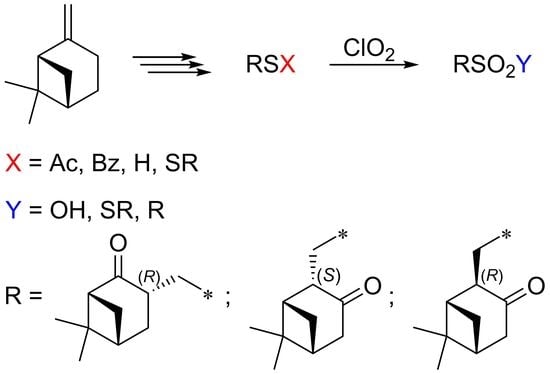
4.2. General Procedure
(1R,5R,E)-3-(Hydroxymethylene)-6,6-dimethylbicyclo [3.1.1]heptan-2-one (3). t-BuOK (15.38 mmol, 1.726 g) was added to a solution of (−)-nopinone
4 (11.4 mmol, 1.575 g) in 20 mL THF cooled to 0 °C. After the dissolution of
t-BuOK, a solution of isoamyl formate (15.38 mmol, 1.726 g) in 10 mL of THF was added dropwise to the mixture. The resulting mixture was stirred for 6 h at room temperature. THF was distilled off, 30 mL of water was added to the dry residue and acidified to pH = 6–7 with 10% HCl solution, and the mixture was extracted with Et
2O (3 × 20 mL). The combined organic layers were washed with brine, dried over Na
2SO
4, and the solvent was distilled off to obtain 1.818 g of ketoenol
3 (yield 96%). Subsequently, compound
3 was used without additional purification. Physicochemical characteristics correspond to the data in the literature [
9].
S-(((1R,3R,5R)-6,6-Dimethyl-2-oxobicyclo[3.1.1]heptan-3-yl)methyl) ethanethioate (5). Two drops of pyridine (~1 mol%) and 3.0 g (0.04 mol) of thioacetic acid were added to 3.0 g (0.02 mol) of 2-norpinanone 2 with stirring. The synthesis time was 5–10 min. The reaction progress was monitored by TLC (eluent–CH2Cl2, developer–vanillin solution in EtOH). After completion of the reaction, a saturated solution of NaHCO3 was added, and the mixture was extracted with CH2Cl2 (3 × 20 mL). The combined organic layers were dried over Na2SO4, and the solvent was removed under reduced pressure. The product was purified by column chromatography (eluent–CH2Cl2). Light-yellow liquid, 79% yield (3.6 g), 98% de,+22.4 (c 0.35, CHCl3). 1H NMR spectrum (CDCl3, δ, ppm, J/Hz): 0.77 (s, 3H, H8), 1.35 (s, 3H, H9), 1.61 (dd, 1H, H4a, J = 13.1, 8.1), 1.72 (d, 1H, H7a, J = 10.5), 2.23–2.31 (m, 1H, H5), 2.36 (s, 3H, CH3Ac), 2.38–2.53 (m, 2H, H4β, H7b), 2.63 (t, 1H, H1, J = 5.1), 2.78–2.92 (m, 1H, H3), 3.00 (dd, 1H, H10a, J = 13.3, 7.84), 3.41 (dd, 1H, H10b, J = 13.5, 5.5). 13C NMR spectrum (CDCl3, δ, ppm): 21.9 (C8), 25.2 (C7), 26.3 (C9), 28.2 (C4), 28.5 (C10), 30.5 (CAc), 40.7 (C5), 42.8 (C3), 43.4 (C6), 57.5 (C1), 195.8 (C2), 213.8 (CAc = O). IR spectrum (KBr, ν, cm−1): 628, 954, 1024, 1109, 1136 st (C–O), 1199, 1253, 1352, 1419, 1460, 1693 st (C=O), 1708 st (C=O), 2872 st, 2927 st; elemental analysis calcd (%) for C12H18O2S: C 63.68, H 8.02, S 14.16; found: C 63.65, H 7.96, S 14.20.
