
Natural Compounds Isolated from Stachybotrys chartarum Are Potent Inhibitors of Human Protein Kinase CK2

 , , and
, , and
Abstract
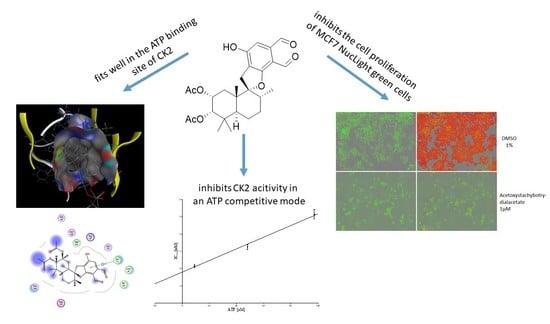
:
1. Introduction
2. Results
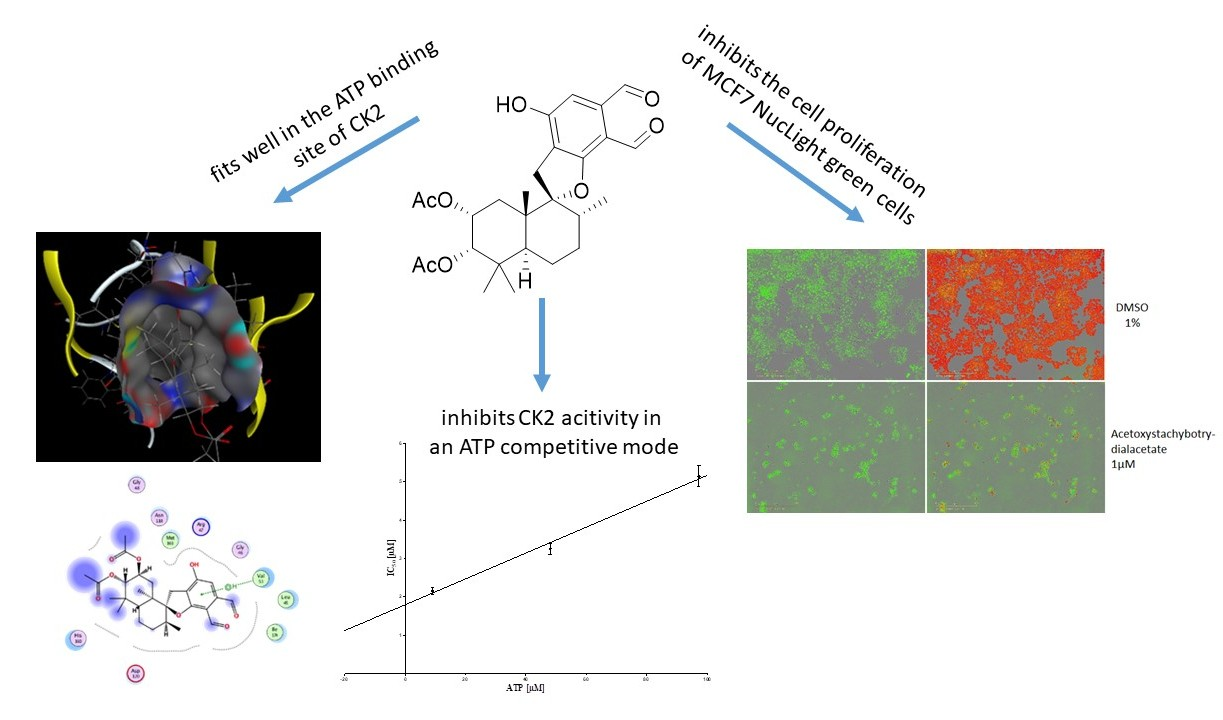
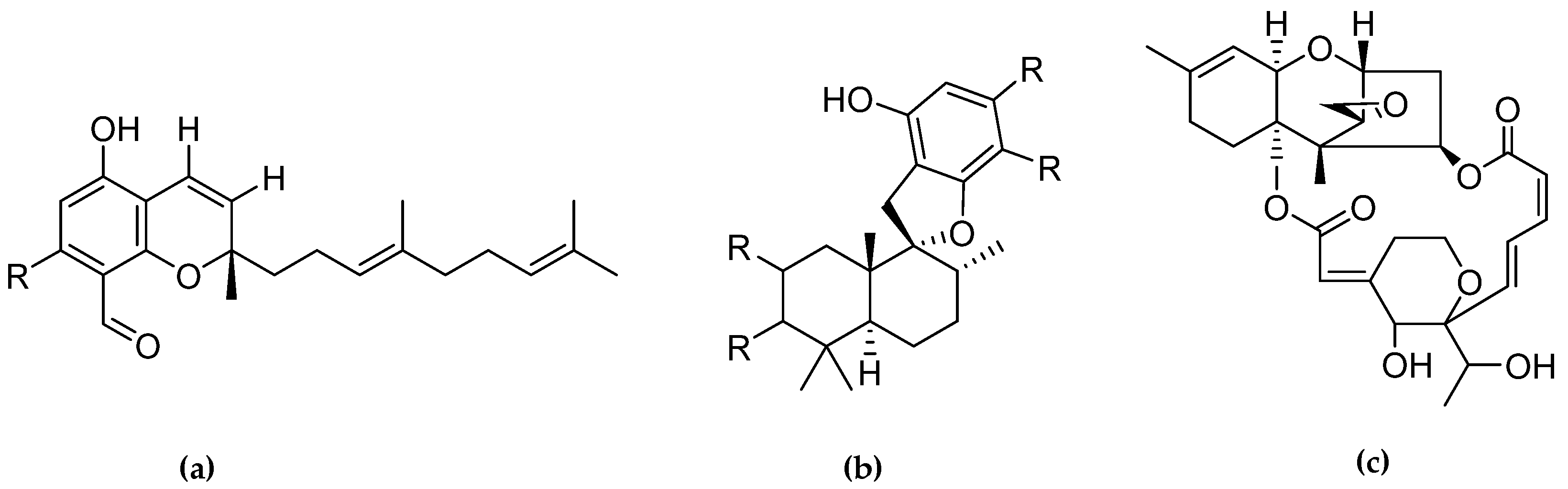
2.1. Inhibition of Human Protein Kinase CK2 by Natural Compounds from Stachybotrys chartarum
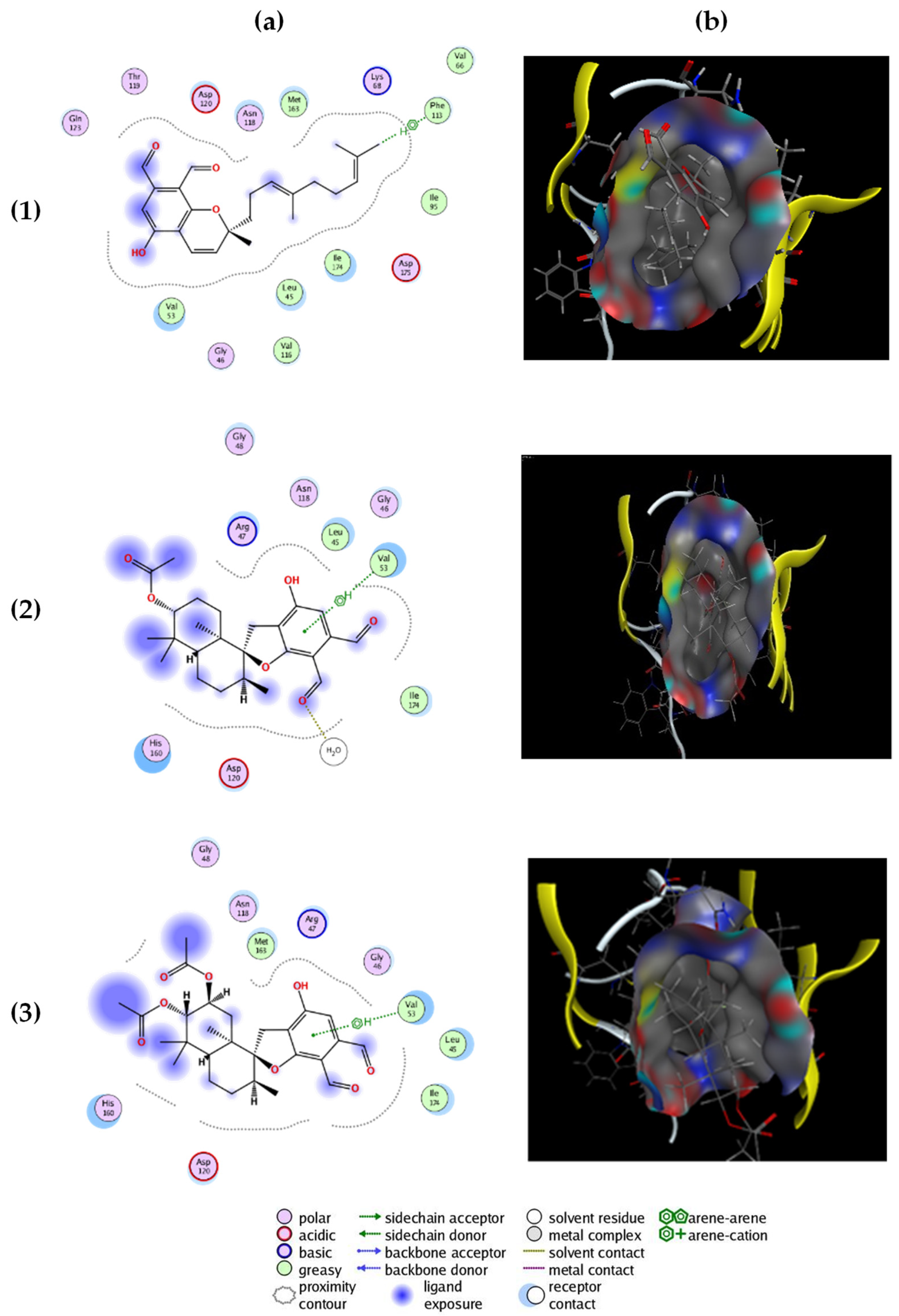
2.2. Molecular Docking
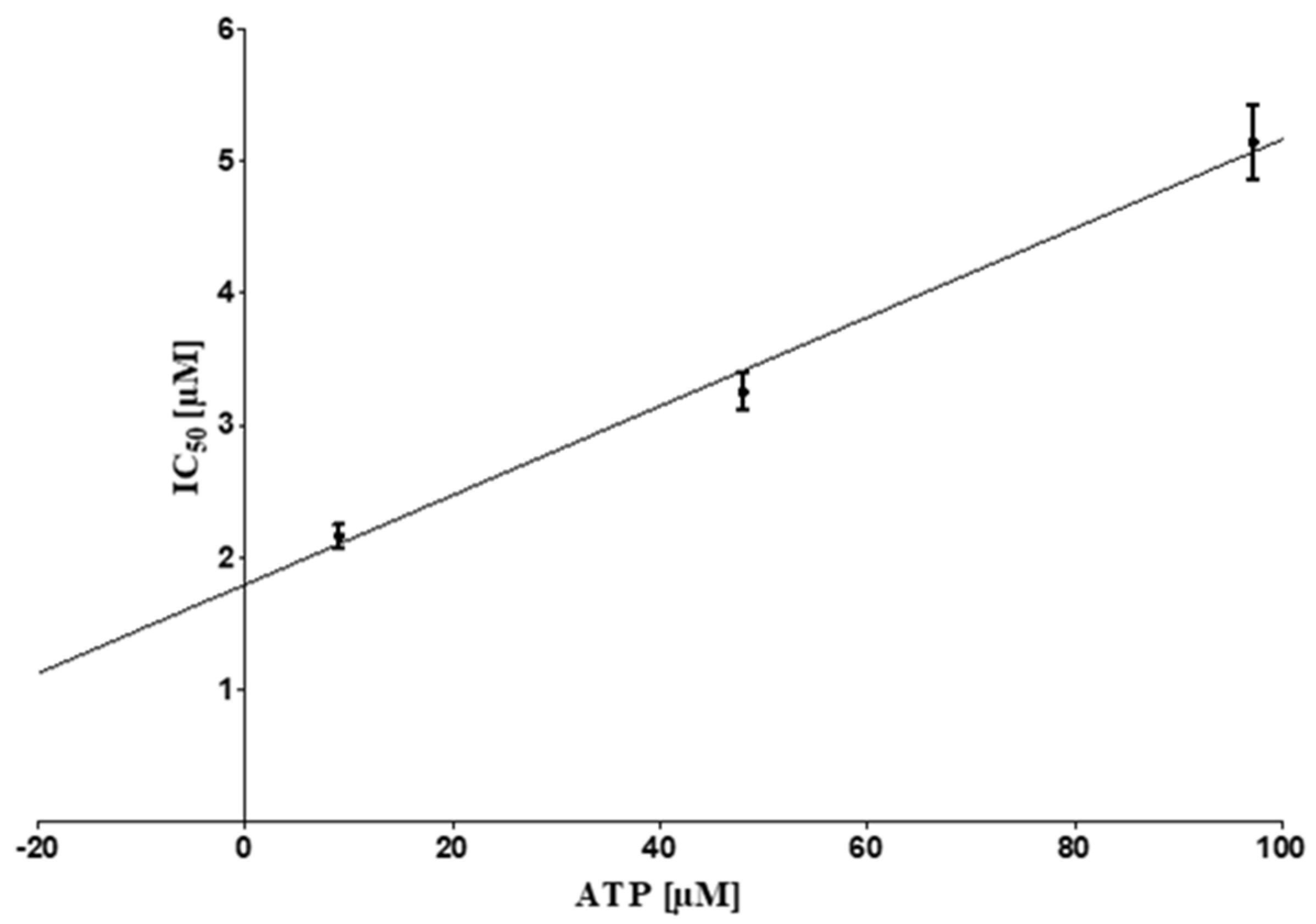
2.3. ATP Competitive Mode of Action
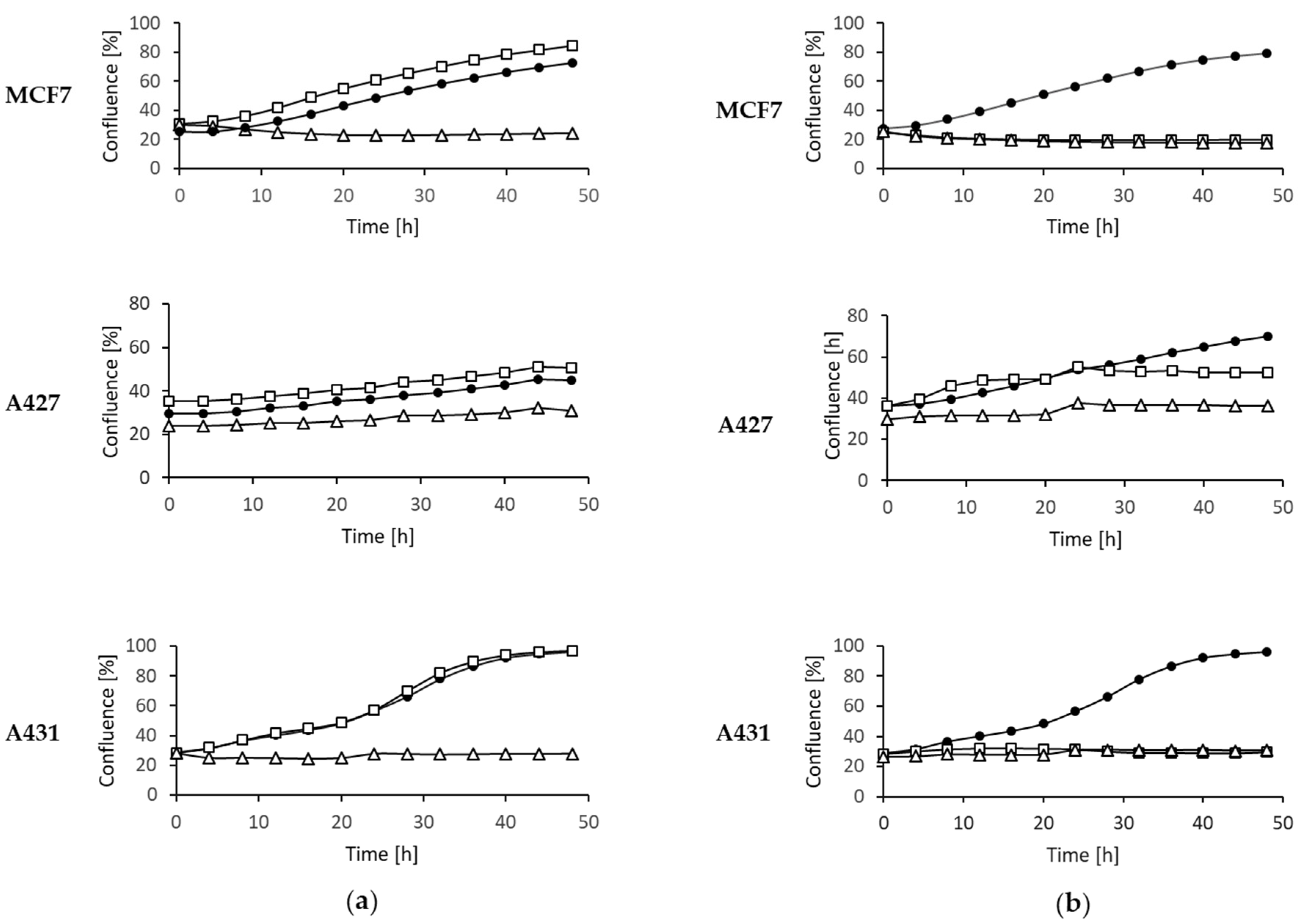
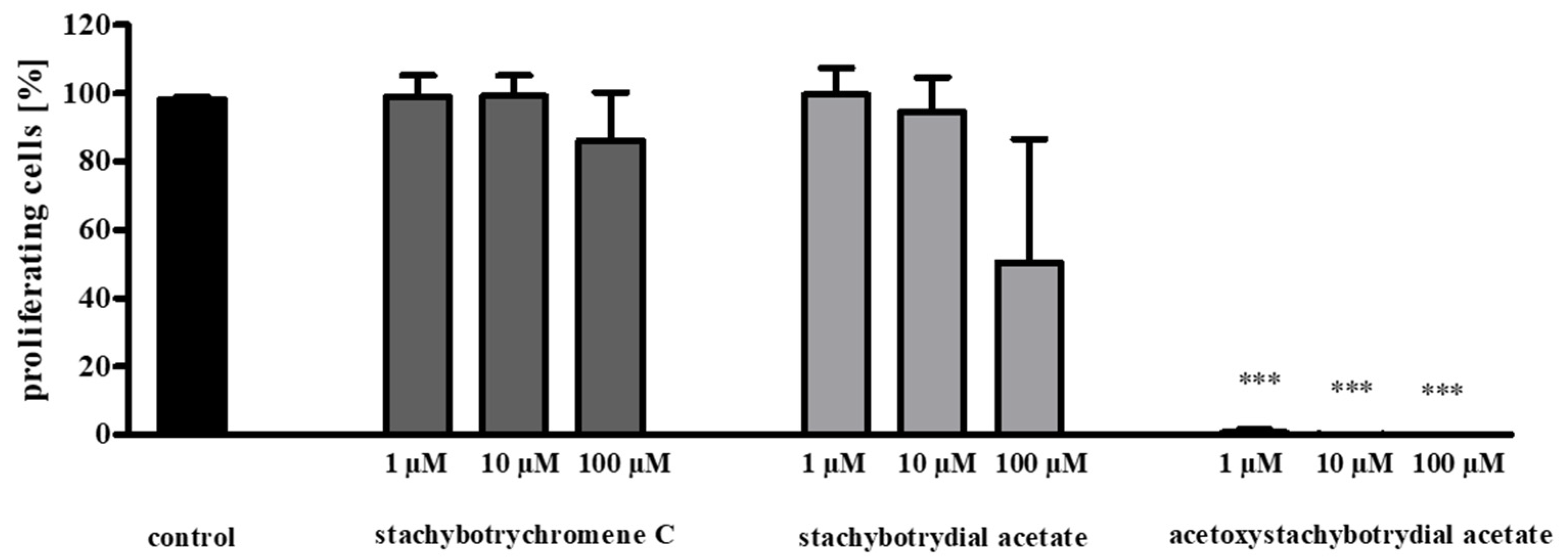
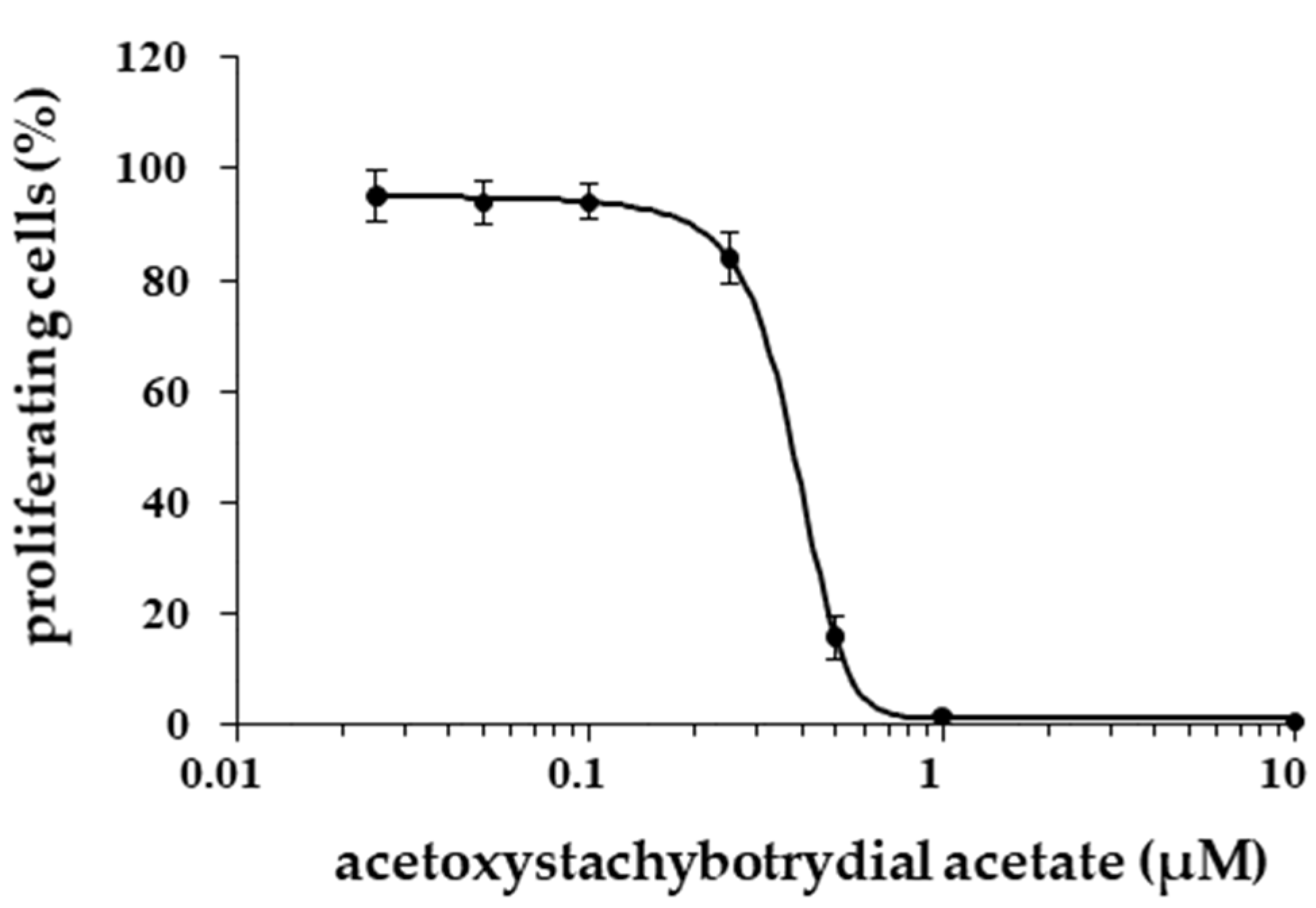
