
Towards Advances in Molecular Understanding of Boric Acid Biocatalyzed Ring-Opening (Co)Polymerization of δ-Valerolactone in the Presence of Ethylene Glycol as an Initiator

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Results and Discussion
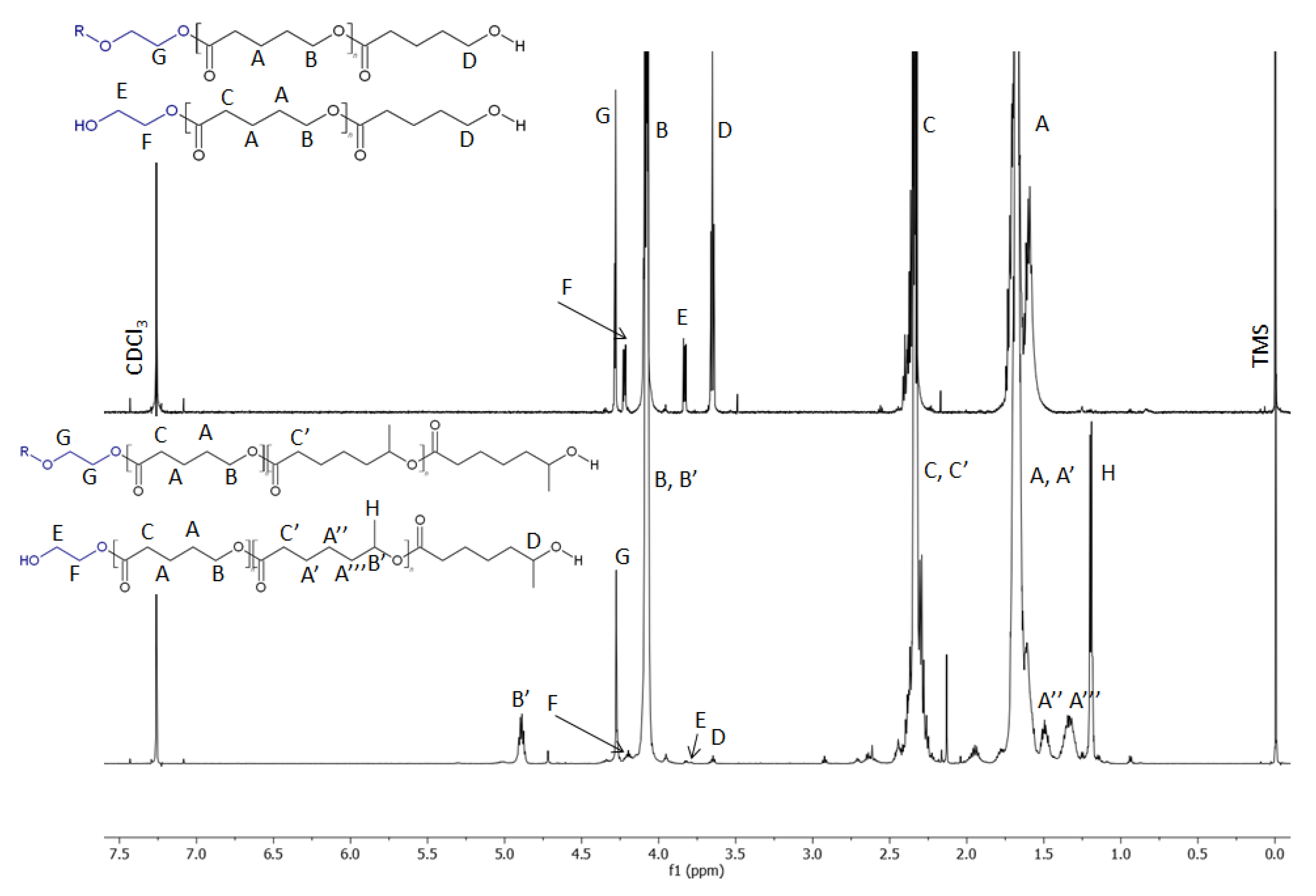
2.1. NMR
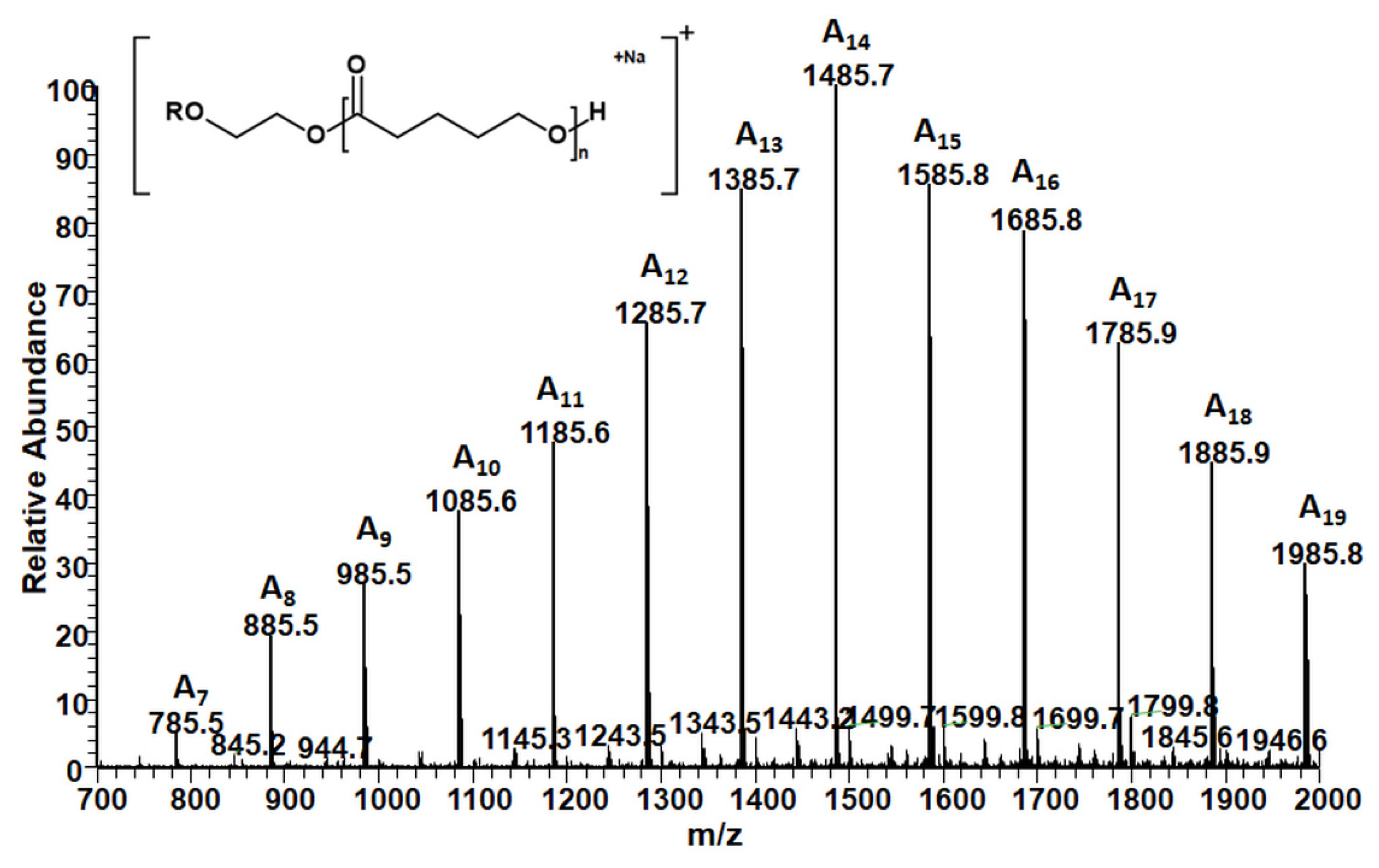
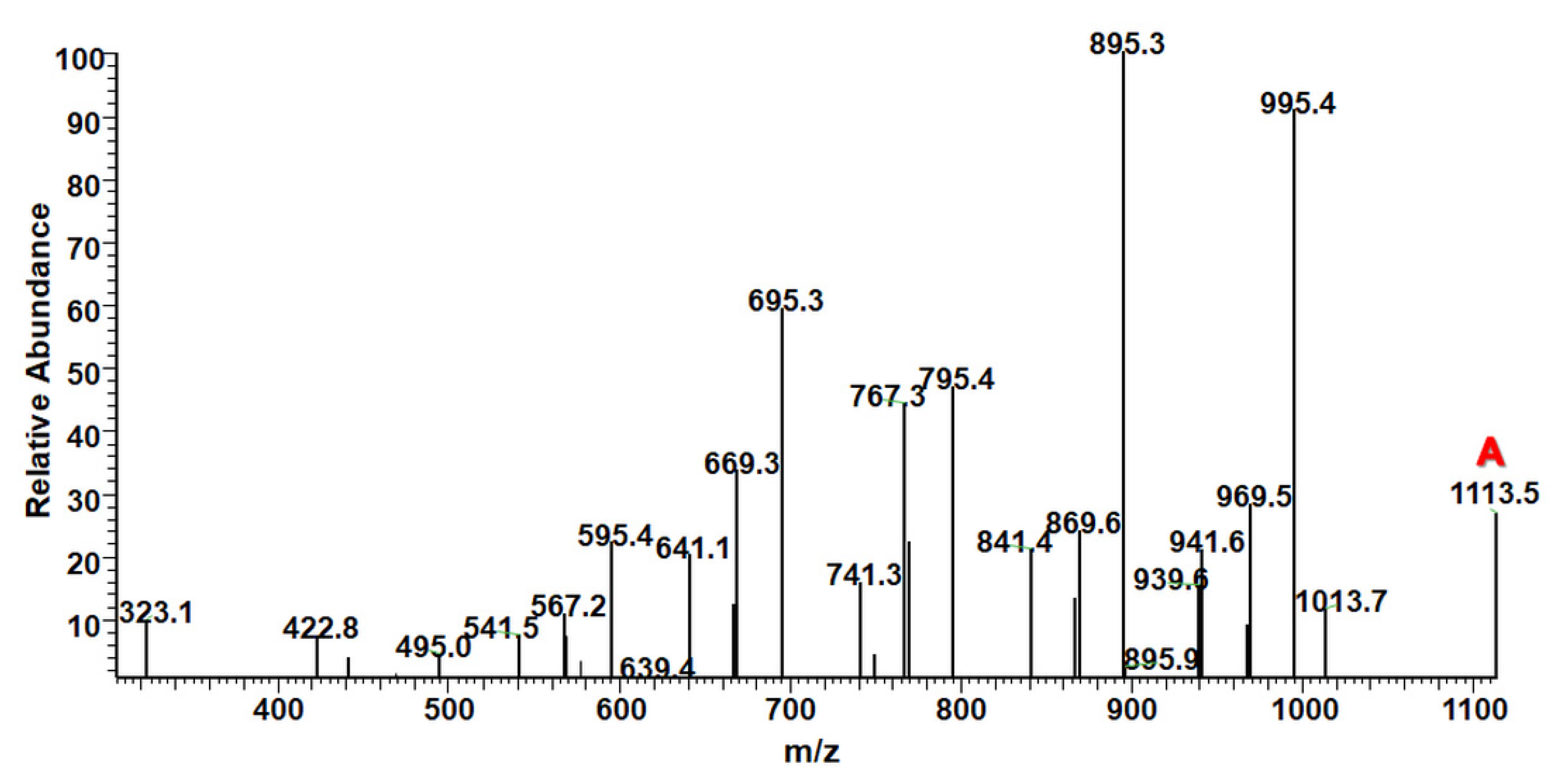
2.2. ESI-MS
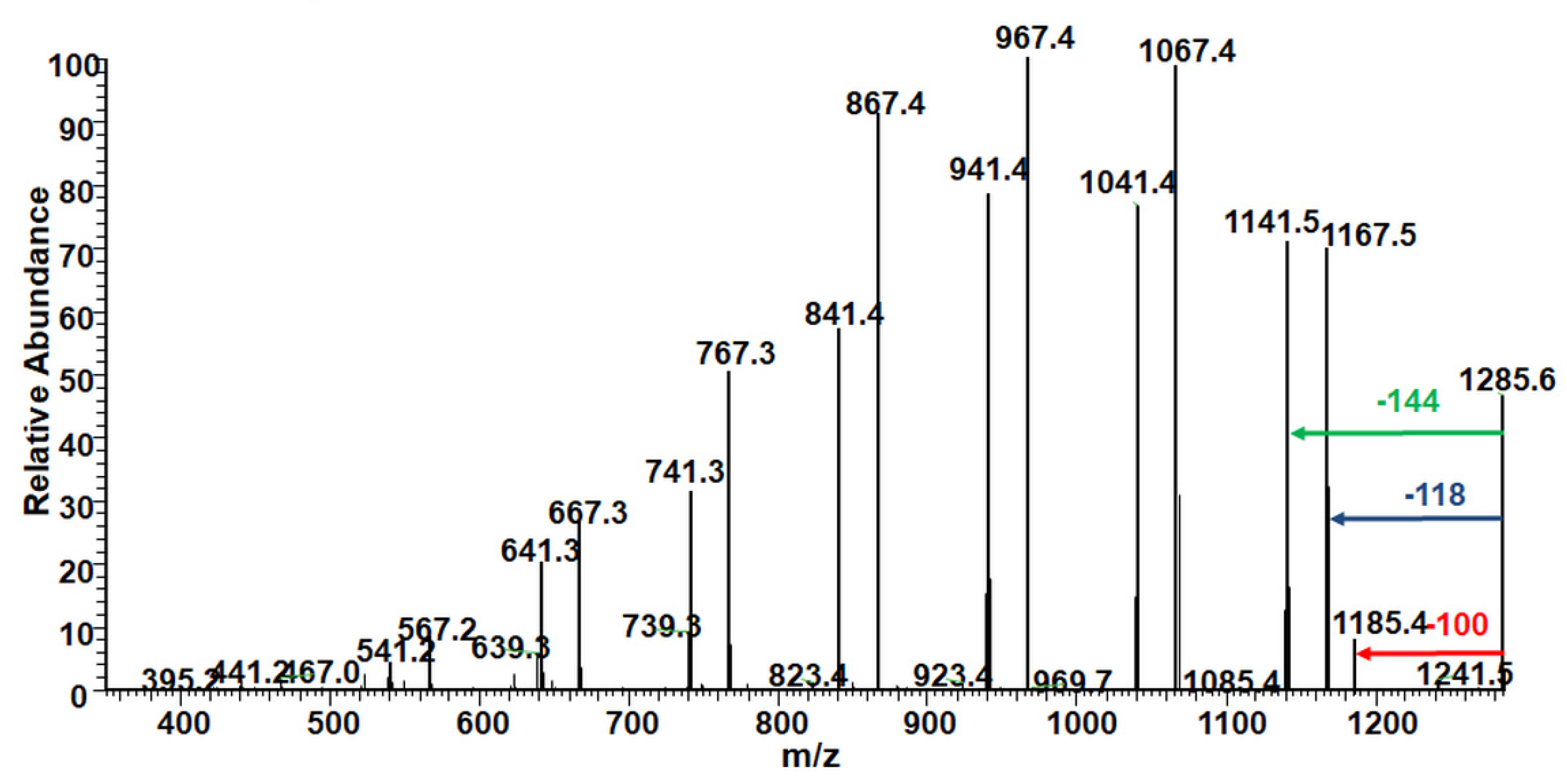
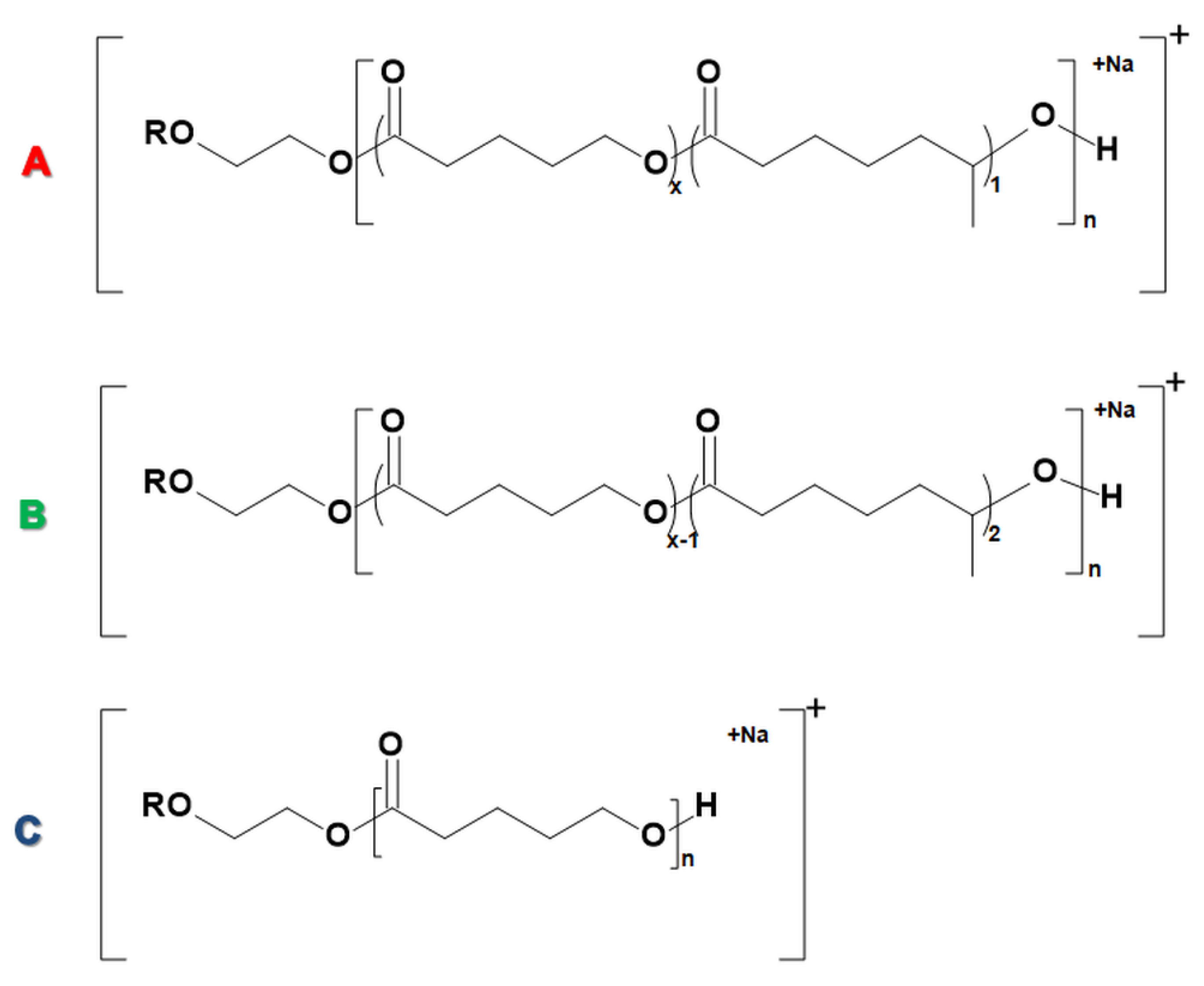
2.2.1. Structural Studies of the Homo-Oligoester PdVL by ESI-MS/MS
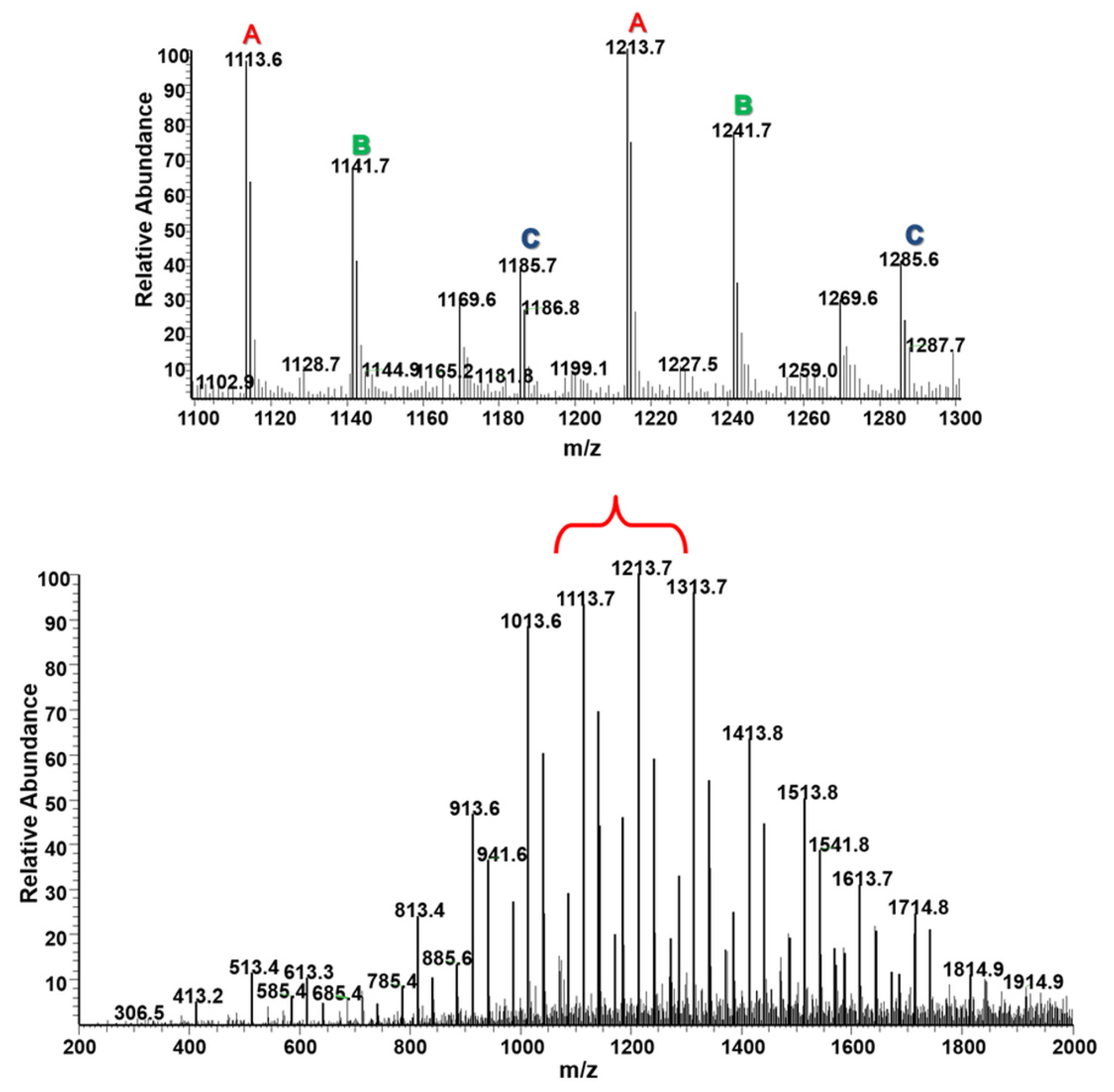
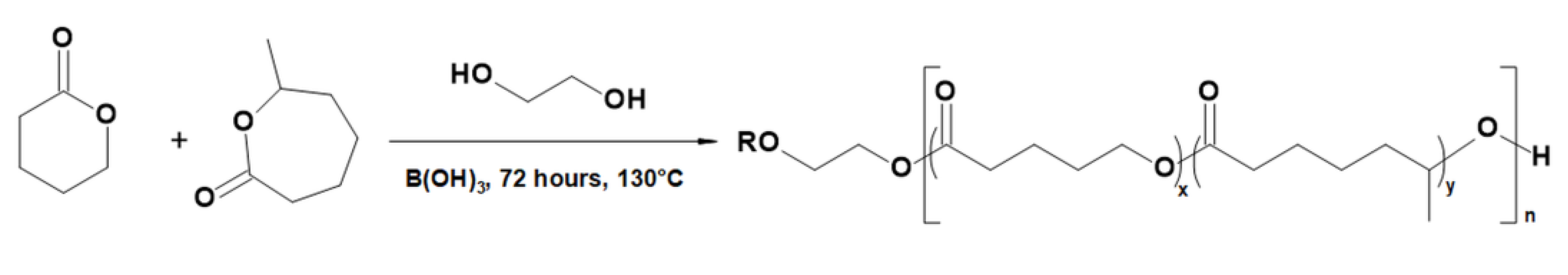
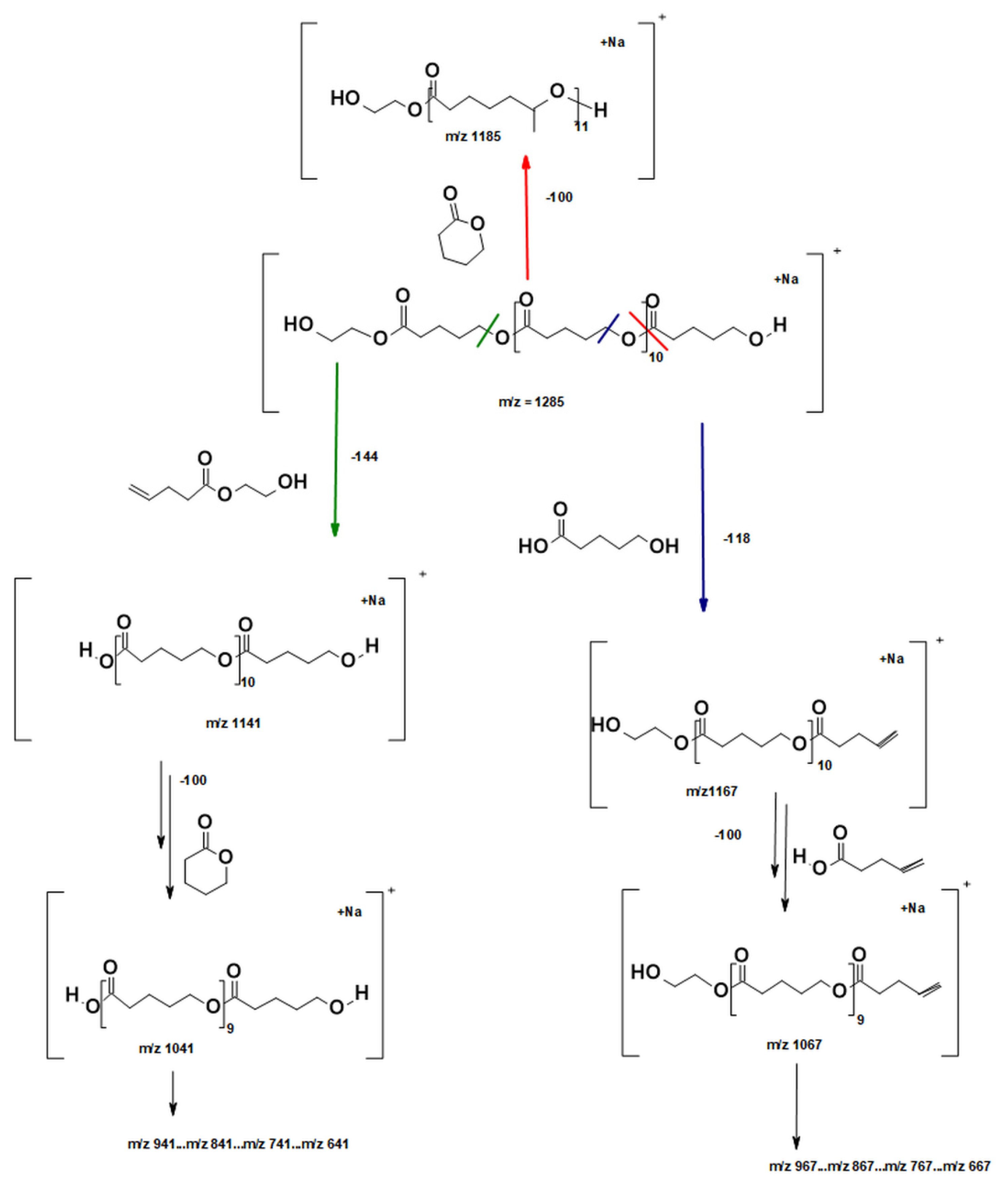
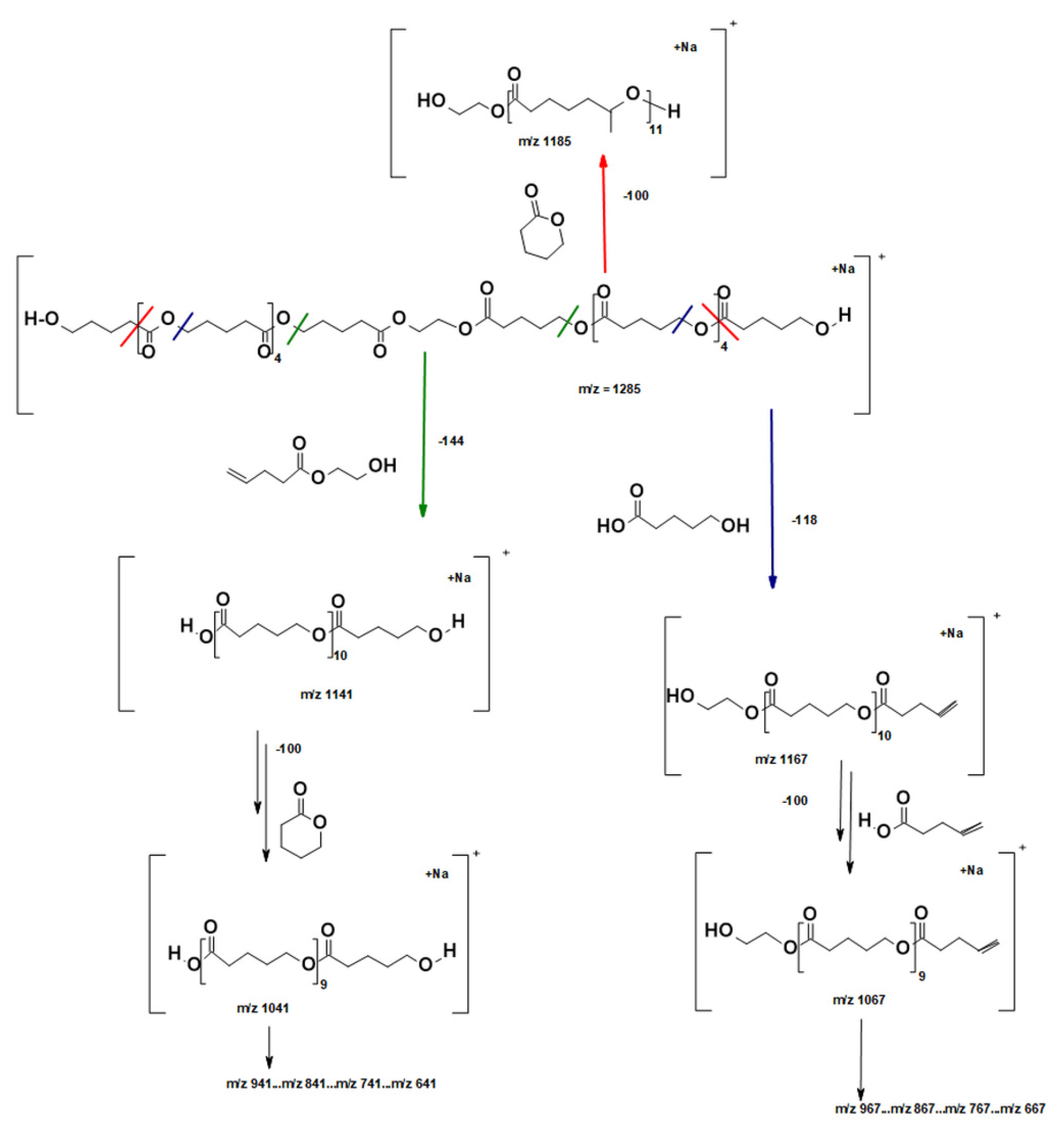
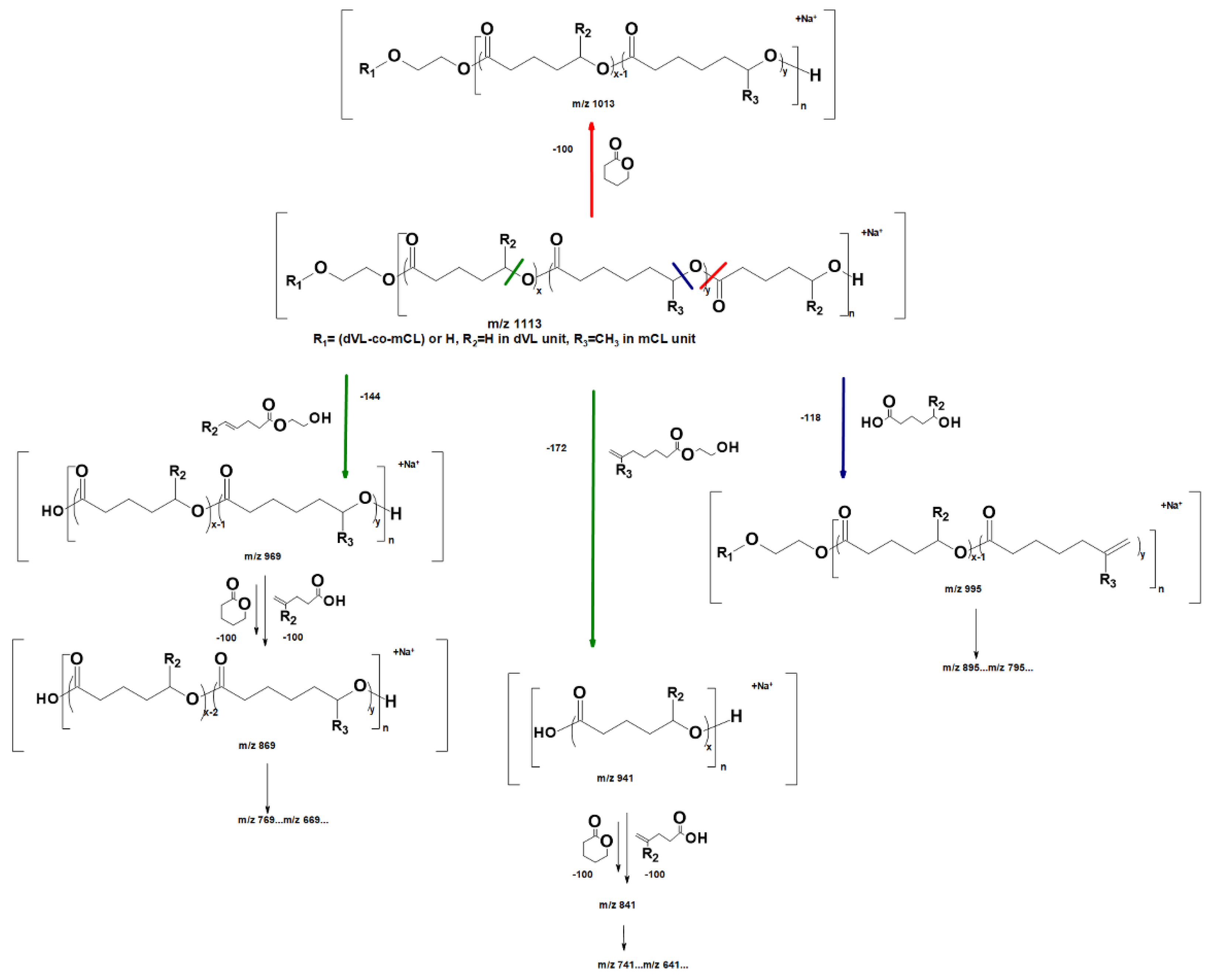
2.2.2. Molecular Level Studies of the Oligoester P(dVL-co-mCL) Performed by ESI-MSn
2.2.3. Fragmentation Studies of P(dVL-co-mCL) Copolymer with One mCL Comonomer Unit
3. Conclusions
4. Materials and Methods
4.1. Gel Permeation Chromatography (GPC)
4.2. Nuclear Magnetic Resonance (NMR)
4.3. Electrospray Ionization Mass Spectrometry (ESI-MSn) Analysis
4.4. Synthesis of 6-Methyl-ε-caprolactone Monomer
4.5. Homopolymerization of δ-Valerolactone
4.6. Copolymerization of δ-Valerolactone with 6-Methyl-ε-caprolactone
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bałakier, T.; Chaładaj, W.; Jurczak, J.; Adamus, G.; Kowalczuk, M. An effective protocol for the synthesis enantiomerically pure 4-substituted oxetane-2-ones. Tetrahedron 2013, 69, 4990–4993. [Google Scholar] [CrossRef]