(1R,3R,5R)-3-(Thiomethyl)-6,6-dimethylbicyclo[3.1.1]heptan-2-one (6). Hydrazine hydrate 0.1 g (2 mmol) was added dropwise to a solution of 0.23 g (1 mmol) of thioacetate 5 in 2.5 mL of THF cooled to 0 °C. The mixture was stirred for 4 h. 1N HCl solution (2 mmol) was added to the reaction mixture to wash from hydrazine hydrate. The mixture was extracted with Et2O (3 × 20 mL) and washed with brine. The extract was dried over Na2SO4, and the solvent was removed under reduced pressure. The product was purified by column chromatography (eluent–CHCl3). Transparent liquid, 75% yield (0.14 g), +2.2 (c 0.27, CHCl3). 1H NMR spectrum (CDCl3, δ, ppm, J/Hz): 0.75 (s, 3H, H8), 1.33 (s, 3H, H9), 1.56–1.62 (m, 1H, H4a), 1.65 (t, 1H, SH, J = 8.6), 1.71 (d, 1H, H7a, J = 9.9), 2.25–2.34 (m, 1H, H5), 2.40–2.54 (m, 3H, H4b, H7b, H10a), 2.54–2.62 (m, 1H, H1), 2.72–2.86 (m, 1H, H3), 3.15 (ddd, 1H, H10b, J = 13.5, 8.9, 4.6). 13C NMR spectrum (CDCl3, δ, ppm): 21.9 (C8), 24.4 (C10), 25.4 (C7), 26.3 (C9), 28.0 (C4), 40.7 (C5), 43.2 (C6), 45.5 (C3), 57.5 (C1), 214.1 (C2). IR spectrum (KBr, ν, cm−1): 472, 534, 628, 954, 1026, 1087, 1172, 1199, 1253, 1315, 1346, 1369, 1386, 1458, 1707 st (C=O), 2567 (SH), 2870 st, 2931 st; elemental analysis calcd (%) for C10H16OS: C 65.17, H 8.75, S 17.40; found: C 65.35, H 8.76, S 17.49.
(1R,1′R,3R,3′R,5R,5′R)-3,3′-(Disulfanediylbis(methylene))bis(6,6-dimethylbicyclo[3.1.1]heptan-2-one) (7). An aqueous solution containing 0.068 g (1 mmol) of ClO2 was added to a solution of 0.184 g (1 mmol) of thiol 6 in 20 mL of chloroform (the volume of the aqueous solution varied depending on the ClO2 concentration; it did not affect the composition and ratio of products). The solvent was distilled off after 30 min. The residue was purified by column chromatography (eluent –CHCl3). White powder, m.p. 97–99 °C, 90% yield (0.150 g), −174.4 (c 0.51, CHCl3). 1H NMR spectrum (CDCl3, δ, ppm, J/Hz): 0.72 (s, 3H, H8), 1.32 (s, 3H, H9), 1.58 (dd, 1H, H4a), 1.74 (d, 1H, H7a, J = 10.6), 2.23–2.32 (m, 1H, H5), 2.40–2.53 (m, 3H, H4b, H7b, H10a), 2.60 (t, 1H, H1, J = 5.28), 2.94–3.07 (m, 1H, H3), 3.52 (dd, 1H, H10b, J = 13.2, 3.9). 13C NMR spectrum (CDCl3, δ, ppm): 22.0 (C8), 25.3 (C7), 26.2 (C9), 28.6 (C4), 38.4 (C10), 40.7 (C5), 41.7 (C3), 42.9 (C6), 57.6 (C1), 214.3 (C2). IR spectrum (KBr, ν, cm−1): 472, 536, 1024, 1197, 1253, 1311, 1705 st (C=O), 2868 st, 2941 st; elemental analysis calcd (%) for C20H30O2S2: C 65.53, H 8.25, S 17.49; found: C 66.05, H 8.36, S 17.30.
