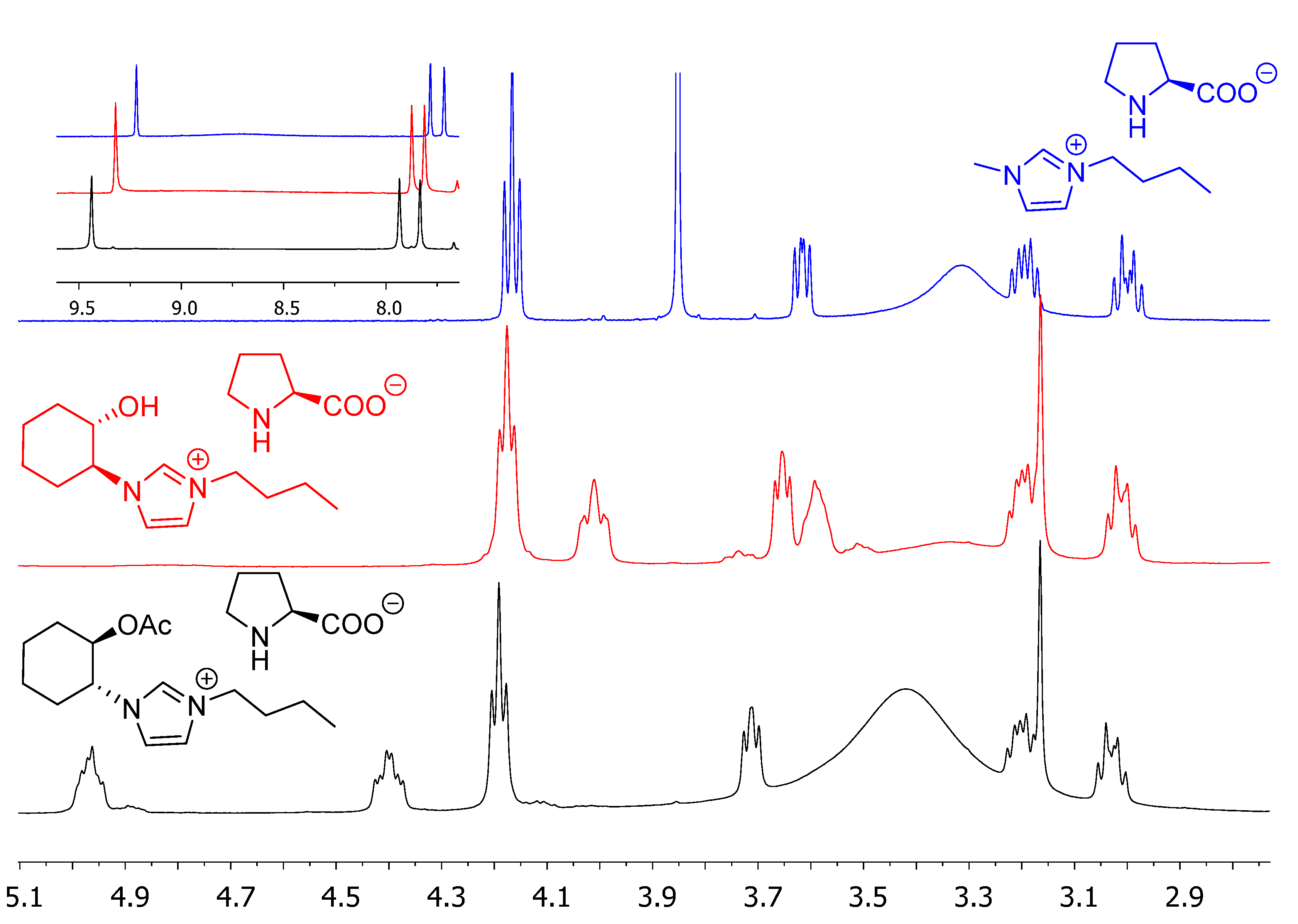
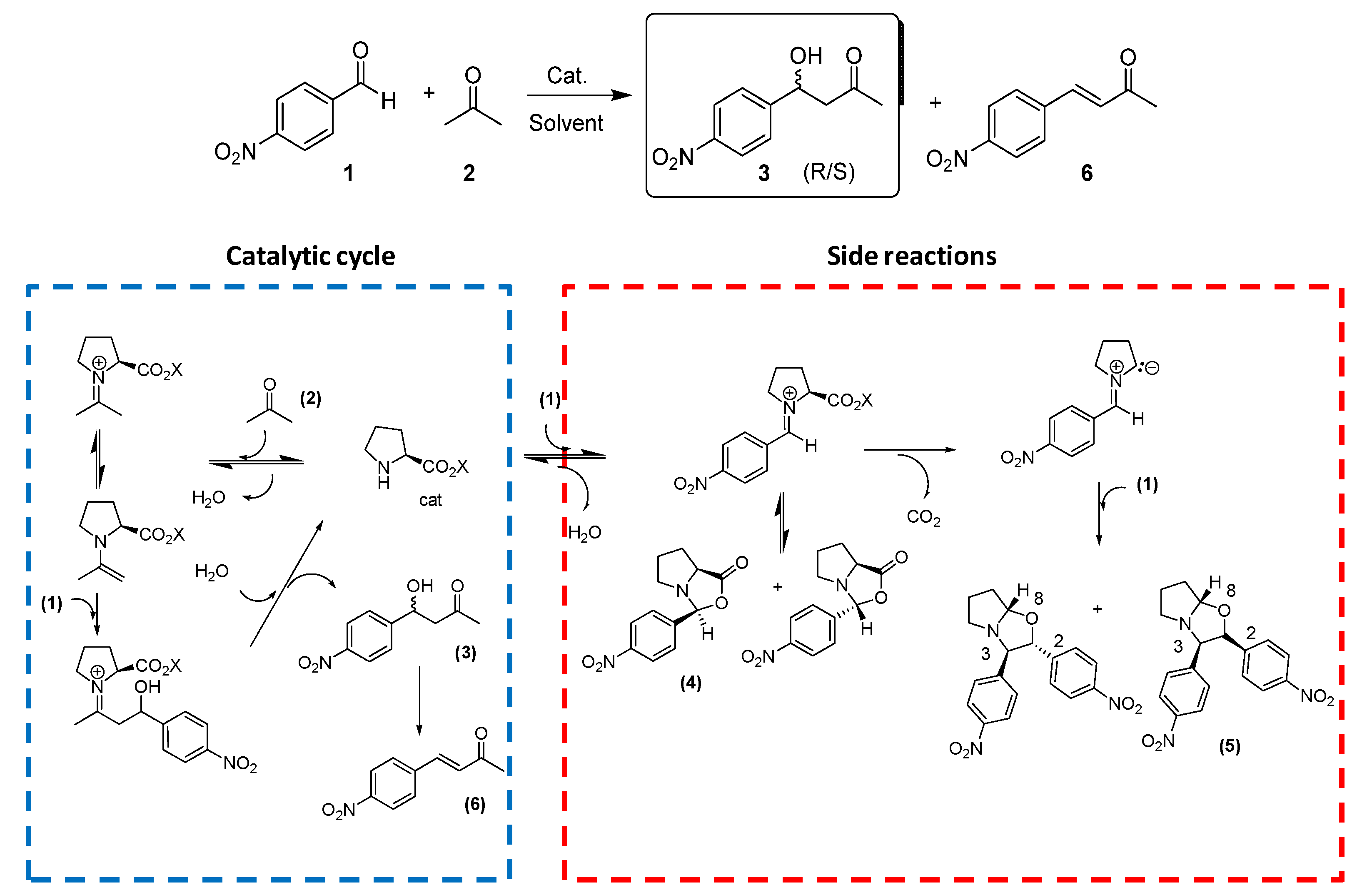
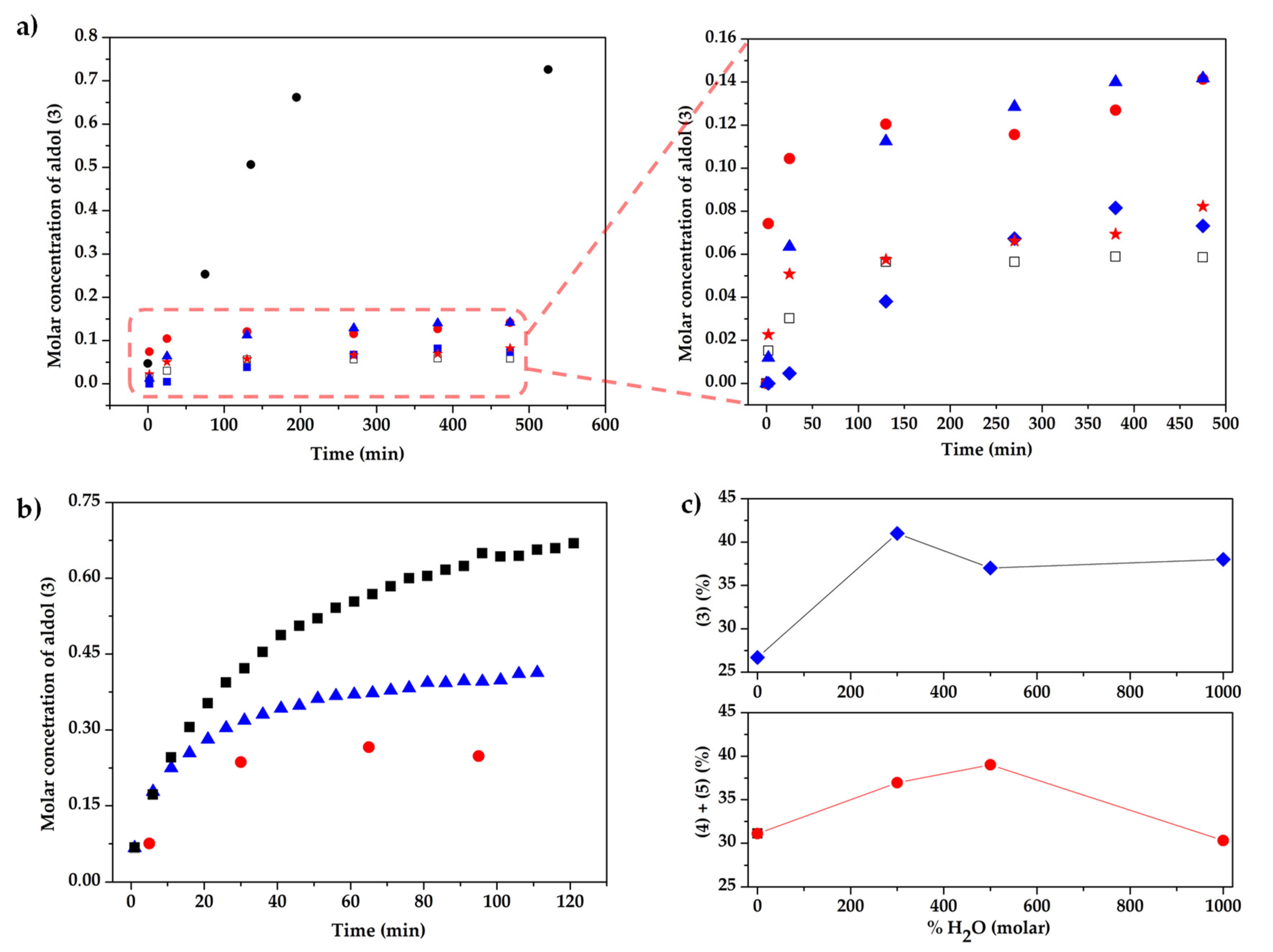
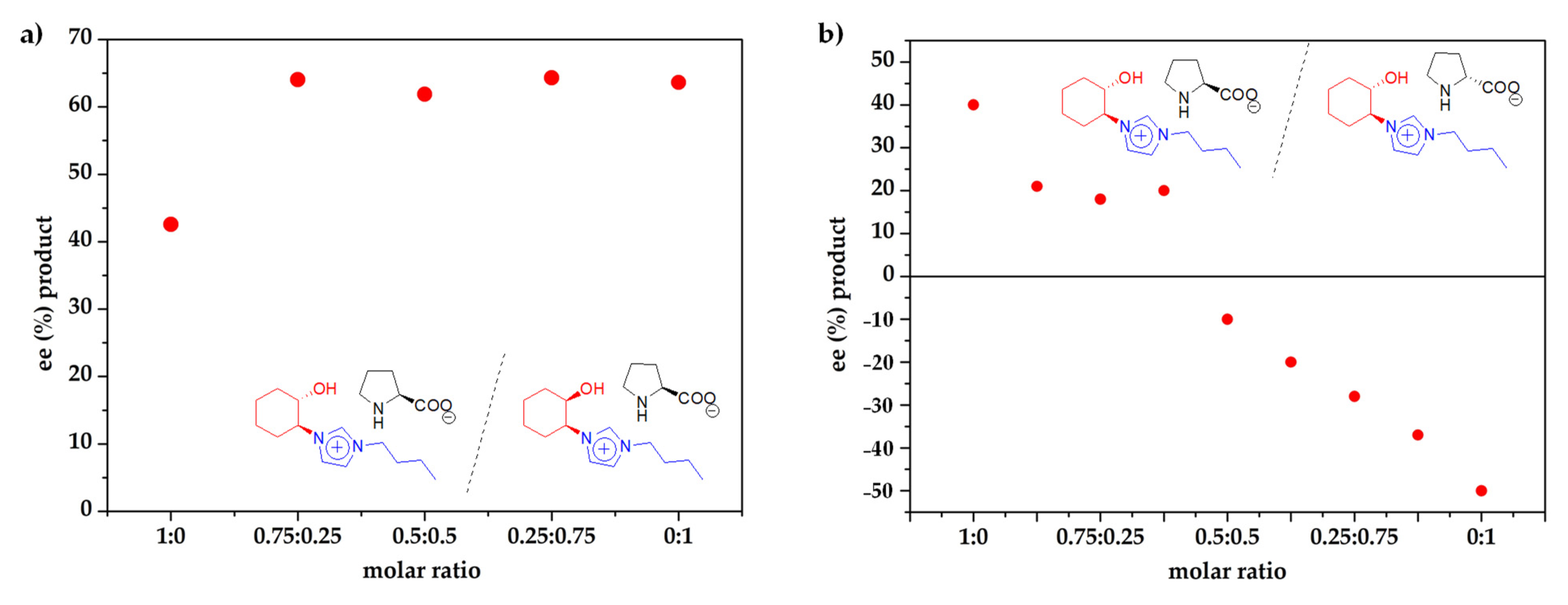
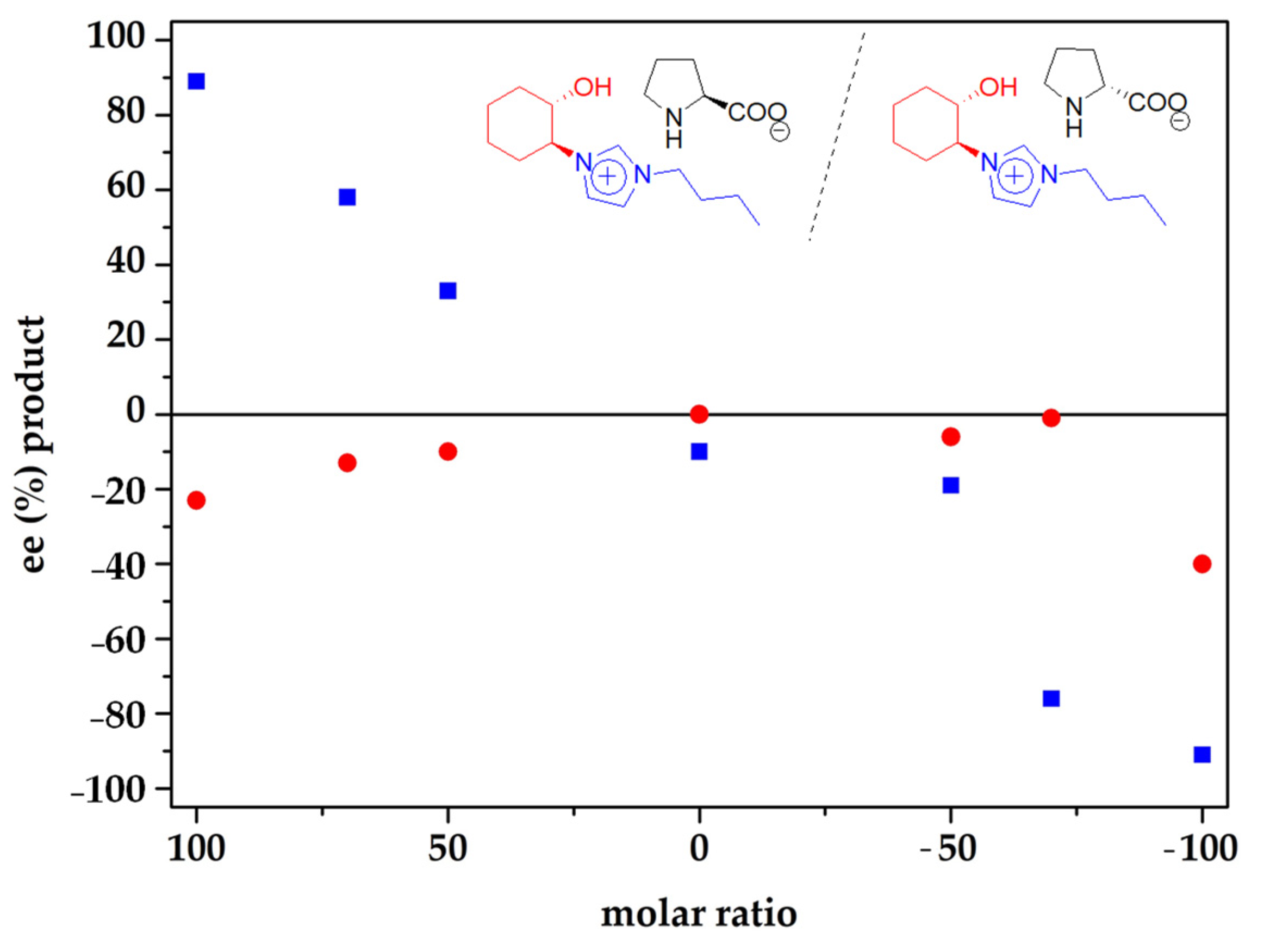
2. Results and Discussion
3. Materials and Methods
3.1. Experimental Section
3.2. General Procedure for the Preparation of Imidazolium Prolinates
3.3. General Procedure for the Aldol Reactions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Han, B.; He, X.H.; Liu, Y.Q.; He, G.; Peng, C.; Li, J.L. Asymmetric organocatalysis: An enabling technology for medicinal chemistry. Chem. Soc. Rev. 2021, 50, 1522–1586. [Google Scholar] [CrossRef] [PubMed]
- Dondoni, A.; Massi, A. Asymmetric organocatalysis: From infancy to adolescence. Angew. Chem. Int. Ed. 2008, 47, 4638–4660. [Google Scholar] [CrossRef]
- Berkessel, A.; Groger, H. Asymmetric Organocatalysis; Wiley: Weinhein, Germany, 2005. [Google Scholar]
- List, B. Enamine catalysis is a powerful strategy for the catalytic generation and use of carbanion equivalents. Acc. Chem. Res. 2004, 37, 548–557. [Google Scholar] [CrossRef]
- Notz, W.; Tanaka, F.; Barbas III, C.F. Enamine-based organocatalysis with proline and diamines: The development of direct catalytic asymmetric aldol, Mannich, Michael, and Diels-Alder reactions. Acc. Chem. Res. 2004, 37, 580–591. [Google Scholar] [CrossRef]
- Alarcon-Matus, E.; Alvarado, C.; Romero-Ceronio, N.; Ramos-Rivera, E.M.; Lobato-Garcia, C.E. Proline-derived Long-aliphatic-chain Amphiphilic Organocatalysts (PDLACAOs) for Asymmetric Reactions in Aqueous Media. Asian J. Org. Chem. 2020, 9, 1667–1687. [Google Scholar] [CrossRef]
- Jimeno, C. Amino Acylguanidines as Bioinspired Catalysts for the Asymmetric Aldol Reaction. Molecules 2021, 26, 826. [Google Scholar] [CrossRef]
- Liu, J.; Wang, L. Recent Advances in Asymmetric Reactions Catalyzed by Proline and Its Derivatives. Synthesis 2017, 49, 960–972. [Google Scholar]
- Trost, B.M.; Brindle, C.S. The direct catalytic asymmetric aldol reaction. Chem. Soc. Rev. 2010, 39, 1600–1632. [Google Scholar] [CrossRef] [Green Version]
