
Expanding the Myxochelin Natural Product Family by Nicotinic Acid Containing Congeners

 , ,
, ,
Abstract
:1. Introduction
2. Results
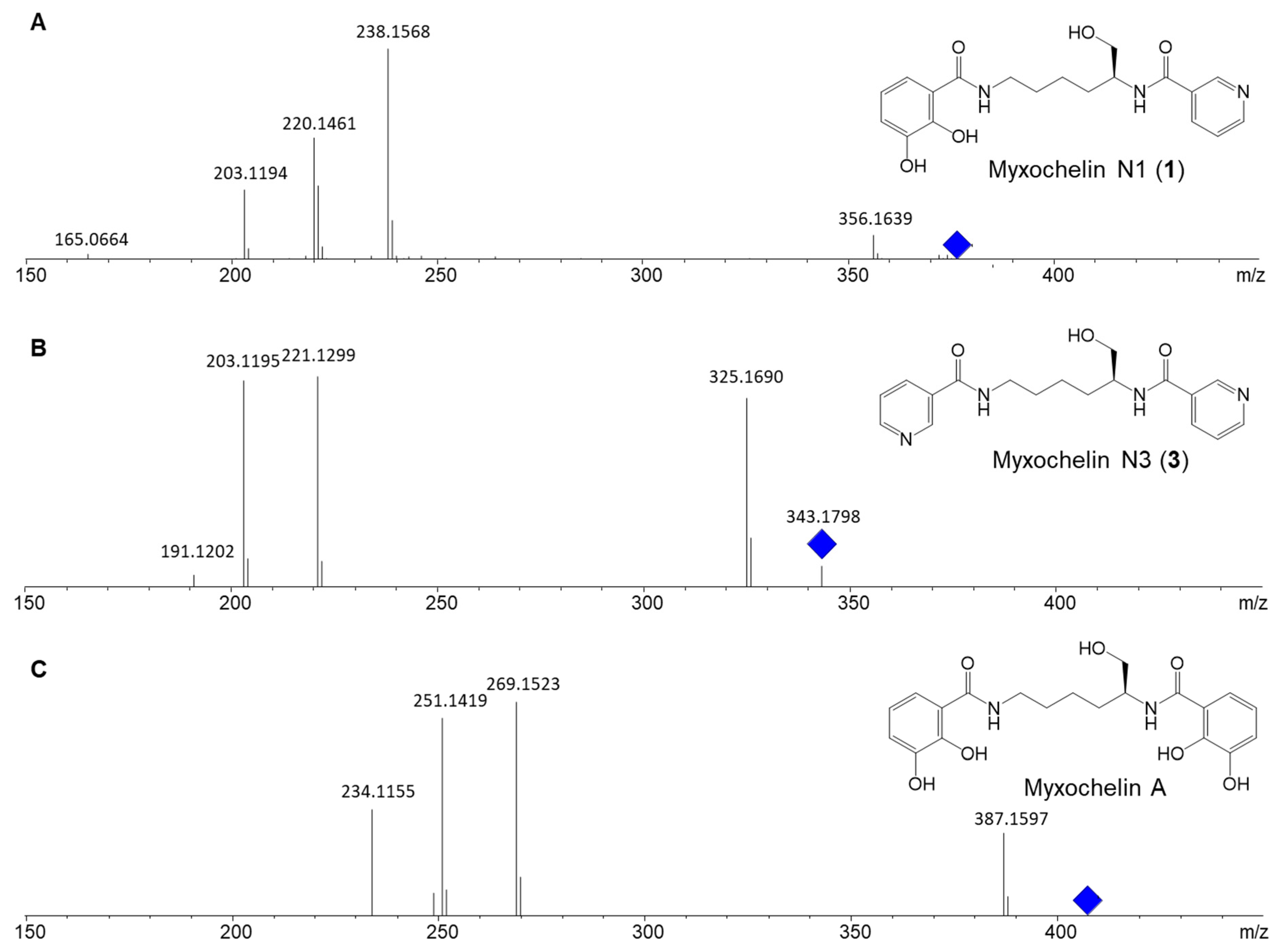
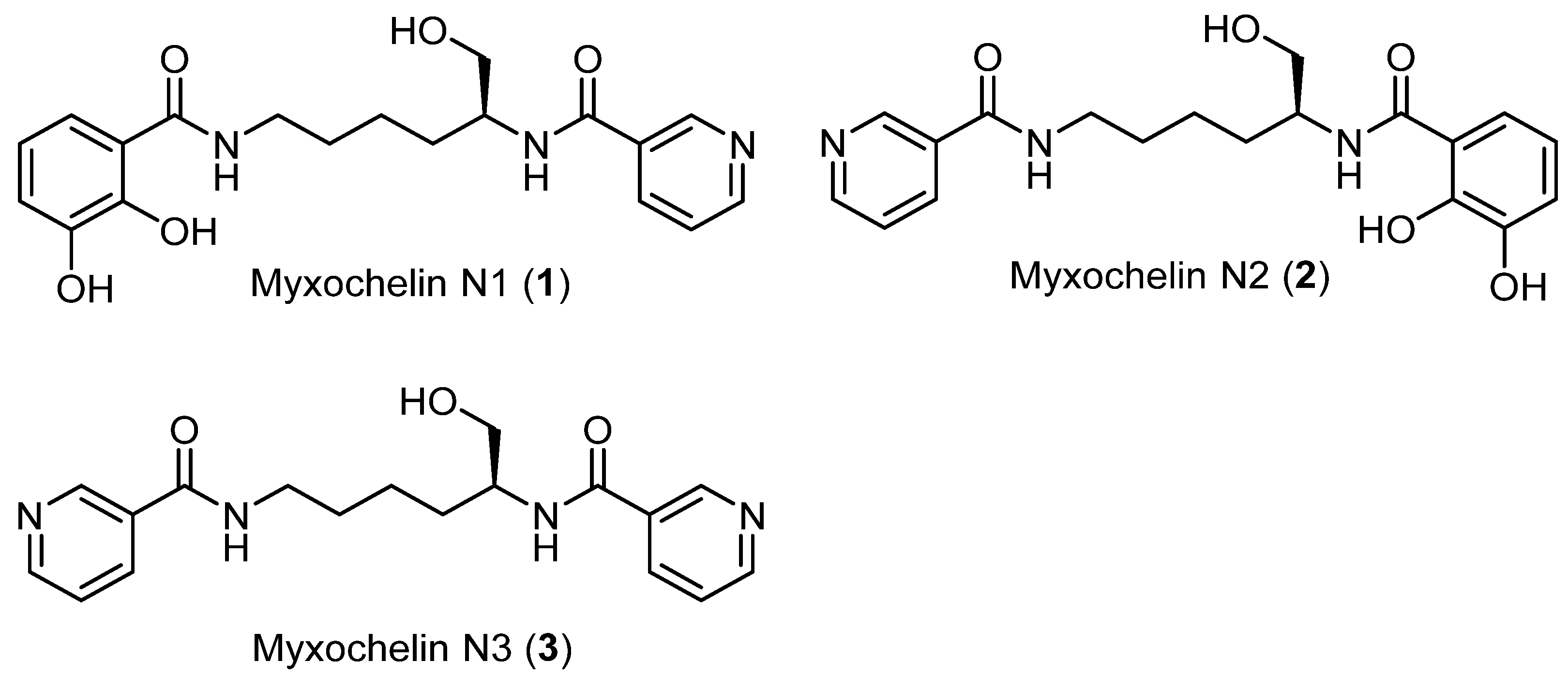
2.1. Discovery of Myxochelin N1
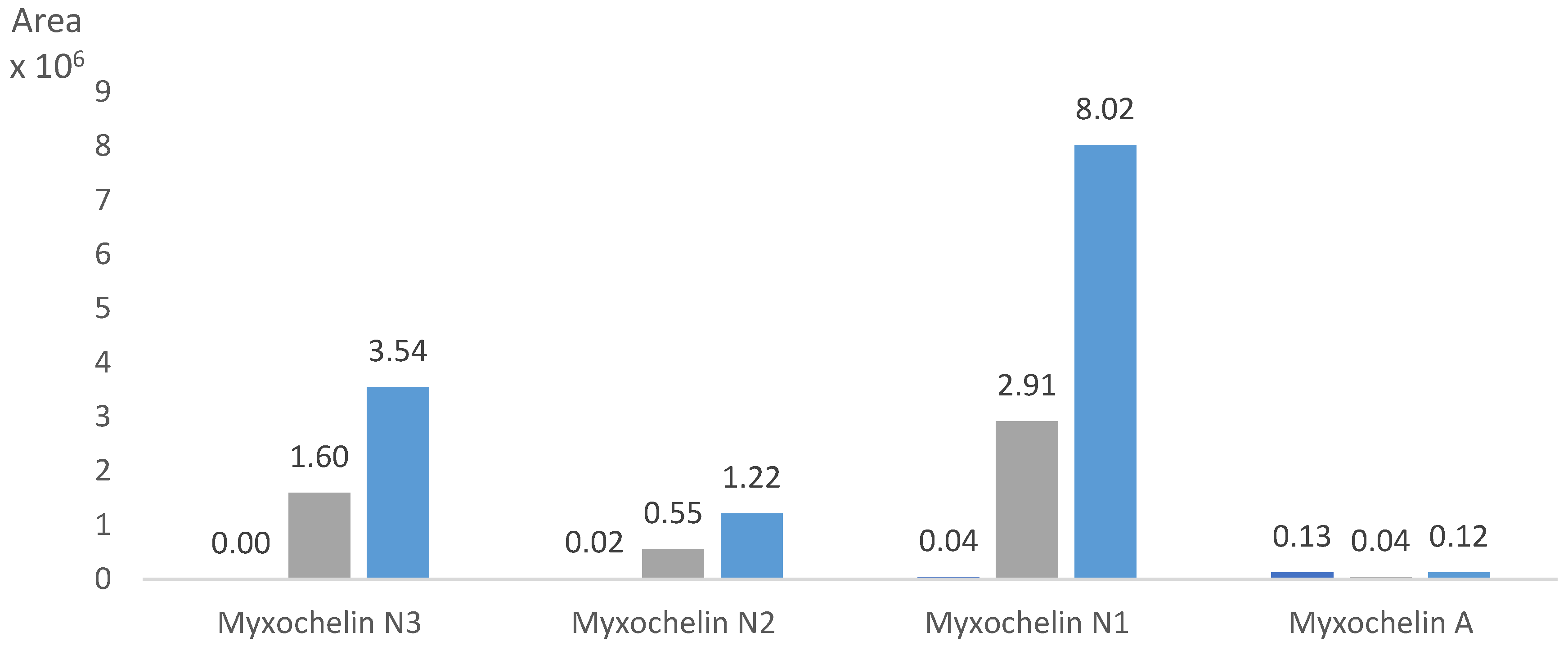
2.2. Precursor-Directed Biosynthesis and Isolation of Myxochelin N1 and N3
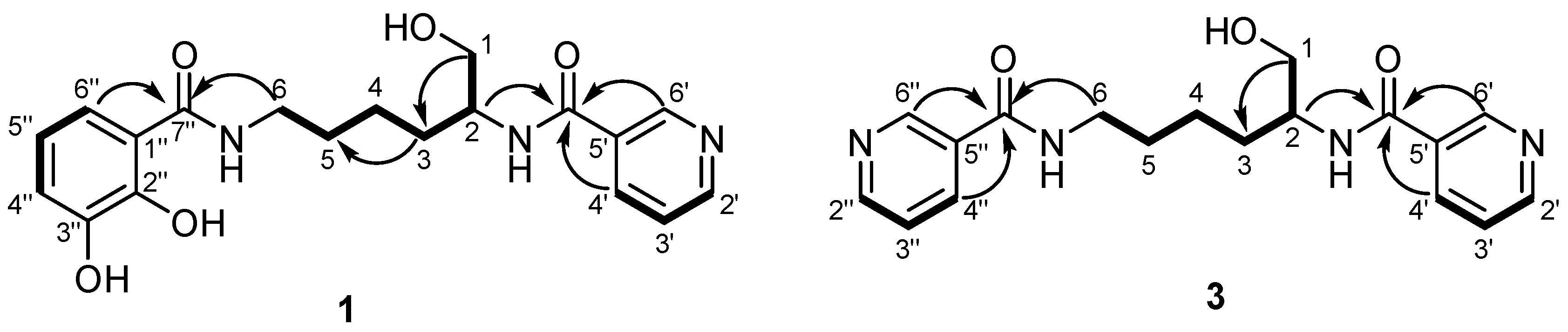
2.3. Structure Elucidation
2.4. Total Synthesis
2.5. Biological Activities
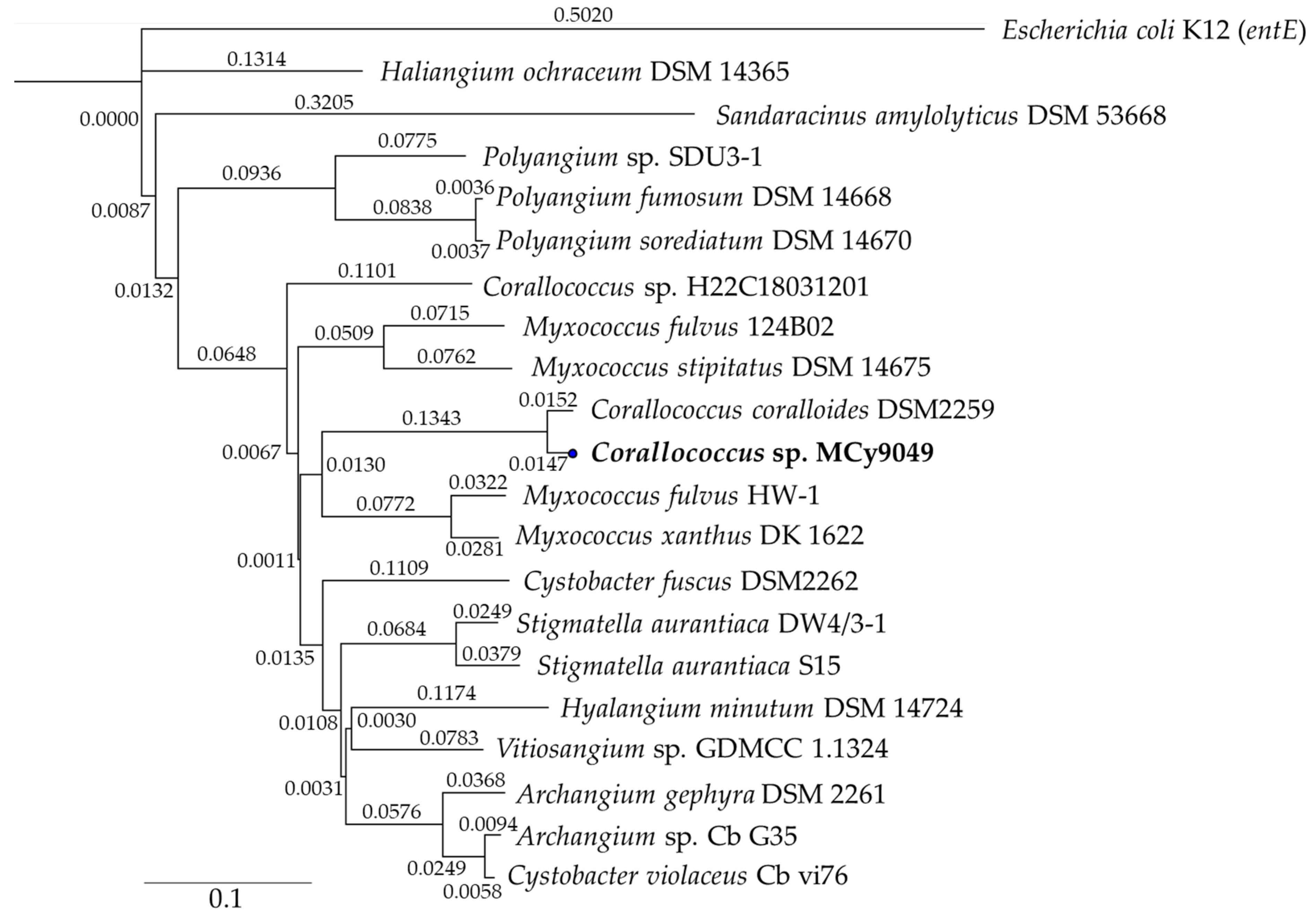
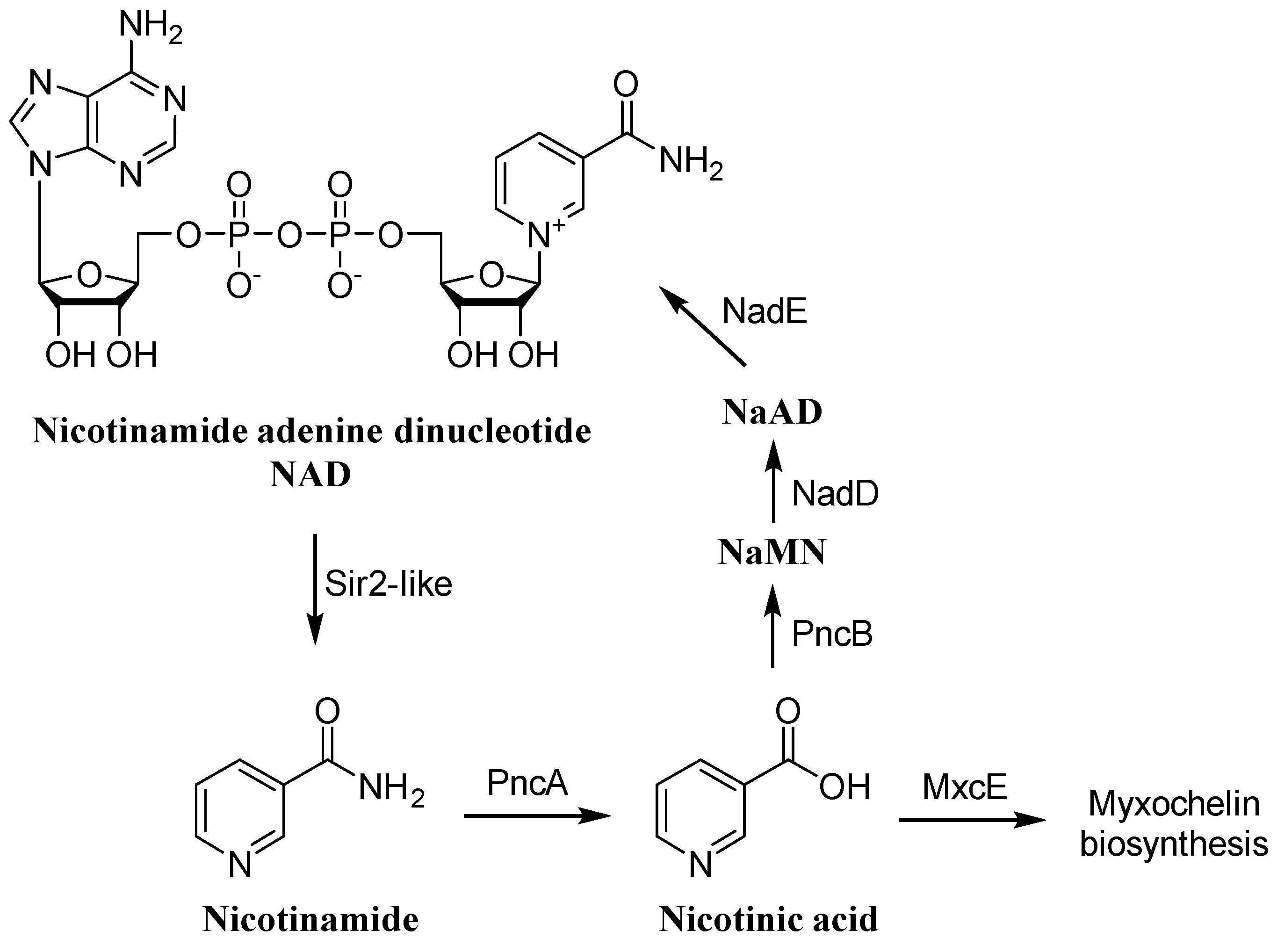
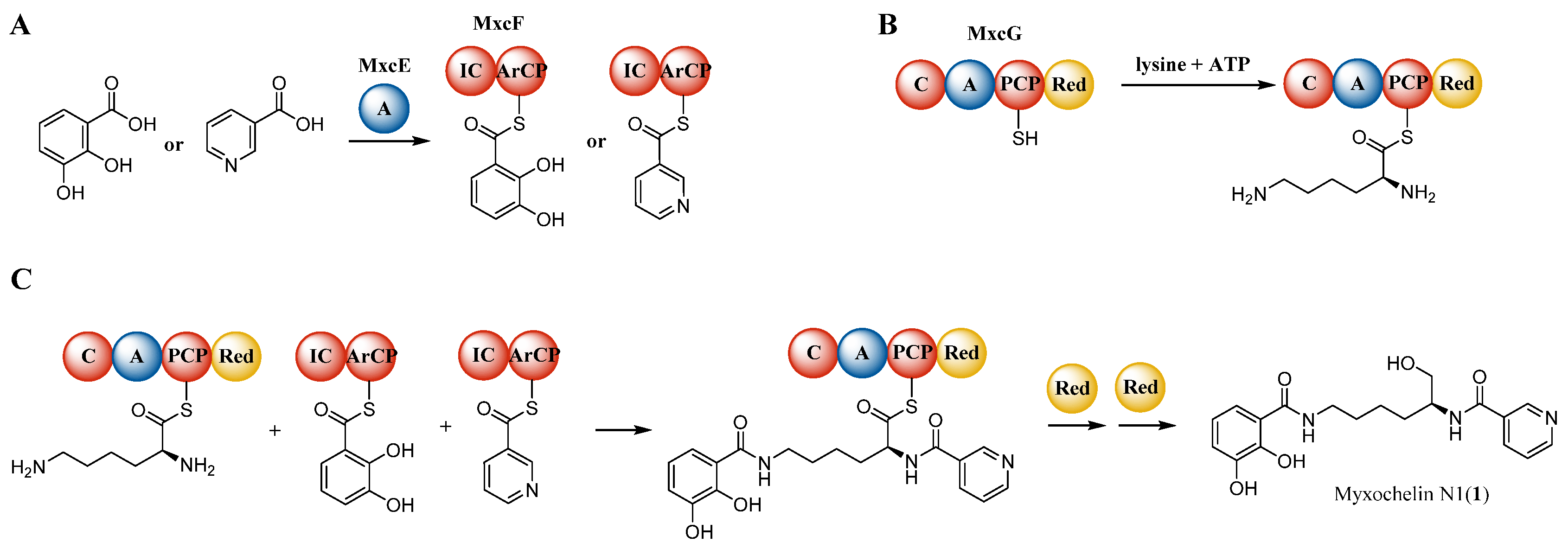
2.6. Biosynthetic Origin of the New Myxochelins 1–3
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Myxobacterial Fermentation and Small-Scale Precursor Feeding
5.2. Analysis of the Secondary Metabolome of Bacterial Crude Extracts
5.3. Compound Isolation