2.4. Inhibition of Tumor Cell Proliferation
3. Discussion
4. Materials and Methods
4.1. The Compounds
4.2. Inhibition of Human CK2 Holoenzyme
4.3. Cultivation of Cancer Cell Lines
4.4. Cell Proliferation
4.5. Cell Viability Assay
4.6. IncuCyte® Cytotoxicity Assay
4.7. LDH Assay
4.8. Computational Study
4.8.1. Database Generation
4.8.2. Protein Structure Preparation and Molecular Docking
4.9. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Pinna, L.A. Protein kinase CK2: A challenge to canons. J. Cell Sci. 2002, 115, 3873–3878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salvi, M.; Sarno, S.; Cesaro, L.; Nakamura, H.; Pinna, L.A. Extraordinary pleiotropy of protein kinase CK2 revealed by weblogo phosphoproteome analysis. BBA-Mol. Cell Res. 2009, 1793, 847–859. [Google Scholar] [CrossRef] [PubMed]
- Vilk, G.; Weber, J.E.; Turowec, J.P.; Duncan, J.S.; Wu, C.; Derksen, R.; Zien, P.; Sarno, S.; Donella-Deana, A.; Lajoie, G.; et al. Protein kinase CK2 catalyzes tyrosine phosphorylation in mammalian cells. Cell. Signal. 2008, 20, 1942–1951. [Google Scholar] [CrossRef] [PubMed]
- Ghavidel, A.; Schultz, M.C. TATA binding protein-associated CK2 transduces DNA damage, signals to the RNA polymerase III transcriptional machinery. Cell 2001, 106, 575–584. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, K.A.; Wang, G.; Unger, G.; Slaton, J.; Ahmed, K. Protein kinase CK2—A key suppressor of apoptosis. Adv. Enzym. Regul. 2008, 48, 179–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cozza, G.; Pinna, L.A. Casein kinases as potential therapeutic targets. Expert Opin. Ther. Targets 2016, 20, 319–340. [Google Scholar] [CrossRef]
- Patridge, E.; Gareiss, P.; Kinch, M.S.; Hoyer, D. An analysis of FDA-approved drugs: Natural products and their derivatives. Drug Discov. Today 2016, 21, 204–207. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the 30 Years from 1981 to 2010. J. Nat. Prod. 2012, 75, 311–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cozza, G. The Development of CK2 Inhibitors: From Traditional Pharmacology to in Silico Rational Drug Design. Pharmaceuticals 2017, 10, 26. [Google Scholar] [CrossRef] [Green Version]
- Cozza, G.; Pinna, L.A.; Moro, S. Kinase CK2 Inhibition: An Update. Curr. Med. Chem. 2013, 20, 671–693. [Google Scholar] [CrossRef] [PubMed]
- Haidar, S.; Bouaziz, Z.; Marminon, C.; Laitinen, T.; Poso, A.; Le Borgne, M.; Jose, J. Development of Pharmacophore Model for Indeno[1,2-b]indoles as Human Protein Kinase CK2 Inhibitors and Database Mining. Pharmaceuticals 2017, 10, 8. [Google Scholar] [CrossRef] [PubMed]