- Meyer, C.C.; Ortiz, E.; Krische, M.J. Catalytic Reductive Aldol and Mannich Reactions of Enone, Acrylate, and Vinyl Heteroaromatic Pronucleophiles. Chem. Rev. 2020, 120, 3721–3748. [Google Scholar] [CrossRef]
- Schoevaart, R.; van Rantwijk, F.; Sheldon, R.A. Stereochemistry of nonnatural aldol reactions catalyzed by DHAP aldolases. Biotechnol. Bioeng. 2000, 70, 349–352. [Google Scholar] [CrossRef]
- Shiflett, M.B. (Ed.) Commercial Applications of Ionic Liquids; Springer-Nature: Berlin/Heidelberg, Germany, 2020. [Google Scholar]
- Montolio, S.; Altava, B.; Garcia-Verdugo, E.; Luis, S.V. Supported ILs and Materials Based on ILs for the Development of Green Synthetic Processes and Procedures. In Green Synthetic Processes and Procedures; Ballini, R., Ed.; RSC: London, UK, 2019. [Google Scholar]
- Sheldon, R.A. Green and sustainable manufacture of chemicals from biomass: State of the art. Green Chem. 2014, 16, 950–963. [Google Scholar] [CrossRef]
- Van Rantwijk, F.; Sheldon, R.A. Biocatalysis in ionic liquids. Chem. Rev. 2007, 107, 2757–2785. [Google Scholar] [CrossRef]
- Flieger, J.; Feder-Kubis, J.; Tatarczak-Michalewska, M. Chiral Ionic Liquids: Structural Diversity, Properties and Applications in Selected Separation Techniques. Int. J. Mol. Sci. 2020, 21, 4253. [Google Scholar] [CrossRef]
- González-Rodríguez, J.; Valls, A.; Arias Abrodo, P.; Gutiérrez Álvarez, M.D.; González-Álvarez, J.; Altava, B.; Luis, S.V. Polymeric Ionic Liquids Derived from L-Valine for the Preparation of Highly Selective Silica-Supported Stationary Phases in Gas Chromatography. Polymers 2020, 12, 2348. [Google Scholar] [CrossRef] [PubMed]
- Karimi, B.; Tavakolian, M.; Akbari, M.; Mansouri, F. Ionic Liquids in Asymmetric Synthesis: An Overall View from Reaction Media to Supported Ionic Liquid Catalysis. ChemCatChem 2018, 10, 3173–3205. [Google Scholar] [CrossRef]
- Gaertner, P.; Bica, K. Applications of Chiral Ionic Liquids. Eur. J. Org. Chem. 2008, 2008, 3235–3250. [Google Scholar]
- Gonzalez, L.; Altava, B.; Bolte, M.; Burguete, M.I.; Garcia-Verdugo, E.; Luis, S.V. Synthesis of Chiral Room Temperature Ionic Liquids from Amino Acids-Application in Chiral Molecular Recognition. Eur. J. Org. Chem. 2012, 2012, 4996–5009. [Google Scholar] [CrossRef] [Green Version]