(1S,2S,5R)-2-(Thiomethyl)-6,6-dimethylbicyclo[3.1.1]heptan-3-one ((2S)-8) was synthesized similarly to thiol 6 from thioacetate (2S)-11. The product was purified by column chromatography (eluent–CH2Cl2: Et2O = 30:1). Transparent liquid, 90% yield, +41.8 (c 0.22, CHCl3). 1H NMR spectrum (CDCl3, δ, ppm, J/Hz): 0.83 (s, 3H, H8), 1.19 (d, 1H, H7a, J = 10.8), 1.33 (s, 3H, H9), 1.56 (t, 1H, SH, J = 8.21), 2.09–2.18 (m, 1H, H5), 2.31–2.41 (m, 1H, H10a), 2.44–2.52 (m, 2H, H1, H2), 2.54–2.29 (m, 1H, H4a), 2.60–2.74 (m, 1H, H4b, H7b), 3.27–3.38 (m, 1H, H10b). 13C NMR spectrum (CDCl3, δ, ppm): 21.7 (C8), 25.6 (C10), 26.7 (C9), 33.8 (C7), 38.5 (C5), 39.0 (C6), 40.6 (C1), 44.5 (C4), 60.2 (C2), 211.6 (C3). IR spectrum (KBr, ν, cm−1): 486, 734, 952, 1047, 1080, 1159, 1201, 1271, 1323, 1409, 1467, 1710 st (C=O), 2567 (SH), 2877 st, 2923 st, 2970 st; elemental analysis calcd (%) for C10H16OS: C 65.17, H 8.75, S 17.40; found: C 65.65, H 8.76, S 17.49.
S-(((1S,2S,5R)-6,6-Dimethyl-3-oxobicyclo[3.1.1]heptan-2-yl)methyl) ethanethioate ((2S)-11). A solution of 3.0 g (0.02 mol) of pinocarvone 9 in 18 mL of THF was cooled with stirring to −65 °C, two drops of pyridine (~1 mol%) were added, and then a solution of 3.0 g (0.04 mol) of thioacetic acid was added in 18 mL THF. The progress of the reaction was monitored by TLC (eluent–petroleum ether: EtOAc = 5:1, developer–vanillin solution in EtOH). The mixture was stirred for 6h, then a saturated NaHCO3 solution was added, and the mixture was extracted with Et2O (3 × 20 mL). The combined organic layers were dried over Na2SO4, and the solvent was removed under reduced pressure. The product was purified by column chromatography (eluent–CH2Cl2: Et2O = 30:1). Light-yellow liquid, 84% yield (3.8g), 92% de, −9.6 (c 0.25, CHCl3). 1H NMR spectrum (CDCl3, δ, ppm, J/Hz): 0.95 (s, 3H, H8), 1.17 (д, 1H, H7a, J = 10.3), 1.33 (c, 3H, H9), 2.11–2.18 (m, 1H, H5), 2.22–2.30 (m, 1H, H1), 2.34 (s, 3H, H12), 2.46–2.60 (m, 2H, H2, H4a), 2.64–2.73 (m, 2H, H4b, H7b), 2.96 (dd, 1H, H10a, J = 13.9, 9.8), 3.47 (dd, 1H, H10b, J = 13.2, 4.6). 13C NMR spectrum (CDCl3, δ, ppm): 21.7 (C8), 26.7 (C9), 30.1 (C10), 30.4 (C12), 33.9 (C7), 38.6 (C5), 39.1 (C6), 41.6 (C1), 44.6 (C4), 56.6 (C2), 195.3 (C11), 211.5 (C3). IR spectrum (KBr, ν, cm−1): 628, 756, 822, 957, 1047, 1134 st, 1199, 1325, 1411, 1469, 1691 st (C=O), 1712 st (C=O), 2877 st, 2929 st, 2968 st; elemental analysis calcd (%) for C12H18O2S: C 63.68, H 8.02, S 14.16; found: C 63.65, H 8.16, S 14.20.
S-(((1S,2R,5R)-6,6-Dimethyl-3-oxobicyclo[3.1.1]heptan-2-yl)methyl) ethanethioate ((2R)-11) was synthesized according to the method[
6] from pinocarvone
9 upon cooling to –50 °C. The product was purified by column chromatography (eluent–CH
2Cl
2: Et
2O = 30:1). The ratio was (2
S)-
11: (2
R)-
11 ≈ 5:1 (
de 69%). The yield of the mixture (2
S)-
11, (2
R)-
11 was 83%. NMR spectra were obtained by subtracting signals from the spectrum of the mixture of thioacetates (2
S)
-11, (2
R)
-11.