- Bull, J.A.; Croft, R.A.; Davis, O.A.; Doran, R.; Morgan, K.F. Oxetanes: Recent advances in synthesis, reactivity, and medicinal chemistry. Chem. Rev. 2016, 116, 12150–12233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adamus, G.; Kurcok, P.; Radecka, I.; Kowalczuk, M. Bioactive oligomers from natural polyhydroxyalkanoates and their synthetic analogues. Polimery 2017, 62, 317–322. [Google Scholar] [CrossRef]
- Adamus, G.; Kwiecień, I.; Maksymiak, M.; Bałakier, T.; Jurczak, J.; Kowalczuk, M. Molecular level structure of novel synthetic analogues of aliphatic biopolyesters as revealed by multistage mass spectrometry. Anal. Chim. Acta 2014, 808, 104–114. [Google Scholar] [CrossRef]
- Kwiecień, I.; Bałakier, T.; Jurczak, J.; Kowalczuk, M.; Adamus, G. Molecular architecture of novel potentially bioactive (co) oligoesters containing pesticide moieties established by electrospray ionization multistage mass spectrometry. Rapid Commun. Mass Spectrom. 2015, 29, 533–544. [Google Scholar] [CrossRef]
- Maksymiak, M.; Bałakier, T.; Jurczak, J.; Kowalczuk, M.; Adamus, G. Bioactive (co) oligoesters with antioxidant properties–synthesis and structural characterization at the molecular level. RSC Adv. 2016, 6, 57751–57761. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Pan, H.; Chen, X. Mass spectrometry for structural characterization of therapeutic antibodies. Mass Spectrom. Rev. 2009, 28, 147–176. [Google Scholar] [CrossRef]