- Gauchot, V.; Gravel, J.; Vidal, M.; Charbonneau, M.; Kairouz, V.; Schmitzer, A.R. Imidazolium and Benzimidazolium Salts: A Veritable Playground for Organic and Supramolecular Chemists. Synlett 2015, 26, 2763–2779. [Google Scholar]
- Ou, W.H.; Huang, Z.Z. An efficient and practical synthesis of chiral imidazolium ionic liquids and their application in an enantioselective Michael reaction. Green Chem. 2006, 8, 731–734. [Google Scholar] [CrossRef]
- Ni, B.; Headley, A.D. Novel imidazolium chiral ionic liquids that contain a urea functionality. Tetrahedron Lett. 2006, 47, 7331–7334. [Google Scholar] [CrossRef]
- Ni, B.; Headley, A.D.; Li, G. Design and synthesis of C-2 substituted chiral imidazolium ionic liquids from amino acid derivatives. J. Org. Chem. 2005, 70, 10600–10602. [Google Scholar] [CrossRef]
- Miao, W.; Chan, T.H. Ionic-Liquid-Supported Organocatalyst: Efficient and Recyclable Ionic-Liquid-Anchored Proline for Asymmetric Aldol Reaction. Adv. Synth. Catal. 2006, 348, 1711–1718. [Google Scholar] [CrossRef]
- Siyutkin, D.E.; Kucherenko, A.S.; Zlotin, S.G. Hydroxy-α-amino acids modified by ionic liquid moieties: Recoverable organocatalysts for asymmetric aldol reactions in the presence of water. Tetrahedron 2009, 65, 1366–1372. [Google Scholar] [CrossRef]
- Kucherenko, A.S.; Perepelkin, V.V.; Zhdankina, G.M.; Kryshtal, G.V.; Srinivasan, E.; Inanib, H.; Zlotin, S.G. Ionic liquid supported 4-HO-Pro-Val derived organocatalysts for asymmetric aldol reactions in the presence of water. Mendeleev Commun. 2016, 26, 388–390. [Google Scholar] [CrossRef]
- Qian, Y.; Zheng, X.; Wang, Y. A Green and Efficient Asymmetric Aldol Reaction Catalyzed by a Chiral Anion Modified Ionic Liquid. Eur. J. Org. Chem. 2010, 3672–3677. [Google Scholar] [CrossRef]
- Fukumuto, K.; Yoshizawa, M.; Ohno, H. Room Temperature Ionic Liquids from 20 Natural Amino Acids. J. Am. Chem. Soc. 2005, 127, 2398–2399. [Google Scholar] [CrossRef] [PubMed]