1H NMR spectrum (CDCl
3, δ, ppm,
J/Hz): 0.88 (s, 3H, H
8), 1.26–1.35 (m, 4H, H
7a, H
9), 2.11–2.18 (m, 1H, H
5), 2.22–2.72 (m, 8H, H
1, H
2, H
4, H
7b, H
12), 2.90–3.00 (m, 1H, H
10a), 3.30–3.38 (m, 1H, H
10b).
13C NMR spectrum (CDCl
3, δ, ppm): 19.7 (C
8), 26.3 (C
9), 28.7 (C
10), 29.1 (C
7), 30.4 (C
12), 38.0 (C
5), 38.3 (C
6), 41.2 (C
1), 44.2 (C
4), 51.7 (C
2), 195.3 (C
11), 211.5 (C
3). Elemental analysis calcd (%) for C
12H
18O
2S: C 63.68, H 8.02, S 14.16; found: C 63.65, H 8.16, S 14.20.
S-(((1S,2S,5R)-6,6-Dimethyl-3-oxobicyclo[3.1.1]heptan-2-yl)methyl) benzothioate ((2S)-12) was obtained by the reaction of pinocarvone 9 with thiobenzoic acid similarly to thioacetate (2S)-11. Transparent liquid, 68% yield, de 93%, +3.5 (c 0.23, CHCl3). 1H NMR spectrum (CDCl3, δ, ppm, J/Hz): 1.01 (s, 3H, H8), 1.19 (d, 1H, H7a, J = 10.3), 1.34 (s, 3H, H9), 2.10–2.19 (m, 1H, H5), 2.34 (t, 1H, H1, J = 6.2), 2.52–2.78 (m, 4H, H2, H4a, H4b, H7b), 3.16 (dd, 1H, H10a, J = 13.6, 10.0), 3.69 (dd, 1H, H10b, J = 13.9, 4.7), 7.45 (t, 2H, HAr, J = 7.7), 7.53–7.62 (m, 1H, HAr), 7.98 (d, 2H, HAr, J = 8.7). 13C NMR spectrum (CDCl3, δ, ppm): 21.7 (C8), 26.7 (C9), 29.9 (C10), 33.9 (C7), 38.6 (C5), 39.1 (C6), 41.7 (C1), 44.7 (C4), 56.7 (C2), 127.1 (CAr), 128.5 (CAr), 133.3 (CAr), 136.8 (CAr), 191.4 (CAr), 211.5 (C3). Elemental analysis calcd (%) for C17H20O2S: C 70.80, H 6.99, S 11.12; found: C 70.65, H 7.06, S 11.20.
(1S,1′S,2S,2′S,5R,5′R)-2,2′-(Disulfanediylbis(methylene))bis(6,6-dimethylbicyclo[3.1.1]heptan-3-one) ((2S)-13) was obtained similarly to disulfide 7 from thiol (2S)-8. White powder, m.p. 93–95 °C, 90% yield, +202.6 (c 0.23, CHCl3). 1H NMR spectrum (CDCl3, δ, ppm, J/Hz): 0.88 (s, 3H, H8), 1.26 (d, 1H, H7a, J = 10.6), 1.37 (s, 3H, H9), 2.18 (dd, 1H, H5, J = 5.9, 2.8), 2.42–2.58 (m, 2H, H1, H10a), 2.58–2.82 (m, 4H, H2, H4, H7b), 3.53 (dd, 1H, H10b, J = 13.3, 3.6). 13C NMR spectrum (CDCl3, δ, ppm): 21.9 (C8), 26.7 (C9), 33.9 (C7), 38.7 (C5), 38.9 (C10), 39.1 (C6), 40.9 (C1), 44.6 (C4), 55.7 (C2), 212.1 (C3). IR spectrum (KBr, ν, cm−1): 470, 489, 1047, 1078, 1157, 1999, 1321, 1409, 1467, 1710 st (C=O), 2883 st, 2924 st, 2972 st; elemental analysis calcd (%) for C20H30O2S2: C 65.53, H 8.25, S 17.49; found: C 65.85, H 8.30, S 17.40.