- Ray, A.; Bristow, T.; Whitmore, C.; Mosely, J. Online reaction monitoring by mass spectrometry, modern approaches for the analysis of chemical reactions. Mass Spectrom. Rev. 2018, 37, 565–579. [Google Scholar] [CrossRef] [PubMed]
- Hanton, S.D. Mass spectrometry of polymers and polymer surfaces. Chem. Rev. 2001, 101, 527–570. [Google Scholar] [CrossRef]
- Peacock, P.M.; McEwen, C.N. Mass spectrometry of synthetic polymers. Anal. Chem. 2004, 76, 3417–3428. [Google Scholar] [CrossRef] [PubMed]
- Kowalczuk, M.; Adamus, G. Mass spectrometry for the elucidation of the subtle molecular structure of biodegradable polymers and their degradation products. Mass Spectrom. Rev. 2016, 35, 188–198. [Google Scholar] [CrossRef]
- Kowalczuk, M. New vistas in mass spectrometry for sequence analysis of natural and synthetic biodegradable macromolecules. Chim. Oggi Chem. Today 2016, 34, 2. [Google Scholar]
- Alexander, N.E.; Swanson, J.P.; Joy, A.; Wesdemiotis, C. Sequence analysis of cyclic polyester copolymers using ion mobility tandem mass spectrometry. Int. J. Mass Spectrom. 2018, 429, 151–157. [Google Scholar] [CrossRef]