- Porcar, R.; Ríos-Lombardía, N.; Busto, E.; Gotor-Fernández, V.; Gotor, V.; García-Verdugo, E.; Burguete, M.I.; Luis, S.V. Chemoenzymatic synthesis of optically active 2-(2- or 4-substituted-1H-imidazol-1-yl)cycloalcanols. Chiral additives for (L)-proline. Catal. Sci. Technol. 2013, 3, 2596–2601. [Google Scholar] [CrossRef]
- González, L.; Escorihuela, J.; Altava, B.; Burguete, M.I.; Luis, S.V. Chiral Room Temperature Ionic Liquids as Enantioselective Promoters for the Asymmetric Aldol Reaction. Eur. J. Org. Chem. 2014, 2014, 5356–5363. [Google Scholar] [CrossRef]
- Porcar, R.; Burguete, M.I.; Lozano, P.; Garcia-Verdugo, E.; Luis, S.V. Supramolecular Interactions Based on Ionic Liquids for Tuning of the Catalytic Efficiency of (L)-Proline. ACS Sustain. Chem. Eng. 2016, 4, 6062–6071. [Google Scholar] [CrossRef]
- Hollfelder, F.; Kirby, A.J.; Tawfik, D.S. Efficient Catalysis of Proton Transfer by Synzymes. J. Am. Chem. Soc. 1997, 119, 9578–9579. [Google Scholar] [CrossRef]
- Haimov, A.; Cohen, H.; Neumann, R. Alkylated polyethyleneimine/polyoxometalate synzymes as catalysts for the oxidation of hydrophobic substrates in water with hydrogen peroxide. J. Am. Chem. Soc. 2004, 126, 11762–11763. [Google Scholar] [CrossRef]
- Roux, Y.; Ricoux, R.; Avenier, F.; Mahy, J.P. Bio-inspired electron-delivering system for reductive activation of dioxygen at metal centres towards artificial flavoenzymes. Nat. Commun. 2015, 6, 8509–8517. [Google Scholar] [CrossRef] [Green Version]
- Esteve, F.; Altava, B.; Burguete, M.I.; Bolte, M.; Garcia-Verdugo, E.; Luis, S.V. Pseudopeptidic macrocycles as cooperative minimalistic synzyme systems for the remarkable activation and conversion of CO2 in the presence of the chloride anion. Green Chem. 2020, 22, 4697–4705. [Google Scholar] [CrossRef]
- Andres, S.; Escuder, B.; Domenech, A.; Garcia-Espana, E.; Luis, S.V.; Marcelino, V.; Llinares, J.M.; Ramirez, J.A.; Soriano, C. CO2 fixation and activation by metal complexes of small polyazacyclophanes. J. Phys. Org. Chem. 2001, 14, 495–500. [Google Scholar] [CrossRef]