S-(((1R,3R,5R)-6,6-Dimethyl-2-oxobicyclo[3.1.1]heptan-3-yl)methyl) ((1R,3R,5R)-6,6-dimethyl-2-oxobicyclo[3.1.1]heptan-3-yl)methanesulfonothioate (14). 0.027 g (0.1 mmol) of VO(acac)2 was added to a solution of 0.366 g (1 mmol) of disulfide 7 in 40 mL of acetonitrile with stirring, then an aqueous solution of 0.068 g (1 mmol) of ClO2 was added dropwise. After 1 h, the reaction mixture was partially evaporated until a precipitate formed (target product). The precipitate was filtered off. Light-yellow waxy powder, 74% yield (0.295 g), +0.1 (c 0.26, CHCl3). 1H NMR spectrum (CDCl3, δ, ppm, J/Hz): 0.74, 0.76 (2s, 6H, H8, H8′), 1.37 (s, 6H, H9′, H9), 1.73–1.88 (m, 4H, H4a, H4′a, H7a, H7′a), 2.27–2.36 (m, 2H, H5, H5′), 2.53–2.77 (m, 6H, H1, H1′, H4b, H4′b, H7b, H7′b), 3.05–3.29 (m, 4H, H3, H3′, H10a, H10′a), 3.58–3.70 (m, 1H, H10′b), 4.32 (d, 1H, H10b, J = 11.9). 13C NMR spectrum (CDCl3, δ, ppm): 22.0, 22.1 (C8, C8′), 25.1, 25.2 (C7, C7′), 26.1, 26.3 (C9′, C9), 28.4 (C4′), 29.9 (C4), 35.6 (C10′), 38.6 (C3), 40.6, 40.7 (C5, C5′), 43.4 (C3′), 43.8 (C6, C6′), 57.2, 57.4 (C1, C1′), 62.4 (C10), 211.1, 213.4 (C2, C2′). IR spectrum (KBr, ν, cm−1): 472, 489, 534, 611, 1024, 1130 st (SO2), 1321 st (SO2), 1460, 1708 st (C=O), 2872 st, 2945 st; elemental analysis calcd (%) for C20H30O4S2: C 60.27, H 7.59, S 16.09; found: C 60.25, H 7.66, S 16.20.
S-(((1S,2S,5R)-6,6-Dimethyl-3-oxobicyclo[3.1.1]heptan-2-yl)methyl) ((1S,2S,5R)-6,6-dimethyl-3-oxobicyclo[3.1.1]heptan-2-yl)methanesulfonothioate ((2S)-15) was obtained similarly to thiosulfonate 14 from disulfide (2S)-13. The ratio (2S)-13: ClO2 = 1:2. The synthesis time was 1h. Light-yellow powder, m.p. 134–137 °C, 81% yield, +9.2 (c 0.24, CHCl3). 1H NMR spectrum (CDCl3, δ, ppm, J/Hz): 0.88, 0.90 (2s, 6H, H8, H8′), 1.26–1.32 (m, 1H, H7a, H7′a), 1.35 (s, 6H, H9′, H9), 2.13–2.21 (m, 2H, H5, H5′), 2.41 (t, 1H, H1′, J = 6.3), 2.51–2.79 (m, 7H, H1, H4, H4′, H7b, H7′b), 2.82–2.91 (m, 1H, H2′), 2.98 (m, 2H, H2, H10′a), 3.13–3.23 (m, 1H, H10a), 3.80 (dd, 1H, H10′b, J = 13.6, 5.6), 4.10 (d, 1H, H10b, J = 13.9). 13C NMR spectrum (CDCl3, δ, ppm): 21.8, 21.9 (C8, C8′), 26.4, 26.6 (C9′, C9), 33.7, 34.1 (C7, C7′), 37.2 (C10′), 38.4, 38.7 (C5, C5′), 38.8, 39.2 (C6, C6′), 42.3, 42.7 (C1, C1′), 44.5, 44.6 (C4, C4′), 51.4 (C2), 56.4 (C2′), 62.5 (C10), 209.5, 212.3 (C3, C3′). IR spectrum (KBr, ν, cm−1): 474, 545, 1045, 1128 st (SO2), 1321 st (SO2), 1409, 1467, 1712 st (C=O), 2872 st, 2924 st, 2933 st; elemental analysis calcd (%) for C20H30O4S2: C 60.27, H 7.59, S 16.09; found: C 60.55, H 7.66, S 16.10.
