
Halogen Interactions in Halogenated Oxindoles: Crystallographic and Computational Investigations of Intermolecular Interactions

 , , and
, , and
Abstract
:1. Introduction
2. Results and Discussion
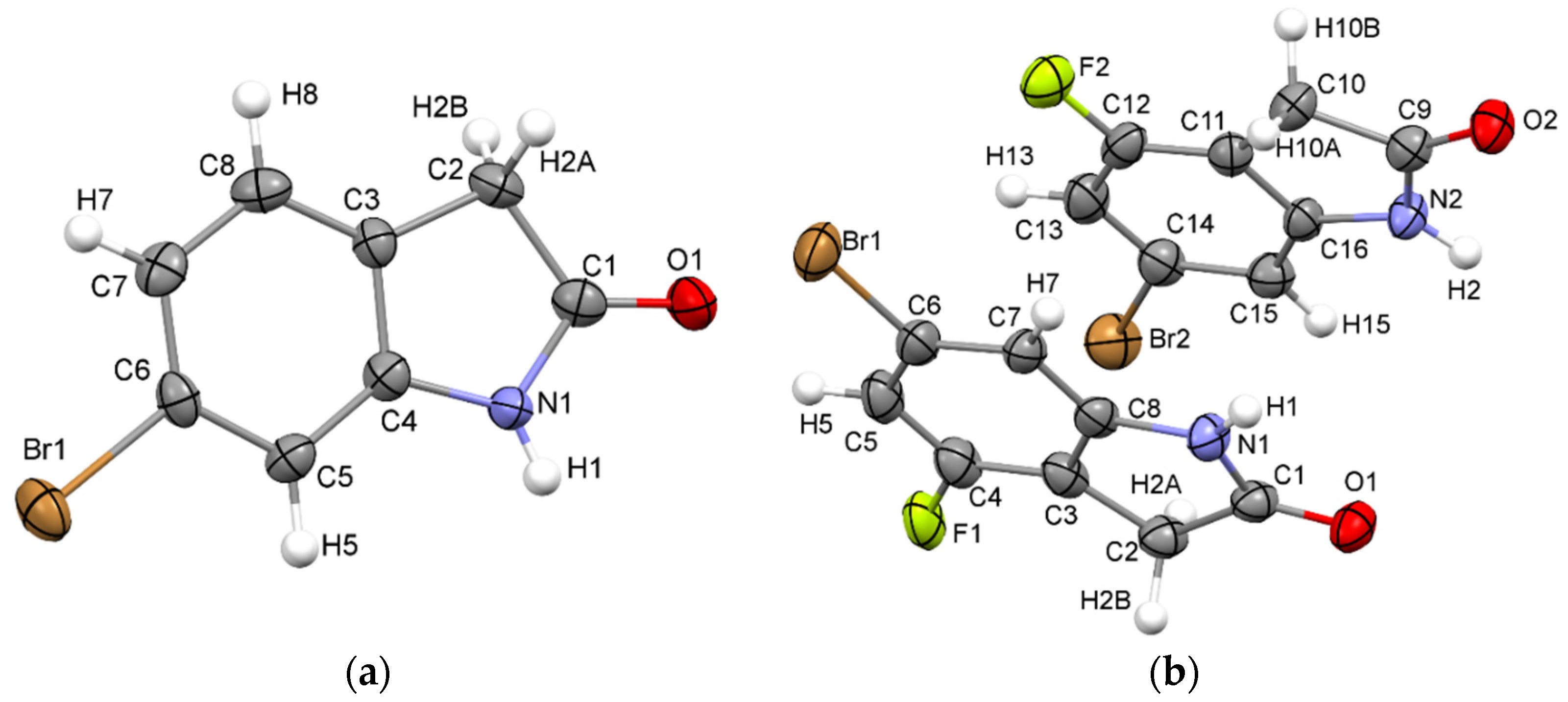
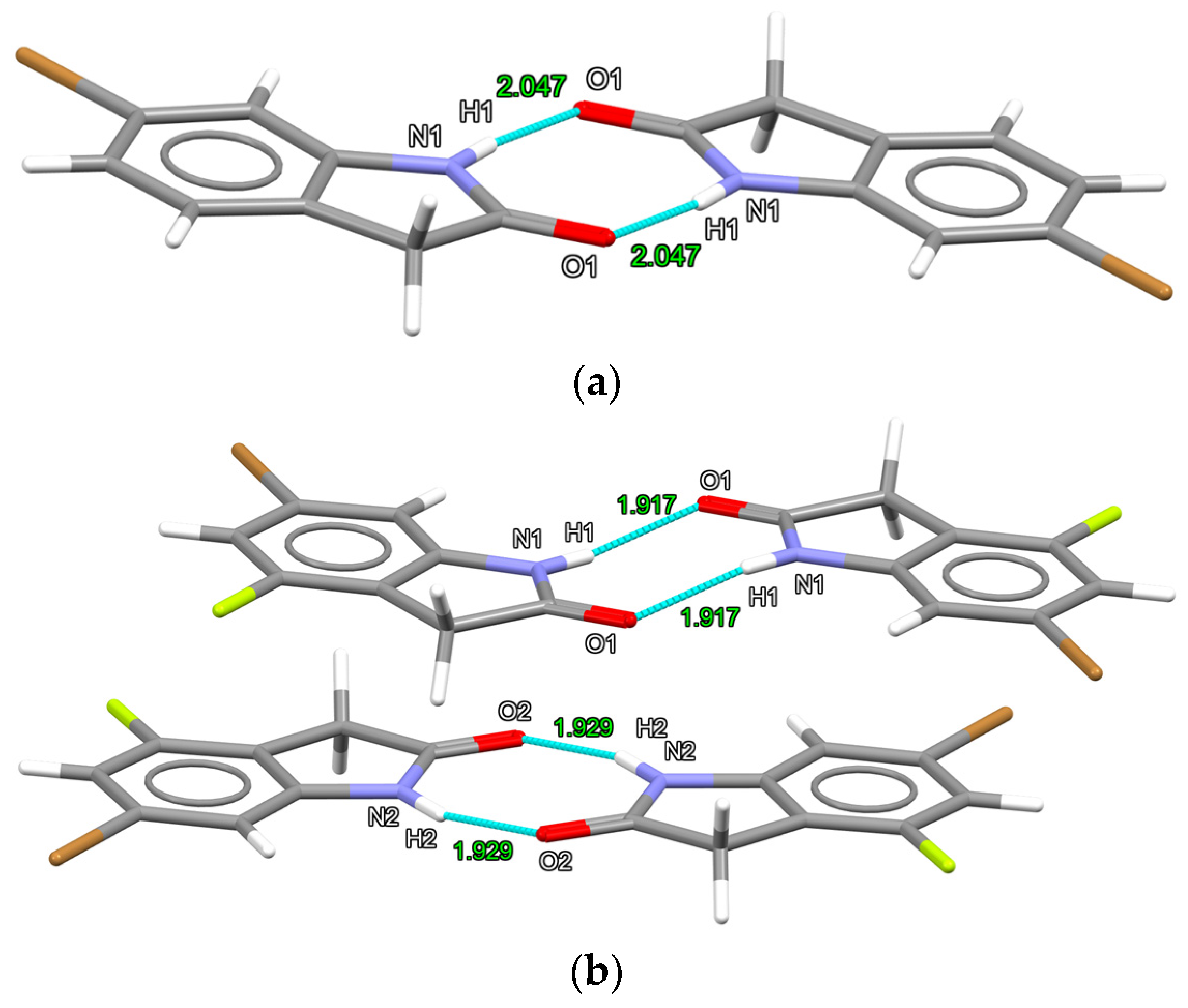
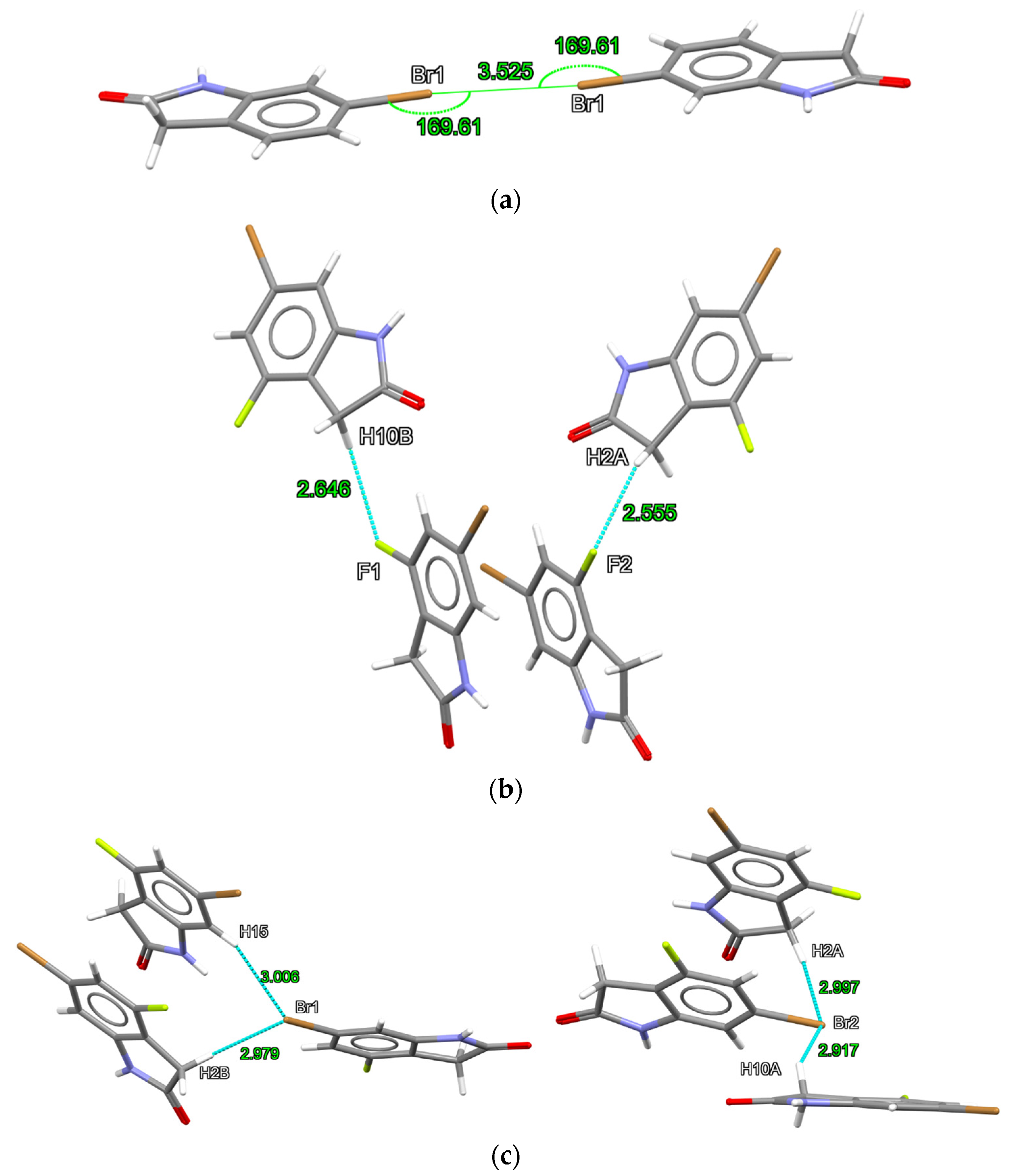
2.1. X-ray Structures of 6-Bromooxindole (1) and 6-Bromo-4-fluoro-indolin-2-one (2)