- Ríos-Lombardía, N.; Busto, E.; Gotor-Fernández, V.; Gotor, V.; Porcar, R.; García-Verdugo, E.; Luis, S.V.; Alfonso, I.; Garcia-Granda, S.; Menéndez-Velázquez, A. From salts to ionic liquids by systematic structural modifications: A rational approach towards the efficient modular synthesis of enantiopure imidazolium salts. Chem. Eur. J. 2010, 16, 836–847. [Google Scholar] [CrossRef] [Green Version]
- Busto, E.; Gotor-Fernández, V.; Ríos-Lombardía, N.; García-Verdugo, E.; Alfonso, I.; García-Granda, S.; Menéndez-Velázquez, A.; Burguete, M.I.; Luis, S.V.; Gotor, V. Simple and straightforward synthesis of novel enantiopure ionic liquids via efficient enzymatic resolution of (±)-2-(1H-imidazol-1-yl)cyclohexanol. Tetrahedron Lett. 2007, 48, 5251–5254. [Google Scholar] [CrossRef]
- Lombardo, M.; Pasi, F.; Easwar, S.; Trombini, C. An improved protocol for the direct asymmetric aldol reaction in ionic liquids, catalysed by onium ion-tagged prolines. Adv. Synth. Catal. 2007, 349, 2061–2065. [Google Scholar] [CrossRef]
- Gruttadauria, M.; Riela, S.; Aprile, C.; Lo Meo, P.; D’Anna, F.; Noto, R. Supported ionic liquids. New recyclable materials for the L-proline-catalyzed aldol reaction. Adv. Synth. Catal. 2006, 348, 82–92. [Google Scholar] [CrossRef] [Green Version]
- Zotova, N.; Franzke, A.; Armstrong, A.; Blackmond, D.G. Clarification of the Role of Water in Proline-Mediated Aldol Reactions. J. Am. Chem. Soc. 2007, 129, 15100–15101. [Google Scholar] [CrossRef] [PubMed]
- Nyberg, A.I.; Usano, A.; Pihko, P.M. Proline-Catalyzed Ketone-Aldehyde Aldol Reactions are Accelerated by Water. Synlett 2004, 2004, 1891–1896. [Google Scholar] [CrossRef]
- Pihko, P.M.; Laurikainen, K.M.; Usano, A.; Nyberg, A.I.; Kaavi, J.A. Effect of additives on the proline-catalyzed ketone-aldehyde aldol reactions. Tetrahedron 2006, 62, 317–328. [Google Scholar] [CrossRef]
- Jimeno, C. Water in asymmetric organocatalytic systems: A global perspective. Org. Biomol. Chem. 2016, 14, 6147–6164. [Google Scholar] [CrossRef] [PubMed]
- North, M.; Villuendas, P. A Chiral solvent effect in asymmetric organocatalysis. Org. Lett. 2010, 12, 2378–2381. [Google Scholar] [CrossRef]
- Kalow, J.A.; Doyle, A.G. Mechanistic investigations of cooperative catalysis in the enantioselective fluorination of epoxides. J. Am. Chem. Soc. 2011, 133, 16001–16012. [Google Scholar] [CrossRef] [PubMed]