- Arslan, H.; Adamus, G.; Hazer, B.; Kowalczuk, M. Electrospray ionisation tandem mass spectrometry of poly (R, S)-3-hydroxybutanoic acid] telechelics containing primary hydroxy end groups. Rapid Commun. Mass Spectrom. 1999, 13, 2433–2438. [Google Scholar] [CrossRef]
- Jedliński, Z.; Adamus, G.; Kowalczuk, M.; Schubert, R.; Szewczuk, Z.; Stefanowicz, P. Electrospray tandem mass spectrometry of poly (3-hydroxybutanoic acid) end groups analysis and fragmentation mechanism. Rapid Commun. Mass Spectrom. 1998, 12, 357–360. [Google Scholar] [CrossRef]
- Alli, A.; Hazer, B.; Adamus, G.; Kowalczuk, M. Telechelic polyhydroxyalkanoates/polyhydroxybutyrates (PHAs/PHBs). In Handbook of Telechelic Polyesters, Polycarbonates and Polyethers; Pan Stanford Publishing Pte. Ltd.: Sigapore, 2017; pp. 65–114. [Google Scholar]
- Duale, K.; Zięba, M.; Chaber, P.; Di Fouque, D.J.; Memboeuf, A.; Peptu, C.; Radecka, I.; Kowalczuk, M.; Adamus, G. Molecular level structure of biodegradable poly (Delta-Valerolactone) obtained in the presence of boric acid. Molecules 2018, 23, 2034. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Wei, Z.; Leng, X.; Wang, Y.; Li, Y. Boric acid as biocatalyst for living ring-opening polymerization of ε-caprolactone. Polymer 2015, 78, 51–58. [Google Scholar] [CrossRef]
- Song, Q.; Pascouau, C.; Zhao, J.; Zhang, G.; Peruch, F.; Carlotti, S. Ring-opening polymerization of γ-lactones and copolymerization with other cyclic monomers. Prog. Polym. Sci. 2020, 110, 101309. [Google Scholar] [CrossRef]