2.2. Computational Studies of Structures 1 and 2
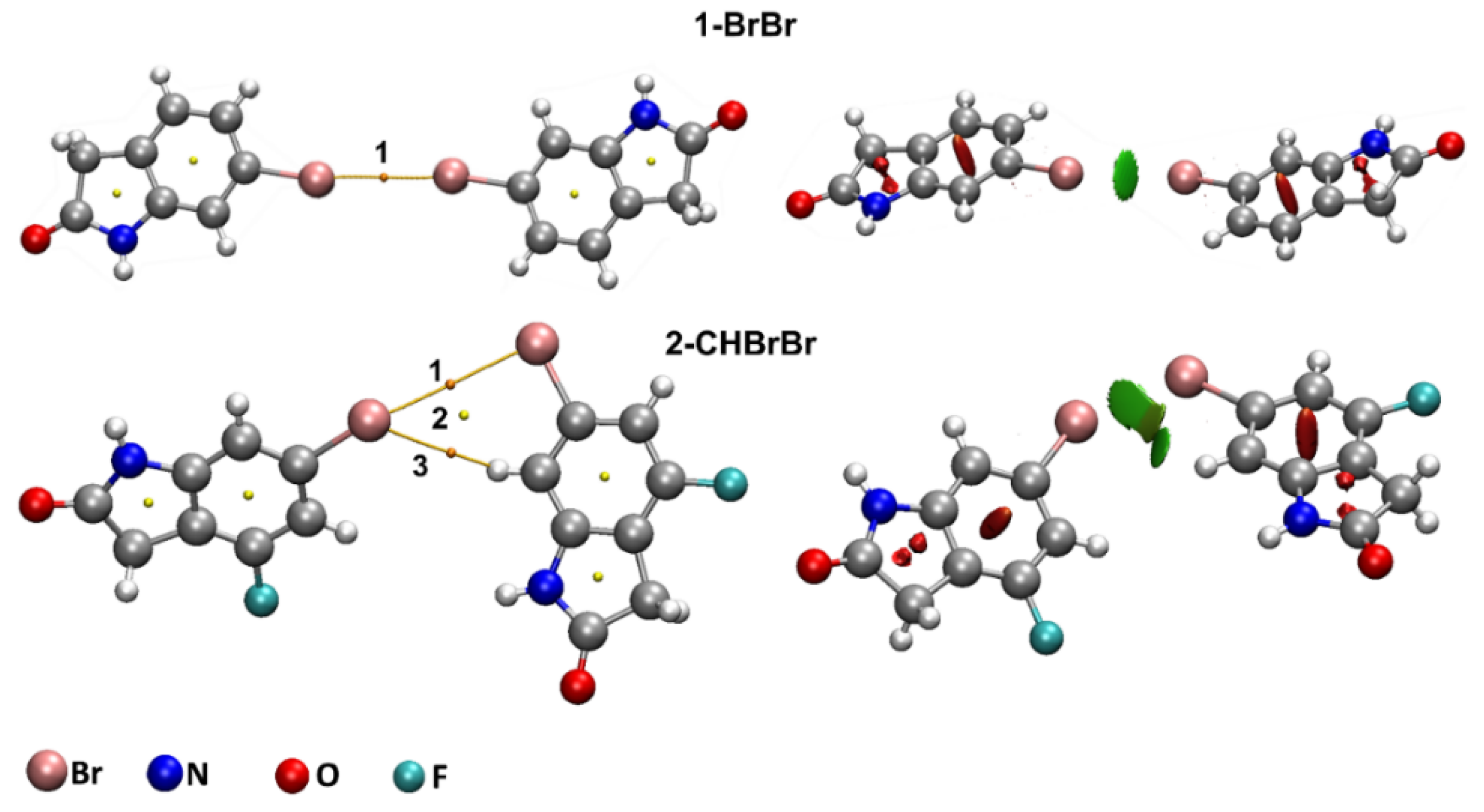
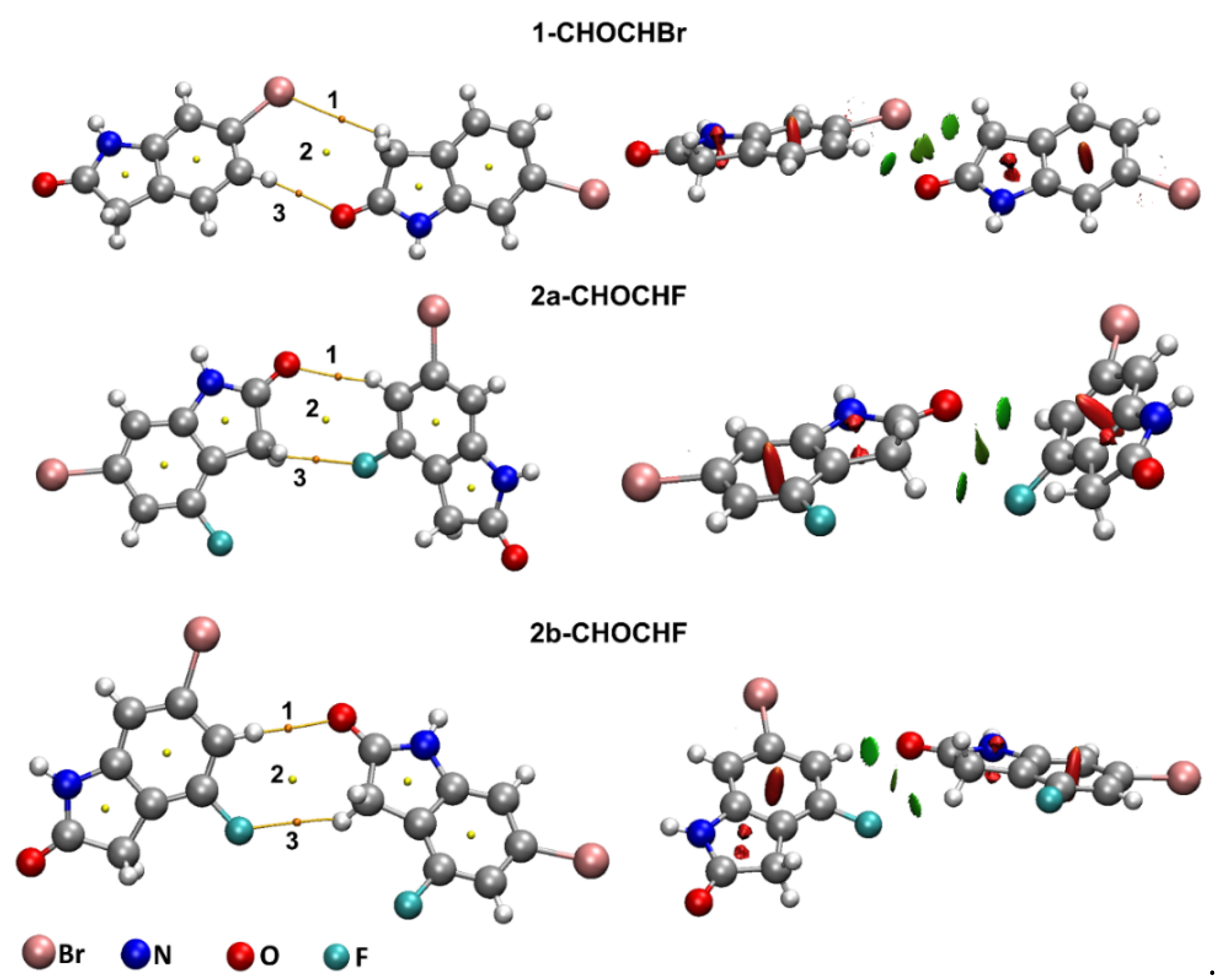
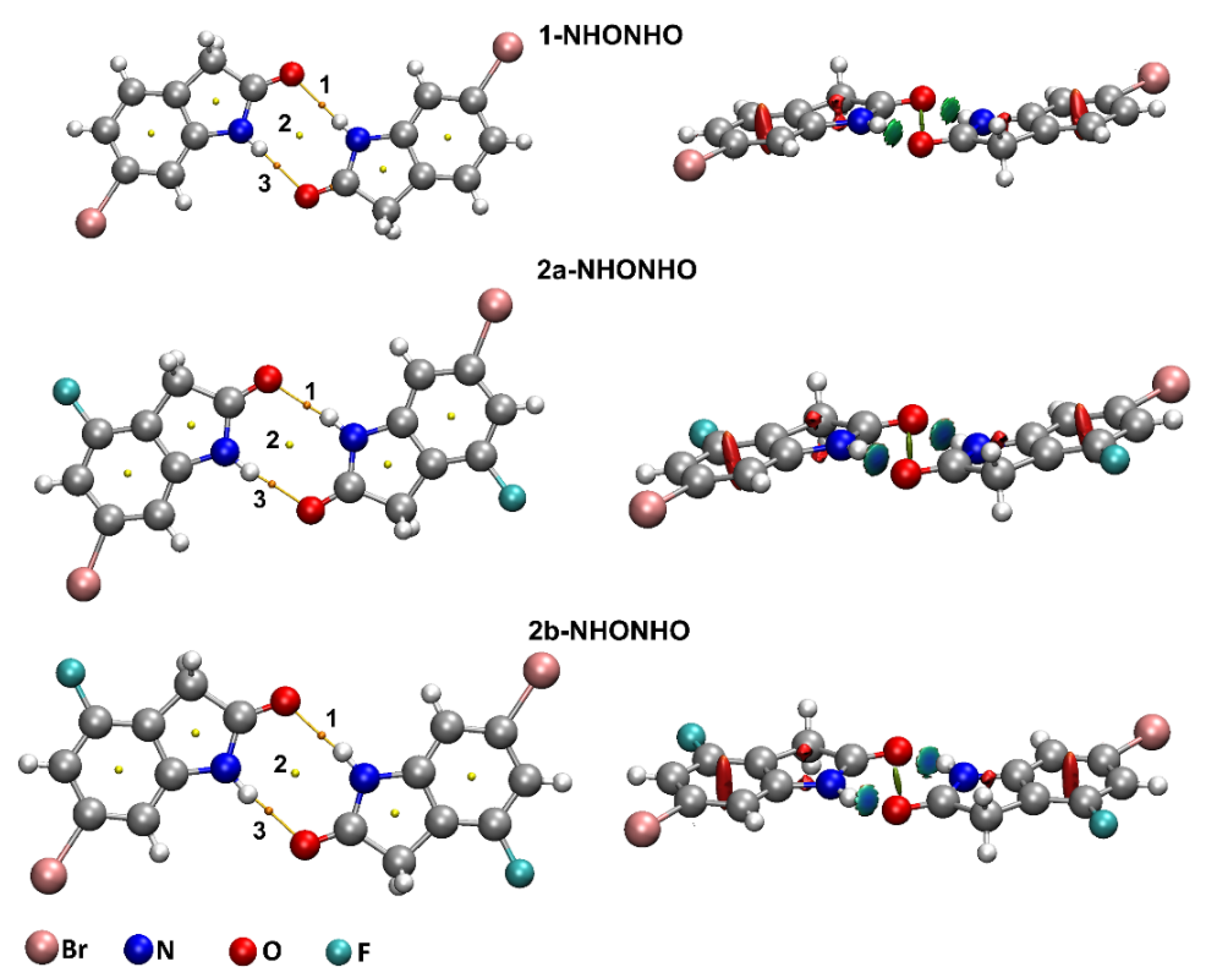
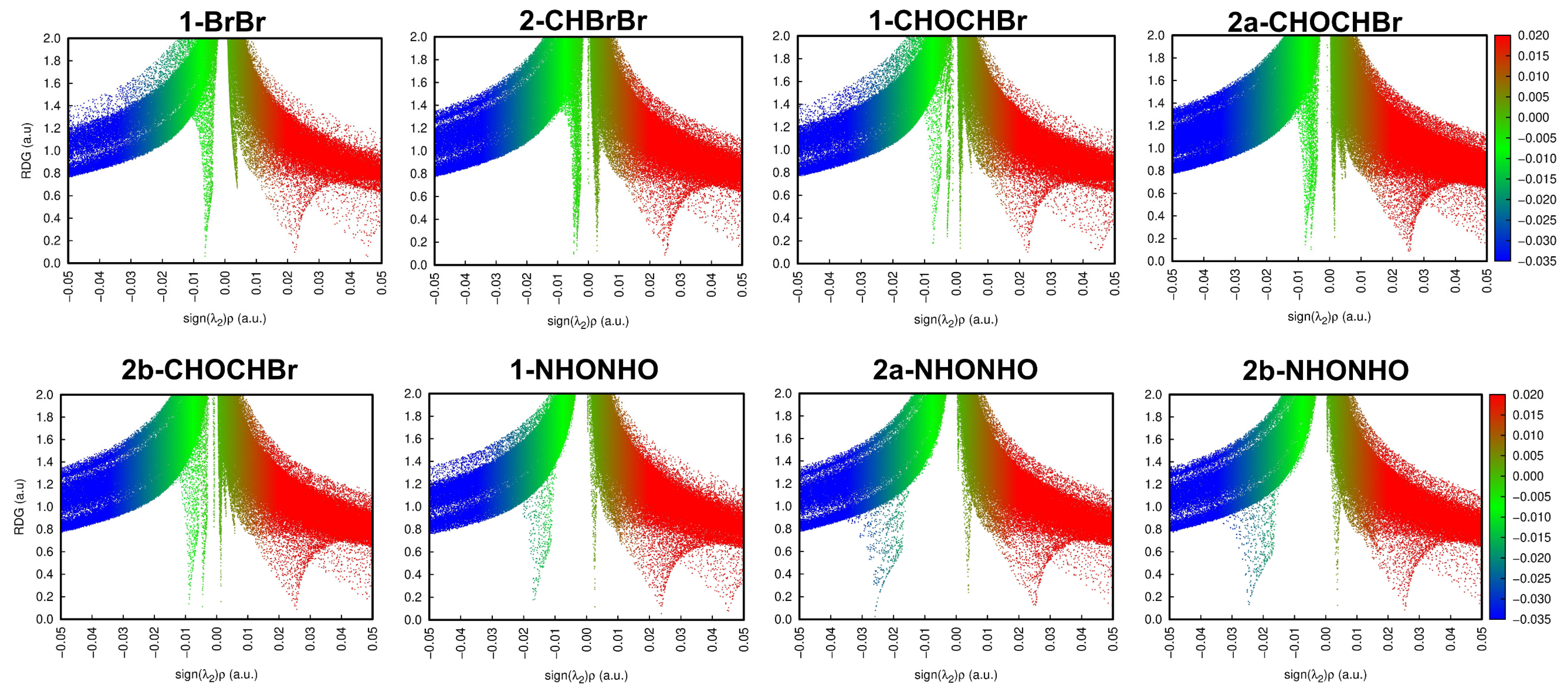