- Haidar, S.; Aichele, D.; Birus, R.; Hielscher, J.; Laitinen, T.; Poso, A.; Jose, J. In Vitro and in Silico Evaluation of Bikaverin as a Potent Inhibitor of Human Protein Kinase CK2. Molecules 2019, 24, 1380. [Google Scholar] [CrossRef] [Green Version]
- Jagels, A.; Lindemann, V.; Ulrich, S.; Gottschalk, C.; Cramer, B.; Hubner, F.; Gareis, M.; Humpf, H.U. Exploring Secondary Metabolite Profiles of Stachybotrys spp. by LC-MS/MS. Toxins 2019, 11, 133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jagels, A.; Hovelmann, Y.; Zielinski, A.; Esselen, M.; Kohler, J.; Hubner, F.; Humpf, H.U. Stachybotrychromenes A–C: Novel cytotoxic meroterpenoids from Stachybotrys sp. Mycotoxin Res. 2018, 34, 179–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olgen, S.; Gotz, C.; Jose, J. Synthesis and biological evaluation of 3-(substituted-benzylidene)-1,3-dihydro-indolin derivatives as human protein kinase CK2 and p60(c-Src) tyrosine kinase inhibitors. Biol. Pharm. Bull. 2007, 30, 715–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gratz, A.; Gotz, C.; Jose, J. A CE-based assay for human protein kinase CK2 activity measurement and inhibitor screening. Electrophoresis 2010, 31, 634–640. [Google Scholar] [CrossRef]
- Available online: https://www.rcsb.org/pdb/explore/explore.do?structureId=3C13 (accessed on 23 April 2018).