- Steinman, N.Y.; Bentolila, N.Y.; Domb, A.J. Effect of molecular weight on gelling and viscoelastic properties of poly (caprolactone)–b-Poly (ethylene glycol)–b-Poly (caprolactone)(PCL–PEG–PCL) Hydrogels. Polymers 2020, 12, 2372. [Google Scholar] [CrossRef] [PubMed]
- Lecomte, P.; Jérôme, C. Recent developments in ring-opening polymerization of lactones. Synth. Biodegrad. Polym. 2011, 245, 173–217. [Google Scholar]
- Yuntawattana, N.; McGuire, T.M.; Durr, C.B.; Buchard, A.; Williams, C.K. Indium phosphasalen catalysts showing high isoselectivity and activity in racemic lactide and lactone ring opening polymerizations. Catal. Sci. Technol. 2020, 10, 7226–7239. [Google Scholar] [CrossRef]
- Dubois, P.; Jérǒme, R.; Teyssié, P. Aluminium alkoxides: A family of versatile initiators for the ring-opening polymerization of lactones and lactides. In Proceedings of the 6th International Symposium on Ring-Opening and Cyclopolymerization, Boston, MA, USA, 22–27 April 1990; Volume 42, pp. 103–116. [Google Scholar]
- Piao, L.; Dai, Z.; Deng, M.; Chen, X.; Jing, X. Synthesis and characterization of PCL/PEG/PCL triblock copolymers by using calcium catalyst. Polymer 2003, 44, 2025–2031. [Google Scholar] [CrossRef]
- Dube, M.A.; Salehpour, S. Applying the principles of green chemistry to polymer production technology. Macromol. React. Eng. 2014, 8, 7–28. [Google Scholar] [CrossRef]