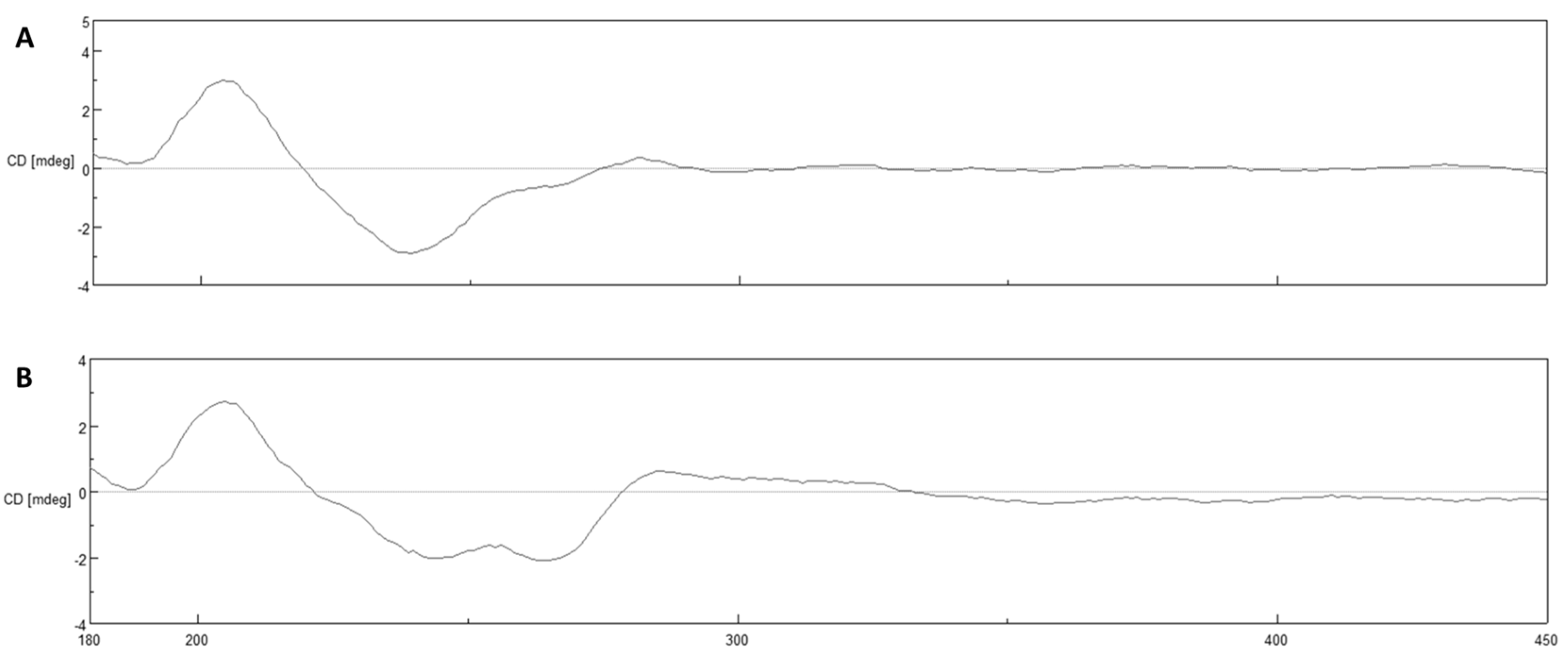
5.4. Structure Elucidation, Chiroptical and Circular Dichroism Measurements
5.5. Chemistry
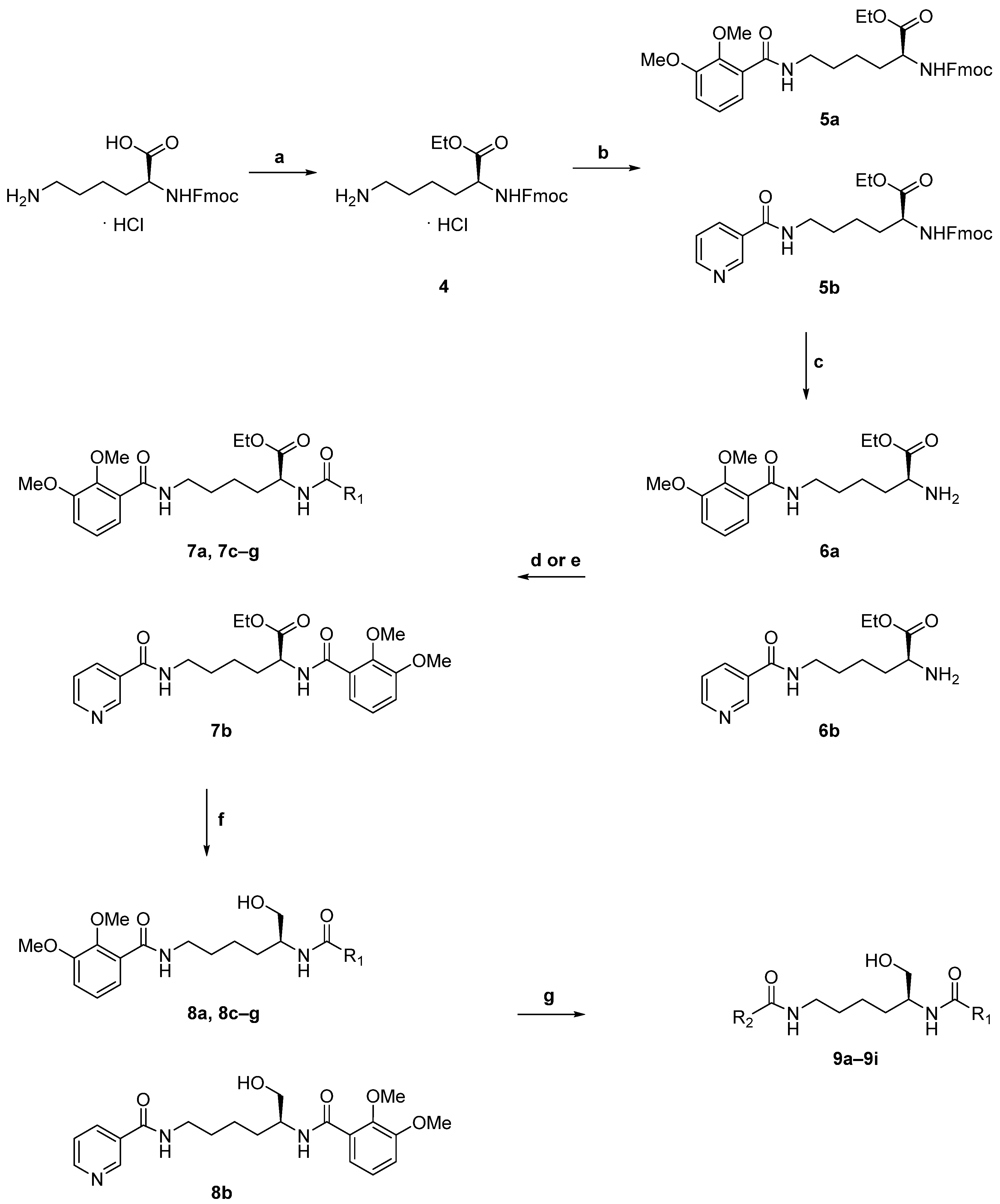
5.5.1. General Procedure for Synthetic Step a
5.5.2. General Procedure for Synthetic Step b
5.5.3. General Procedure for Synthetic Step c
5.5.4. General Procedure for Synthetic Step d
5.5.5. General Procedure for Synthetic Step e
5.5.6. General Procedure for Synthetic Step f
5.5.7. General Procedure for Synthetic Step g
5.6. Characterization of Synthetic Intermediates and Final Products
5.7. Reaction Monitoring and Purification of Synthetic Compounds
5.8. Bioactivity
5.8.1. Assessment of Antimicrobial Activities (MIC and Synergy)
5.8.2. Cytotoxic Activity (IC50)
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Muñoz-Dorado, J.; Marcos-Torres, F.J.; García-Bravo, E.; Moraleda-Muñoz, A.; Pérez, J. Myxobacteria: Moving, killing, feeding, and surviving together. Front. Microbiol. 