- Mashiko, T.; Kumagai, N.; Shibasaki, M. Managing highly coordinative substrates in asymmetric catalysis: A catalytic asymmetric amination with a lanthanum-based ternary catalyst. J. Am. Chem. Soc. 2009, 131, 14990–14999. [Google Scholar] [CrossRef] [PubMed]
- Ding, K.; Ishii, A.; Mikami, K. Super high throughput screening (SHTS) of chiral ligands and activators: Asymmetric activation of chiral diol–Zinc catalysts by chiral nitrogen activators for the enantioselective addition of diethylzinc to aldehydes. Angew. Chem. Int. Ed. 1999, 38, 497–501. [Google Scholar] [CrossRef]
- Kobayashi, S.; Kawamura, M. Catalytic enantioselective 1,3-dipolar cycloadditions between nitrones and alkenes using a novel heterochiral Ytterbium(III) catalyst. J. Am. Chem. Soc. 1998, 120, 5840–5841. [Google Scholar] [CrossRef]
- Satyanarayana, T.; Abraham, S.; Kagan, H.B. Nonlinear Effects in Asymmetric Catalysis. Angew. Chem. Int. Ed. 2009, 48, 456–494. [Google Scholar] [CrossRef] [PubMed]
- Blackmond, D.G. Kinetic Aspects of Nonlinear Effects in Asymmetric Catalysis. Acc. Chem. Res. 2000, 33, 402–411. [Google Scholar] [CrossRef] [PubMed]
- Blackmond, D.G.; Rosner, T.; Neugebauer, T.; Reetz, M.T. Kinetic Influences on Enantioselectivity for Non-Diastereopure Catalyst Mixtures. Angew. Chem. Int. Ed. 1999, 38, 2196–2199. [Google Scholar] [CrossRef]
- Bolm, C.; Muñiz, K.; Hildebrand, J.P. Planar−Chiral Ferrocenes in Asymmetric Catalysis: The Impact of Stereochemically Inhomogeneous Ligands. Org. Lett. 1999, 1, 491–493. [Google Scholar] [CrossRef]
- McCleland, B.W.; Nugent, W.A.; Finn, M.G. Mechanistic Studies of the Zirconium-Triisopropanolamine-Catalyzed Enan-tioselective Addition of Azide to Cyclohexene Oxide. J. Org. Chem. 1998, 63, 6656–6666. [Google Scholar] [CrossRef]
- Zarotti, P.; Knöpfel, T.F.; Aschwanden, P.; Carreira, E.M. Nonlinear Effects with Diastereomeric Ligand Mixtures in Enan-tioselective, Catalytic Additions of Terminal Alkynes Involving Copper−PINAP Complexes. ACS Catal. 2012, 2, 1232–1234. [Google Scholar] [CrossRef]