- Schnitzler, A.; Gratz, A.; Bollacke, A.; Weyrich, M.; Kuckländer, U.; Wünsch, B.; Götz, C.; Niefind, K.; Jose, J. A π-Halogen Bond of Dibenzofuranones with the Gatekeeper Phe113 in Human Protein Kinase CK2 Leads to Potent Tight Binding Inhibitors. Pharmaceuticals 2018, 11, 23. [Google Scholar] [CrossRef] [Green Version]
- Zuman, P. Reactions of orthophthalaldehyde with nucleophiles. Chem. Rev. 2004, 104, 3217–3238. [Google Scholar] [CrossRef]
- D’Hollander, A.C.A.; Westwood, N.J. Assessment of the regioselectivity in the condensation reaction of unsymmetrical o-phthaldialdehydes with alanine. Tetrahedron 2018, 74, 224–239. [Google Scholar] [CrossRef] [Green Version]
- Martínez, R.; Geronimo, B.D.; Pastor, M.; Zapico, J.M.; Coderch, C.; Panchuk, R.; Skorokhyd, N.; Maslyk, M.; Ramos, A.; de Pascual-Teresa, B. Multitarget Anticancer Agents Based on Histone Deacetylase and Protein Kinase CK2 inhibitors. Molecules 2020, 25, 1497. [Google Scholar] [CrossRef] [Green Version]
- Sasaoka, M.; Wada, Y.; Hasumi, K. Stachybotrydial selectively enhances fibrin binding and activation of Glu-plasminogen. J. Antibiot. 2007, 60, 674–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawadjoon, S.; Kittakoop, P.; Isaka, M.; Kirtikara, K.; Madla, S.; Thebtaranonth, Y. Antiviral and antiplasmodial spirodihydrobenzofuran terpenes from the fungus Stachybotrys nephrospora. Planta Med. 2004, 70, 1085–1087. [Google Scholar] [CrossRef] [PubMed]
- Hasumi, K.; Hasegawa, K.; Kitano, Y. Isolation and absolute configuration of SMTP-0, a simplest congener of the SMTP family nonlysine-analog plasminogen modulators. J. Antibiot. 2007, 60, 463–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, L.Y.; Chiu, S.M.; Oleinick, N.L. Staurosporine-induced death of MCF-7 human breast cancer cells: A distinction between caspase-3-dependent steps of apoptosis and the critical lethal lesions. Exp. Cell Res. 2003, 283, 135–145. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Molecular Operating Environment (MOE); Chemical Computing Group Inc.: Montreal, QC, Canada, 2013.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Name | Chemical Structure | % Inhibition at 10 µM (IC50 µM) a |
|---|---|---|
| Stachybotrychromene A |  | 26 (n.d.) |
| Stachybotrychromene B |  | 47 (n.d.) |
| Stachybotrychromene C |  | 95 (0.32 ± 0.20) |
| Stachybotrydial acetate |  | 96 (0.69 ± 0.15) |
| Acetoxystachybotrydial acetate |  | 87 (1.86 ± 0.36) |
| Stachybotrydial |  | 72 (4.43 ± 0.30) |
| Stachybotrysin B |  | 50 (13.42 ± 2.40) |
| Stachybotrylactam acetate |  | 44 (n.d.) |
| L-671 |  | 42 (n.d.) |
| Stachybonoid D |  | 47 (n.d.) |
| Stachybotrysin C |  | 37 (n.d.) |
| Stachybotryamide |  | 21 (n.d.) |
| Satratoxin G |  | 12 (n.d.) |
| Satratoxin H |  | 10 (n.d.) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haidar, S.; Jürgens, F.M.; Aichele, D.; Jagels, A.; Humpf, H.-U.; Jose, J. Natural Compounds Isolated from Stachybotrys chartarum Are Potent Inhibitors of Human Protein Kinase CK2. Molecules 2021, 26, 4453. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26154453
Haidar S, Jürgens FM, Aichele D, Jagels A, Humpf H-U, Jose J. Natural Compounds Isolated from Stachybotrys chartarum Are Potent Inhibitors of Human Protein Kinase CK2. Molecules. 2021; 26(15):4453. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26154453
Chicago/Turabian StyleHaidar, Samer, Franziska M. Jürgens, Dagmar Aichele, Annika Jagels, Hans-Ulrich Humpf, and Joachim Jose. 2021. "Natural Compounds Isolated from Stachybotrys chartarum Are Potent Inhibitors of Human Protein Kinase CK2" Molecules 26, no. 15: 4453. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26154453