2016, 7, 781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, P.; Dey, A.; Vassallo, C.N.; Wall, D. How Myxobacteria Cooperate. J. Mol. Biol. 2015, 427, 3709–3721. [Google Scholar] [CrossRef] [Green Version]
- Herrmann, J.; Fayad, A.A.; Müller, R. Natural products from myxobacteria: Novel metabolites and bioactivities. Nat. Prod. Rep. 2017, 34, 135–160. [Google Scholar] [CrossRef] [PubMed]
- Baumann, S.; Herrmann, J.; Raju, R.; Steinmetz, H.; Mohr, K.I.; Hüttel, S.; Harmrolfs, K.; Stadler, M.; Müller, R. Cystobactamids: Myxobacterial topoisomerase inhibitors exhibiting potent antibacterial activity. Angew. Chem. Int. Ed. 2014, 53, 14605–14609. [Google Scholar] [CrossRef] [PubMed]
- Gerth, K.; Bedorf, N.; Irschik, H.; Höfle, G.; Reichenbach, H. The soraphens: A family of novel antifungal compounds from Sorangium cellulosum (Myxobacteria). I. Soraphen A1α: Fermentation, isolation, biological properties. J. Antibiot. 1994, 47, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Held, J.; Gebru, T.; Kalesse, M.; Jansen, R.; Gerth, K.; Müller, R.; Mordmüller, B. Antimalarial activity of the myxobacterial macrolide chlorotonil A. Antimicrob. Agents Chemother. 2014, 58, 6378–6384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Höfle, G.; Bedorf, N.; Steinmetz, H.; Schomburg, D.; Gerth, K.; Reichenbach, H. Epothilone A and B—Novel 16-Membered macrolides with cytotoxic activity: Isolation, crystal structure, and conformation in solution. Angew. Chem. Int. Ed. Engl. 1996, 35, 1567–1569. [Google Scholar] [CrossRef]
- Weissman, K.J.; Müller, R. A brief tour of myxobacterial secondary metabolism. Bioorg. Med. Chem. 2009, 17, 2121–2136. [Google Scholar] [CrossRef] [PubMed]