- Chaudhuri, M.K.; Hussain, S.; Kantam, M.L.; Neelima, B. Boric acid: A novel and safe catalyst for aza-Michael reactions in water. Tetrahedron Lett. 2005, 46, 8329–8331. [Google Scholar] [CrossRef]
- Pal, R. Boric acid in organic synthesis: Scope and recent developments. Ark. Online J. Org. Chem. 2018, 2018, 346–371. [Google Scholar] [CrossRef] [Green Version]
- Pathan, S.K.; Mahaparale, P.; Deshmukh, S.; Une, H.; Arote, R.; Sangshetti, J. Boric Acid: A Versatile catalyst in organic synthesis. In Applications of Nanotechnology for Green Synthesis; Springer: Berlin/Heidelberg, Germany, 2020; pp. 457–483. [Google Scholar]
- Ozer, D.; Köse, D.A.; Şahin, O.; Oztas, N.A. Synthesis and characterization of boric acid mediated metal-organic frameworks based on trimesic acid and terephthalic acid. J. Mol. Struct. 2017, 1141, 261–267. [Google Scholar] [CrossRef]
- Saka, C.; Balbay, A. Influence of process parameters on enhanced hydrogen evolution from alcoholysis of sodium borohydride with a boric acid catalyst. Int. J. Hydrog. Energy 2020, 45, 16193–16200. [Google Scholar] [CrossRef]
- Mylavarapu, R.K.; Gcm, K.; Kolla, N.; Veeramalla, R.; Koilkonda, P.; Bhattacharya, A.; Bandichhor, R. Boric acid catalyzed amidation in the synthesis of active pharmaceutical ingredients. Org. Process. Res. Dev. 2007, 11, 1065–1068. [Google Scholar] [CrossRef]