- Ay, S.; Ziegert, R.E.; Zhang, H.; Nieger, M.; Rissanen, K.; Fink, K.; Kubas, A.; Gschwind, R.M.; Bräse, S. NMR-Spectroscopic and Solid-State Investigations of Cometal-Free Asymmetric Conjugate Addition: A Dinuclear Paracyclophaneimine Zinc Methyl Complex. J. Am. Chem. Soc. 2010, 132, 12899–12905. [Google Scholar] [CrossRef] [PubMed]
- Binder, C.M.; Bautista, A.; Zaidlewicz, M.; Krzemiński, M.P.; Oliver, A.; Singaram, B. Dual Stereoselectivity in the Dialkylzinc Reaction Using (-)-β-Pinene Derived Amino Alcohol Chiral Auxiliaries. J. Org. Chem. 2009, 74, 2337–2343. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.K.; Costa, A.M.; Walsh, P.J. Substrate Dependence of Nonlinear Effects: Mechanistic Probe and Practical Applications. J. Am. Chem. Soc. 2001, 123, 5378–5379. [Google Scholar] [CrossRef]
- Buono, F.; Walsh, P.J.; Blackmond, D.G. Rationalization of Anomalous Nonlinear Effects in the Alkylation of Substituted Benzaldehydes. J. Am. Chem. Soc. 2002, 124, 13652–13653. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Imidazolium Salts | Conversion (%) 2 | Selectivity (%) 2 | Yield (%) 2 | ee (%) 3 |
|---|---|---|---|---|---|
| 1 4 | - | 10 | 99 | 10 | 50 |
| 2 | [BMIM][l-Pro] | 45 | 95 | 43 | 60 |
| 3 | (R,R)-trans-Cy6-OAc-Im-Bu-l-Pro | 73 | 94 | 69 | 66 |
| 4 | (S,S)-trans-Cy6-OH-Im-Bu-l-Pro | 68 | 93 | 63 | 70 |
| Entry | Catalyst | Conv. (%) 2 | Yield (%) 2 | Side Products (4 + 5)(%) 3 | ee (%) 4 |
|---|---|---|---|---|---|
| 1 5 | l-Pro | 70 | 9 | 32 | 36 |
| 2 | [BMIM][l-Pro] | 63 | 11 | 23 | 56 |
| 3 | (S,S)-trans-Cy6-OH-Im-Bu-l-Pro | 70 | 17 | 26 | 73 |
| 4 | (±)-trans-Cy6-OH-Im-Bu-l-Pro | 64 | 9 | 27 | 62 |
| 5 | (R,R)-trans-Cy6-OAc-Im-Bu-l-Pro | 80 | 17 | 32 | 81 |
| 6 6 | l-Pro + (R,R)-trans-Cy6-OAc-Im-Bu-Cl | 99 | 73 | 14 | 70 |
| Entry | Imidazolium Salts | Conversion (%) 2 | Selectivity (%) 2 | ee (%) 3 |
|---|---|---|---|---|
| 1 | [BMIM][l-Pro] | >99 | 91 | 20 |
| 2 | (R,R)-trans-Cy6-OAc-Im-Bu-l-Pro | >99 | 98 | 66 |
| 3 | (S,S)-trans-Cy6-OH-Im-Bu-l-Pro | >99 | 79 | 43 |
| 4 | (R,S)-cis-Cy6-OH-Im-Bu-l-Pro | >99 | 95 | 64 |
| 5 | (±)-trans-Cy6-OH-Im-Bu-l-Pro | >99 | 96 | 64 |
| 6 | (R,R)-trans-Cy5-OAc-Im-Bu-l-Pro | >99 | 90 | 68 |