(1S,1′S,2S,2′S,5R,5′R)-2,2′-(Sulfonylbis(methylene))bis(6,6-dimethylbicyclo[3.1.1]heptan-3-one) ((2S)-16). An aqueous solution containing 0.135 g (2 mmol) of ClO2 was added to a solution of 0.184 g (1 mmol) of thiol (2S)-8 in 20 mL of acetonitrile with stirring. After 30 min, it was extracted with chloroform (3 × 20 mL), and the organic fraction was evaporated. The product was purified by column chromatography (eluent—petroleum ether: Et2O = 3:1). The products were obtained in a mixture (2S)-16: (2R)-16 = 1:5 (de 67%). NMR spectra were obtained by subtracting signals from the spectrum of a mixture of sulfones (2S)-16, (2R)-16. 1H NMR spectrum (CDCl3, δ, ppm, J/Hz): 0.84 (s, 3H, H8), 1.32–1.36 (m, 4H, H7a, H9), 2.12–2.19 (m, 1H, H5), 2.42–2.59 (m, 4H, H1, H4a, H4b, H7b), 2.85–2.92 (m, 1H, H10a), 3.01–3.13 (m, 1H, H2), 3.77–3.86 (m, 1H, H10b). 13C NMR spectrum (CDCl3, δ, ppm): 21.9 (C8), 26.5 (C9), 33.8 (C7), 38.5 (C5), 38.8 (C6), 43.4 (C1), 44.5 (C4), 50.1 (C2), 55.8 (C10), 210.1 (C3). IR spectrum (KBr, ν, cm−1): 459, 481, 763, 1049, 1138 st (SO2), 1201, 1292 st (SO2), 1321, 1411, 1467, 1714 st (C=O), 2877 st, 2926 st, 2981 st; MS (ESI, 5 kV): m/z (%): 389.47 (100) [M+23]+, 367.29 (83) [M+1]+; elemental analysis calcd (%) for C20H30O4S: C 65.54, H 8.25, S 8.75; found: C 65.65, H 8.30, S 8.69.
(1S,1′S,2R,2′R,5R,5′R)-2,2′-(Sulfonylbis(methylene))bis(6,6-dimethylbicyclo[3.1.1]heptan-3-one) ((2R)-16) was obtained similarly to sulfone (2S)-16 or during storage of (S)-thiosulfonate (2S)-15 for 1–3 weeks. White powder (recrystallized from EtOH), m.p. 168–171 °C, 58% yield, −31.0 (c 0.2, CHCl3). 1H NMR spectrum (CDCl3, δ, ppm, J/Hz): 1.01 (s, 3H, H8), 1.14 (d, 1H, H7a, J = 11.2), 1.40 (s, 3H, H9), 2.15–2.24 (m, 1H, H5), 2.47–2.66 (m, 3H, H1, H4a, H7b), 2.71–2.84 (m, 1H, H4b), 3.01 (dd, 1H, H10a, J = 14.1, 9.9), 3.29 (d, 1H, H2, J = 9.9), 3.63 (dd, 1H, H10b, J = 14.1, 2.4). 13C NMR spectrum (CDCl3, δ, ppm): 19.8 (C8), 26.2 (C9), 29.0 (C7), 37.8 (C5), 39.3 (C6), 41.1 (C1), 44.2 (C4), 46.7 (C2), 53.9 (C10), 210.3 (C3). IR spectrum (KBr, ν, cm−1): 459, 481, 763, 1049, 1138 st (SO2), 1201, 1292 st (SO2), 1321, 1411, 1467, 1714 st (C=O), 2877 st, 2926 st, 2981 st; MS (ESI, 5 kV): m/z (%): 389.47 (100) [M+23]+, 367.29 (83) [M+1]+; elemental analysis calcd (%) for C20H30O4S: C 65.54, H 8.25, S 8.75; found: C 65.65, H 8.30, S 8.79.