- Houston, T.A.; Wilkinson, B.L.; Blanchfield, J.T. Boric acid catalyzed chemoselective esterification of α-hydroxycarboxylic acids. Org. Lett. 2004, 6, 679–681. [Google Scholar] [CrossRef] [PubMed]
- Sitko, M.; Szelwicka, A.; Wojewódka, A.; Skwarek, A.; Tadasiewicz, D.; Schimmelpfennig, L.; Dziuba, K.; Morawiec-Witczak, M.; Chrobok, A. Perdecanoic acid as a safe and stable medium-chain peracid for Baeyer–Villiger oxidation of cyclic ketones to lactones. RSC Adv. 2019, 9, 30012–30018. [Google Scholar] [CrossRef] [Green Version]
- Wesdemiotis, C.; Solak, N.; Polce, M.J.; Dabney, D.E.; Chaicharoen, K.; Katzenmeyer, B.C. Fragmentation pathways of polymer ions. Mass Spectrom. Rev. 2011, 30, 523–559. [Google Scholar] [CrossRef]
- Adamus, G.; Hakkarainen, M.; Höglund, A.; Kowalczuk, M.; Albertsson, A.C. MALDI-TOF-MS reveals the molecular level structures of different hydrophilic-hydrophobic polyether-esters. Biomacromolecules 2009, 10, 1540–1546. [Google Scholar] [CrossRef] [PubMed]
- Adamus, G. Molecular Level Structure of (R,S)-3-hydroxybutyrate/(R,S)-3-hydroxy-4-ethoxybutyrate copolyesters with dissimilar architecture. Macromolecules 2009, 42, 4547–4557. [Google Scholar] [CrossRef]
- Martinka Maksymiak, M.; Zięba, M.; Orchel, A.; Musiał-Kulik, M.; Kowalczuk, M.; Adamus, G. Bioactive (Co)oligoesters as potential delivery systems of p-anisic acid for cosmetic purposes. Materials 2020, 13, 4153. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Mn,NMR (g/mol) | Mn,GPC (g/mol) | Mw/Mn |