- Kunze, B.; Bedorf, N.; Kohl, W.; Höfle, G.; Reichenbach, H. Myxochelin A, a new iron-chelating compound from Angiococcus disciformis (Myxobacterales). Production, isolation, physico-chemical and biological properties. J. Antibiot. 1989, 42, 14–17. [Google Scholar] [CrossRef] [Green Version]
- Gaitatzis, N.; Kunze, B.; Müller, R. In vitro reconstitution of the myxochelin biosynthetic machinery of Stigmatella aurantiaca Sg a15: Biochemical characterization of a reductive release mechanism from nonribosomal peptide synthetases. Proc. Natl. Acad. Sci. USA 2001, 98, 11136–11141. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Weissman, K.J.; Müller, R. Myxochelin biosynthesis: Direct evidence for two- and four-electron reduction of a carrier protein-bound thioester. J. Am. Chem. Soc. 2008, 130, 7554–7555. [Google Scholar] [CrossRef] [PubMed]
- Korp, J.; König, S.; Schieferdecker, S.; Dahse, H.-M.; König, G.M.; Werz, O.; Nett, M. Harnessing Enzymatic Promiscuity in Myxochelin Biosynthesis for the Production of 5-Lipoxygenase Inhibitors. ChemBioChem 2015, 16, 2445–2450. [Google Scholar] [CrossRef]
- Sester, A.; Winand, L.; Pace, S.; Hiller, W.; Werz, O.; Nett, M. Myxochelin- and Pseudochelin-Derived Lipoxygenase Inhibitors from a Genetically Engineered Myxococcus xanthus Strain. J. Nat. Prod. 2019, 82, 2544–2549. [Google Scholar] [CrossRef] [PubMed]
- Gaitatzis, N.; Kunze, B.; Müller, R. Novel insights into siderophore formation in myxobacteria. ChemBioChem 2005, 6, 365–374. [Google Scholar] [CrossRef]
- Panter, F.; Popoff, A.; Garcia, R.; Krug, D.; Müller, R. Characterization of the Vitamin K2 derived Myxoquinones and their distribution among myxobacteria. Microorganisms. Manuscript in preparation.