| 7 | (S,S)-trans-Cy5-OH-Im-Bu-l-Pro | >99 | 90 | 60 |
| Entry | Catalyst | Yield (%) 2 | Selectivity anti:syn 2 | eeanti (%) 3 | eesyn (%) 3 |
|---|---|---|---|---|---|
| 1 | [BMIM][l-Pro] | 86 | 86:14 | 90 | −3 |
| 2 | (R,R)-trans-Cy6-OAc-Im-Bu-l-Pro | 79 | 75:25 | 86 | −74 |
| 3 | (S,S)-trans-Cy6-OH-Im-Bu-l-Pro | 80 | 82:18 | 90 | −23 |
| 4 | (R,R)-trans-Cy5-OAc-Im-Bu-l-Pro | 99 | 90:10 | 95 | 73 |
| 5 | (S,S)-trans-Cy5-OH-Im-Bu-l-Pro | 99 | 90:10 | 96 | 79 |

| Entry | R1 | Catalyst | Yield (%) 2 | Selectivity anti:syn 2 | eeanti (%) 3 | eesyn (%) 3 |
|---|---|---|---|---|---|---|
| 1 | H | l-proline | 78 | 64:36 | 41 | n.d |
| (R,R)-trans-Cy6-OAc-Im-Bu-l-Pro | 89 | 76:24 | 88 | 19 | ||
| (S,S)-trans-Cy6-OH-Im-Bu-l-Pro | 79 | 76:24 | 99 | 71 | ||
| 2 | Cl | l-proline | 63 | 63:37 | 54 | n.d |
| (R,R)-trans-Cy6-OAc-Im-Bu-l-Pro | 95 | 81:19 | 93 | 31 | ||
| (S,S)-trans-Cy6-OH-Im-Bu-l-Pro | 92 | 79:21 | 68 | 57 | ||
| 3 | OMe | l-proline | - | - | - | - |
| (R,R)-trans-Cy6-OAc-Im-Bu-l-Pro | 61 | 69:31 | 86 | 47 | ||
| (S,S)-trans-Cy6-OH-Im-Bu-l-Pro | 60 | 68:32 | 54 | 37 | ||
| 4 | F | l-proline | 38 | 64:36 | 65 | 60 |
| (R,R)-trans-Cy6-OAc-Im-Bu-l-Pro | 90 | 77:23 | 92 | 51 | ||
| (S,S)-trans-Cy6-OH-Im-Bu-l-Pro | 93 | 73:27 | 47 | 27 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Porcar, R.; García-Verdugo, E.; Altava, B.; Burguete, M.I.; Luis, S.V. Chiral Imidazolium Prolinate Salts as Efficient Synzymatic Organocatalysts for the Asymmetric Aldol Reaction. Molecules 2021, 26, 4190. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26144190
Porcar R, García-Verdugo E, Altava B, Burguete MI, Luis SV. Chiral Imidazolium Prolinate Salts as Efficient Synzymatic Organocatalysts for the Asymmetric Aldol Reaction. Molecules. 2021; 26(14):4190. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26144190
Chicago/Turabian StylePorcar, Raúl, Eduardo García-Verdugo, Belén Altava, Maria Isabel Burguete, and Santiago V. Luis. 2021. "Chiral Imidazolium Prolinate Salts as Efficient Synzymatic Organocatalysts for the Asymmetric Aldol Reaction" Molecules 26, no. 14: 4190. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26144190