(1R,1′R,3R,3′R,5R,5′R)-3,3′-(Sulfonylbis(methylene))bis(6,6-dimethylbicyclo[3.1.1]heptan-2-one) (17) was synthesized similarly to sulfone (2S)-16 from thiol 6 or by storage of (S)-thiosulfonate 14 for 1–3 weeks. Product was isolated by column chromatography (petroleum ether: Et2O = 7:1). White powder (recrystallized from EtOH), m.p. 148–150 °C, 54% yield (0.099 g), +10.0 (c 0.1, CHCl3). 1H NMR spectrum (CDCl3, δ, ppm, J/Hz): 0.73 (s, 3H, H8), 1.36 (s, 3H, H9), 1.75–1.85 (m, 2H, H4a, H7a), 2.23–2.34 (m, 1H, H5), 2.55 (ddd, 1H, H7b, J = 10.9, 5.9, 5.6), 2.68–2.77 (m, 2H, H1, H4b), 2.96 (dd, 1H, H10a, J = 13.8, 9.7), 3.28–3.40 (m, 1H, H3), 4.01 (dd, 1H, H10b, J = 13.5, 2.9). 13C NMR spectrum (CDCl3, δ, ppm): 22.1 (C8), 25.1 (C7), 26.1 (C9), 30.0 (C4), 37.1 (C3), 40.7 (C5), 43.3 (C6), 54.8 (C10), 57.3 (C1), 211.6 (C2). IR spectrum (KBr, ν, cm−1): 468, 503, 1024, 1136 m (SO2), 1201, 1282 m (SO2), 1303, 1319, 1462, 1708 st (C=O), 2873 m, 2926 st, 2953 st; elemental analysis calcd (%) for C20H30O4S: C 65.54, H 8.25, S 8.75; found: C 65.65, H 8.30, S 8.80.
((1S,2S,5R)-6,6-Dimethyl-3-oxobicyclo[3.1.1]heptan-2-yl)methanesulfonic acid ((2S)-18) (pyridine solvate). An aqueous solution containing 0.135 g (2 mmol) of ClO2 was added to a solution of 0.184 g (1 mmol) of thiol (2S)-8 in 20 mL of pyridine with stirring. After 1 h, the reaction mixture was evaporated and dissolved in Et2O. The precipitate containing C5H5N·HCl was removed. The solution containing the solvate of acid (2S)-18 with pyridine was distilled off. Viscous liquid, 95% yield (0.60 g). 1H NMR spectrum (CDCl3, δ, ppm, J/Hz): 0.80 (s, 3H, H8), 1.23 (d, 1H, H7a, J = 10.6), 1.29 (s, 3H, H9), 2.05–2.13 (m, 1H, H5), 2.55–2.75 (m, 4H, H1, H4, H7b), 2.82 (dd, 1H, H10b, J = 14.1, 8.8), 3.05 (d, 1H, H2, J = 8.8), 3.62 (dd, 1H, H10b, J = 13.3, 3.6). 13C NMR spectrum (CDCl3, δ, ppm): 21.8 (C8), 26.5 (C9), 33.7 (C7), 38.5 (C5), 38.7 (C6), 42.5 (C1), 44.5 (C4), 52.9 (C10), 53.0 (C2), 212.1 (C3). IR spectrum (KBr, ν, cm−1): 478, 520, 603, 682, 754, 968, 997, 1035 st (SO2), 1163 st, 1999 st, 1226 st (SO2), 1386, 1485, 1714 st (C=O), 2883 st, 2924 st, 2972 st, 3439 st (OH); MS (ESI, 5 kV): m/z (%): 231.14 (100) [M–H]−; elemental analysis calcd (%) for C10H16O4S·5C5H5N: C 66.96, H 6.58, N 11.16, S 5.11; found: C 66.85, H 6.66, N 11.08, S 5.20.