- Miyanaga, S.; Obata, T.; Onaka, H.; Fujita, T.; Saito, N.; Sakurai, H.; Saiki, I.; Furumai, T.; Igarashi, Y. Absolute configuration and antitumor activity of myxochelin A produced by Nonomuraea pusilla TP-A0861. J. Antibiot. 2006, 59, 698–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyanaga, S.; Sakurai, H.; Saiki, I.; Onaka, H.; Igarashi, Y. Synthesis and evaluation of myxochelin analogues as antimetastatic agents. Bioorg. Med. Chem. 2009, 17, 2724–2732. [Google Scholar] [CrossRef]
- Schieferdecker, S.; Nett, M. A fast and efficient method for the preparation of the 5-lipoxygenase inhibitor myxochelin A. Tetrahedron Lett. 2016, 57, 1359–1360. [Google Scholar] [CrossRef]
- Schieferdecker, S.; König, S.; Pace, S.; Werz, O.; Nett, M. Myxochelin-Inspired 5-Lipoxygenase Inhibitors: Synthesis and Biological Evaluation. ChemMedChem 2017, 12, 23–27. [Google Scholar] [CrossRef]
- Li, J.; Sha, Y. A convenient synthesis of amino acid methyl esters. Molecules 2008, 13, 1111–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fields, G.B.; Noble, R.L. Solid phase peptide synthesis utilizing 9-fluorenylmethoxycarbonyl amino acids. Int. J. Pept. Protein Res. 1990, 35, 161–214. [Google Scholar] [CrossRef] [PubMed]
- Schieferdecker, S.; König, S.; Koeberle, A.; Dahse, H.-M.; Werz, O.; Nett, M. Myxochelins target human 5-lipoxygenase. J. Nat. Prod. 2015, 78, 335–338. [Google Scholar] [CrossRef]
- Ahn, J.-W.; Lee, C.-O.; Baek, S.-H. Myxochelin A, a cytotoxic antibiotic from the myxobacterium Angiococcus Disciformis. Orient. Pharm. Exp. Med. 2002, 2, 64–67. [Google Scholar] [CrossRef]
- Hoffmann, T.; Krug, D.; Bozkurt, N.; Duddela, S.; Jansen, R.; Garcia, R.; Gerth, K.; Steinmetz, H.; Müller, R. Correlating chemical diversity with taxonomic distance for discovery of natural products in myxobacteria. Nat. Commun. 2018, 9, 803. [Google Scholar] [CrossRef]
- Röttig, M.; Medema, M.H.; Blin, K.; Weber, T.; Rausch, C.; Kohlbacher, O. NRPSpredictor2—a web server for predicting NRPS adenylation domain specificity. Nucleic Acids Res. 2011, 39, W362–W367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, M.T.; Smith, B.C.; Jackson, M.D.; Denu, J.M. Coenzyme specificity of Sir2 protein deacetylases: Implications for physiological regulation. J. Biol. Chem. 2004, 279, 40122–40129. [Google Scholar] [CrossRef] [Green Version]
- Okazaki, I.J.; Moss, J. Mono-ADP-ribosylation: A Reversible Posttranslational Modification of Proteins. In Advances in pharmacology; August, J.T., Ed.; Academic Press: San Diego, CA, USA, 1996; Volume 35, pp. 247–280. ISBN 9780120329366. [Google Scholar]
- McErlean, M.; Overbay, J.; van Lanen, S. Refining and expanding nonribosomal peptide synthetase function and mechanism. J. Ind. Microbiol. Biotechnol. 2019, 46, 493–513. [Google Scholar] [CrossRef]
- Gholson, R.K. The Pyridine Nucleotide Cycle. Nature 1966, 212, 933–935. [Google Scholar] [CrossRef]
- Rodionov, D.A.; de Ingeniis, J.; Mancini, C.; Cimadamore, F.; Zhang, H.; Osterman, A.L.; Raffaelli, N. Transcriptional regulation of NAD metabolism in bacteria: NrtR family of Nudix-related regulators. Nucleic Acids Res. 2008, 36, 2047–2059. [Google Scholar] [CrossRef] [PubMed]
- Denu, J.M. Linking chromatin function with metabolic networks: Sir2 family of NAD+-dependent deacetylases. Trends Biochem. Sci. 2003, 28, 41–48. [Google Scholar] [CrossRef]
- Subko, K.; Wang, X.; Nielsen, F.H.; Isbrandt, T.; Gotfredsen, C.H.; Ramos, C.; Mackenzie, T.; Vicente, F.; Genilloud, O.; Frisvad, J.C.; et al. Mass Spectrometry guided discovery and design of novel Asperphenamate analogues from Penicilium astrolabium reveals an extraordinary NRPS flexibility. Front. Microbiol. 2021, 11, 3510. [Google Scholar] [CrossRef]
- Itoh, T.; Tokunaga, K.; Matsuda, Y.; Fujii, I.; Abe, I.; Ebizuka, Y.; Kushiro, T. Reconstitution of a fungal meroterpenoid biosynthesis reveals the involvement of a novel family of terpene cyclases. Nat. Chem. 2010, 2, 858–864. [Google Scholar] [CrossRef] [PubMed]