|---|---|---|---|
| 1-PdVL | 1500 | 1500 | 1.07 |
| 2-PdVL | 4400 | 5000 | 1.35 |
| 3-PdVL | 8900 | 9700 | 1.11 |
| 1-P(dVL-co-mCL) | 1200 | 1200 | 1.78 |
| 2-P(dVL-co-mCL) | 3200 | 3700 | 1.83 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duale, K.; Latos, P.; Chrobok, A.; Domiński, A.; Maksymiak, M.M.; Adamus, G.; Kowalczuk, M. Towards Advances in Molecular Understanding of Boric Acid Biocatalyzed Ring-Opening (Co)Polymerization of δ-Valerolactone in the Presence of Ethylene Glycol as an Initiator. Molecules 2021, 26, 4859. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26164859
Duale K, Latos P, Chrobok A, Domiński A, Maksymiak MM, Adamus G, Kowalczuk M. Towards Advances in Molecular Understanding of Boric Acid Biocatalyzed Ring-Opening (Co)Polymerization of δ-Valerolactone in the Presence of Ethylene Glycol as an Initiator. Molecules. 2021; 26(16):4859. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26164859
Chicago/Turabian StyleDuale, Khadar, Piotr Latos, Anna Chrobok, Adrian Domiński, Magdalena Martinka Maksymiak, Grażyna Adamus, and Marek Kowalczuk. 2021. "Towards Advances in Molecular Understanding of Boric Acid Biocatalyzed Ring-Opening (Co)Polymerization of δ-Valerolactone in the Presence of Ethylene Glycol as an Initiator" Molecules 26, no. 16: 4859. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26164859