((1S,2R,5R)-6,6-Dimethyl-3-oxobicyclo[3.1.1]heptan-2-yl)methanesulfonic acid ((2R)-18). An aqueous solution containing 0.135 g (2 mmol) of ClO2 was added to a solution of 0.184 g (1 mmol) of thiol (2S)-8 in 20 mL of acetonitrile with stirring. After 1.5 h, it was extracted with chloroform (3 × 20 mL) and the aqueous fraction was evaporated. The dry residue of the aqueous fraction contained a mixture of acids (2S)-18: (2R)-18 in the ratio of 1:2. The yield of a mixture of acids (2S)-18, (2R)-18 was 0.223 g. NMR spectra were obtained by subtracting signals from the spectrum of a mixture of acids (2S)-18, (2R)-18. 1H NMR spectrum (CDCl3, δ, ppm, J/Hz): 0.90 (s, 3H, H8), 1.18 (d, 1H, H7a, J = 11.3), 1.34 (s, 3H, H9), 2.09–2.19 (m, 1H, H5), 2.39–2.47 (m, 1H, H1), 2.47–2.60 (m, 3H, H4, H7b), 2.87–3.01 (m, 1H, H10a), 3.01–3.10 (m, 1H, H2), 3.28 (d, 1H, H10b, J = 13.9). 13C NMR spectrum (D2O, δ, ppm): 19.1 (C8), 25.5 (C9), 28.4 (C7), 37.3 (C5), 38.4 (C6), 41.2 (C1), 44.2 (C4), 49.2 (C2), 50.7 (C10), 218.0 (C3). IR spectrum (KBr, ν, cm−1): 478, 590, 607, 756, 918, 1039 st (SO2), 1161 st, 1205 st, 1234 st (SO2), 1369, 1408, 1467, 1714 st (C=O), 2877 st, 2924 st, 2964 st, 3327 st (OH); MS (ESI, 5 kV): m/z (%): 231.17 (100) [M–H]−; elemental analysis calcd (%) for C10H16O4S: C 51.71, H 6.94, S 13.80; found: C 51.65, H 7.16, S 13.40.
((1R,3R,5R)-6,6-Dimethyl-2-oxobicyclo[3.1.1]heptan-3-yl)methanesulfonic acid (19) (pyridine solvate) was obtained similarly to acid (2S)-18 from thiol 6. Viscous liquid, 96% yield. 1H NMR spectrum (CDCl3, δ, ppm, J/Hz): 0.44 (s, 3H, H8), 1.06 (s, 3H, H9), 1.49–1.62 (m, 2H, H4a, H7a), 1.95–2.06 (m, 1H, H5), 2.15–2.27 (m, 1H, H7b), 2.33 (t, 1H, H1, J = 5.2), 2.43–2.51 (m, 1H, H4b), 2.55 (dd, 1H, H10a, J = 14.2, 9.6), 2.90–3.04 (m, 1H, H3), 3.50 (d, 1H, H10b, J = 13.9). 13C NMR spectrum (CDCl3, δ, ppm): 21.4 (C8), 24.6 (C7), 25.6 (C9), 29.4 (C4), 39.9 (C3), 40.2 (C5), 42.3 (C6), 51.4 (C10), 56.8 (C1), 127.1, 141.0, 146.1 (CPy), 213.2 (C2). IR spectrum (KBr, ν, cm−1): 528, 609, 682, 756, 999, 1039 (SO2), 1178 (SO2), 1195 (SO2), 1242 (SO2), 1388, 1462, 1705 (C=O), 2873, 2949, 3435 (OH); MS (ESI, 5 kV): m/z (%): 231.14 (100) [M–1]−; elemental analysis calcd (%) for C10H16O4S·4C5H5N: C 65.66, H 6.61, N 10.21, S 5.84; found: C 65.85, H 6.70, N 10.11, S 5.64.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}