- Cutignano, A.; Tramice, A.; de Caro, S.; Villani, G.; Cimino, G.; Fontana, A. Biogenesis of 3-alkylpyridine alkaloids in the marine mollusc Haminoea orbignyana. Angew. Chem. Int. Ed. 2003, 42, 2633–2636. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.M.; Zhou, Q.; Tang, Y.M.; Zhang, Z.; Chen, Y.S.; He, H.Y.; Pan, H.X.; Tang, M.C.; Gao, J.F.; Zhao, S.Y.; et al. Unconventional origin and hybrid system for construction of pyrrolopyrrole moiety in kosinostatin biosynthesis. Chem. Biol. 2013, 20, 796–805. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Throckmorton, K.; Maity, M.; Chevrette, M.G.; Braun, D.R.; Rajski, S.R.; Currie, C.R.; Thomas, M.G.; Bugni, T.S. Bacillibactins E and F from a Marine Sponge-Associated Bacillus sp. J. Nat. Prod. 2020, 84, 136–141. [Google Scholar] [CrossRef]
- Miethke, M.; Klotz, O.; Linne, U.; May, J.J.; Beckering, C.L.; Marahiel, M.A. Ferri-bacillibactin uptake and hydrolysis in Bacillus subtilis. Mol. Microbiol. 2006, 61, 1413–1427. [Google Scholar] [CrossRef]
- Jaehme, M.; Slotboom, D.J. Diversity of membrane transport proteins for vitamins in bacteria and archaea. Biochim. Biophys. Acta 2015, 1850, 565–576. [Google Scholar] [CrossRef] [PubMed]
- Bergeron, R.J.; Gavanaugh, P.F.; Kline, S.J.; Hughes, R.G.; Elliott, G.T.; Porter, C.W. Antineoplastic and antiherpetic activity of spermidine catecholamide iron chelators. Biochem. Biophys. Res. Commun. 1984, 121, 848–854. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 (in CD3OD) | |||
|---|---|---|---|
| No. | δH, Mult. (J in Hz) | δC, Type | HMBC |
| 1 | 3.63, brd (3.4) | 64.4, CH2 | 2, 3 |
| 3.61, brd (3.8) | |||
| 2 | 4.15, m | 53.2, CH | 4, 7′ |
| 3 | 1.63, m | 30.2, CH2 | 1, 2, 4, 5 |
| 4 | 1.49, m | 22.6, CH2 | 2, 5, 6 |
| 5 | 1.72, m | 28.9, CH2 | 3, 6 |
| 6 | 3.39, t (7.0) | 39.4, CH2 | 4, 5, 7″ |
| 2′ | 8.65, brd (8.2) | 151.0, CH | 3′, 4′, 6′ |
| 3′ | 7.49, dd (8.1, 5.0) | 123.6, CH | 2′, 5′ |
| 4′ | 8.20, brd (7.7) | 135.7, CH | 2′, 3′, 6′ |
| 5′ | 130.9, C | ||
| 6′ | 8.96, brd (2.2) | 147.7, CH | 2′, 4′, 7′ |
| 7′ | 166.7, C | ||
| 1″ | 115.3, C | ||
| 2″ | 148.6, C | ||
| 3″ | 145.9, C | ||
| 4″ | 6.90, d (8.0) | 118.1, CH | 2″, 3″, 6″ |
| 5″ | 6.67, t (8.0) | 118.1, CH | 1″, 3″ |
| 6″ | 7.16, d (8.1) | 117.1, CH | 1″, 2″, 4″, 7″ |
| 7″ | 170.1, C |
| 3 (in CD3OD) | |||
|---|---|---|---|
| No. | δH, Mult. (J in Hz) | δC, Type | HMBC |
| 1 | 3.62, dd (10.7, 5.4) | 64.8, CH2 | 2, 3 |
| 3.64, dd (10.6, 5.2) | |||
| 2 | 4.15, m | 53.7, CH | 4, 7′ |
| 3 | 1.64, m | 30.2, CH2 | 1, 2, 4, 5 |
| 4 | 1.49, m | 23.4, CH2 | 2, 5, 6 |
| 5 | 1.74, m | 29.2, CH2 | 3, 6 |
| 6 | 3.42, m | 40.4, CH2 | 4, 5, 7″ |
| 2′/2″ | 8.66, o | 151.7, CH * | 4′, 6′ |
| 3′/3″ | 7.51, dd (7.9, 2.3) | 123.6, CH | 5′ |
| 4′/4″ | 8.20, o | 136.4, CH | 2′, 6′, 7′ |
| 5′/5″ | 131.2, C * | ||
| 6′/6″ | 8.95, o | 148.2, CH | 4′, 7′ |
| 7′/7″ | 167.2, C |
| R1 | R2 | |
|---|---|---|
| 9a = 1 |  |  |
| 9b = 2 | | |
| 9c [12] |  | |
| 9d |  | |
| 9e |  | |
| 9f |  | |
| 9g |  | |
| 9h | |  |
| 9i | | |
| Compound | IC50 [µg/mL] | |
|---|---|---|
| HCT-116 | HepG2 | |
| 9a | >37 | >37 |
| 9b | 29.1 ± 3.0 | >37 |
| 9c | 6.1 ± 0.1 | 6.7 ± 0.9 |
| 9d | 29.5 ± 9.2 | 35.9 ± 0.9 |
| 9e | 17.4 ± 8.0 | 11.3 ± 0.7 |
| 9f | 6.7 ± 2.0 | 10.6 ± 2.6 |
| 9g | 9.2 ± 2.3 | 11.8 ± 1.7 |
| 9h | >37 | >37 |
| 9i | >37 | >37 |
| Doxorubicin | 0.2 | 0.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frank, N.A.; Széles, M.; Akone, S.H.; Rasheed, S.; Hüttel, S.; Frewert, S.; Hamed, M.M.; Herrmann, J.; Schuler, S.M.M.; Hirsch, A.K.H.; et al. Expanding the Myxochelin Natural Product Family by Nicotinic Acid Containing Congeners. Molecules 2021, 26, 4929. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26164929
Frank NA, Széles M, Akone SH, Rasheed S, Hüttel S, Frewert S, Hamed MM, Herrmann J, Schuler SMM, Hirsch AKH, et al. Expanding the Myxochelin Natural Product Family by Nicotinic Acid Containing Congeners. Molecules. 2021; 26(16):4929. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26164929
Chicago/Turabian StyleFrank, Nicolas A., Márió Széles, Sergi H. Akone, Sari Rasheed, Stephan Hüttel, Simon Frewert, Mostafa M. Hamed, Jennifer Herrmann, Sören M. M. Schuler, Anna K. H. Hirsch, and et al. 2021. "Expanding the Myxochelin Natural Product Family by Nicotinic Acid Containing Congeners" Molecules 26, no. 16: 4929. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26164929