2.2.1. QTAIM Analysis of Supramolecular Dimers of 1 and 2
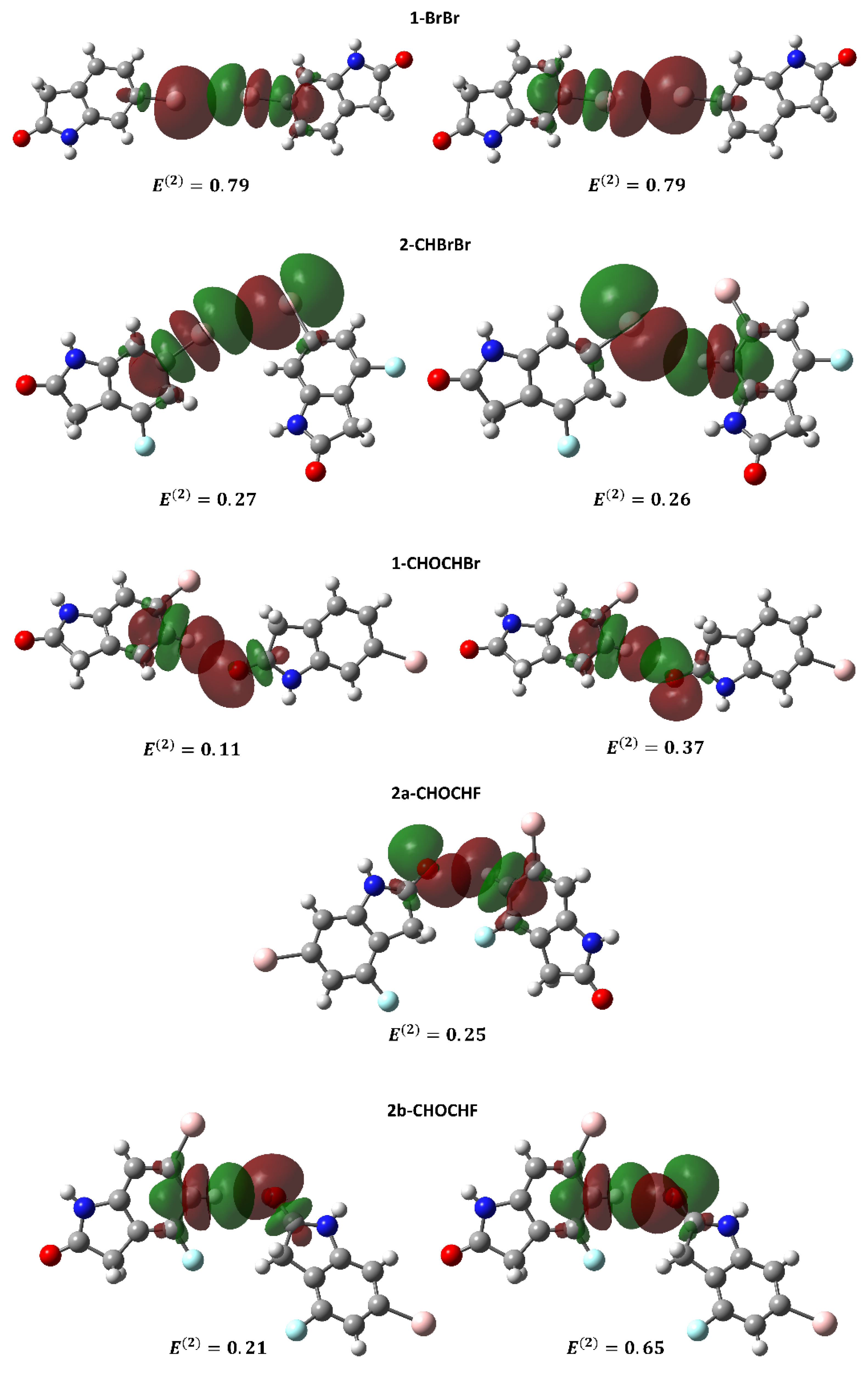
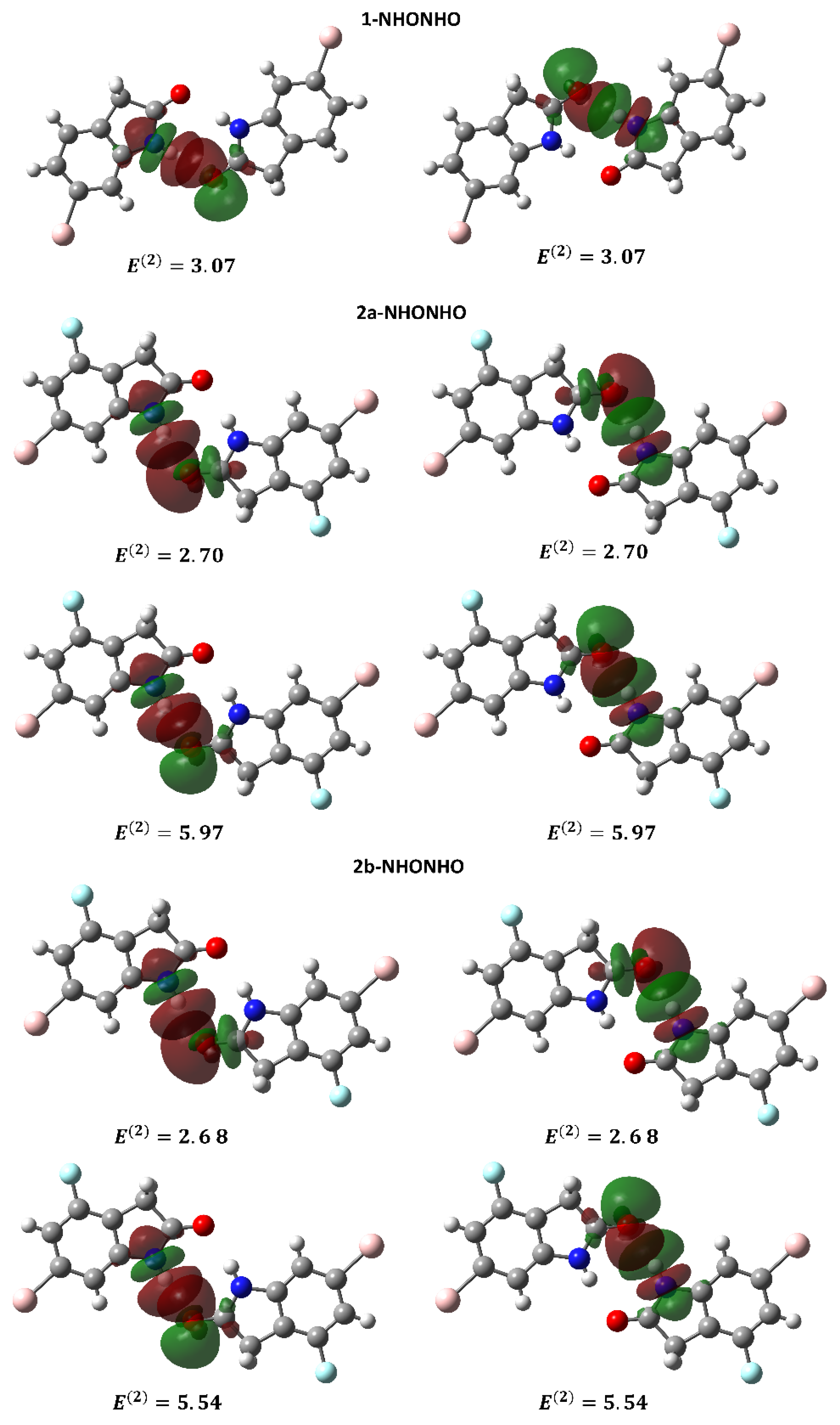
2.2.2. Natural Bond Orbital Analysis of Supramolecular Dimers of 1 and 2
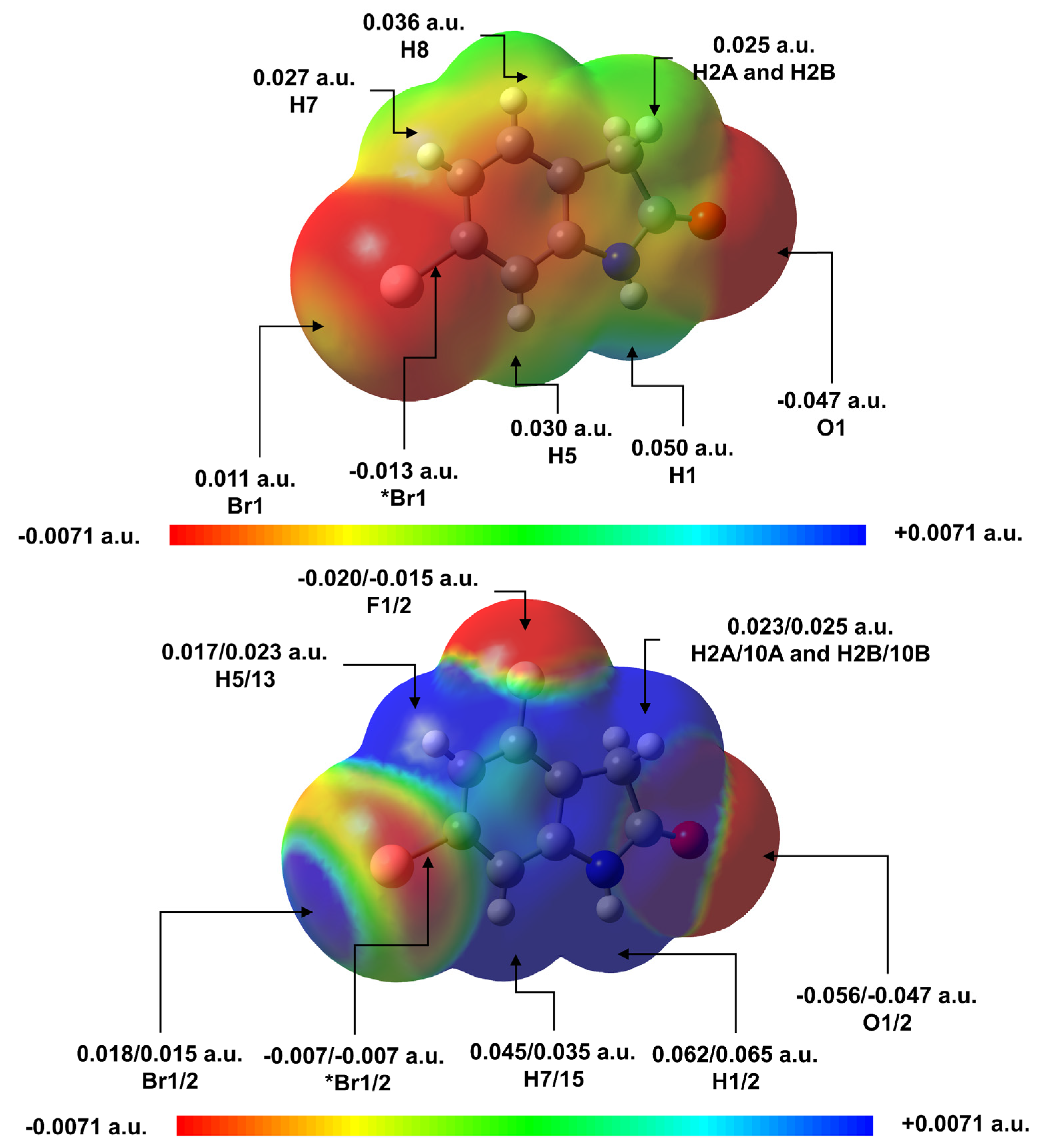
2.2.3. Interaction Energies and MEP Analysis of Supramolecular Dimers of 1 and 2
3. Materials and Methods
3.1. X-ray Crystallography
3.2. Computational Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- De Oliveira, B.G.; Zabardasti, A.; do Rego, D.G.; Pour, M.M. The formation of H···X hydrogen bond, C···X carbon-halide or Si···X tetrel bonds on the silylene-halogen dimers (X = F or Cl): Intermolecular strength, molecular orbital interactions and prediction of covalency. Theor. Chem. Acc. 2020, 139, 1–18. [Google Scholar] [CrossRef]
- Bauzá, A.; Frontera, A. Competition between lone pair-π, halogen-π and triel bonding interactions involving BX3 (X = F, Cl, Br and I) compounds: An ab initio study. Theor. Chem. Acc. 2017, 136, 1–8. [Google Scholar] [CrossRef]
- Karle, I.L.; Butcher, R.J.; Wolak, M.A.; Da Silva Filho, D.A.; Uchida, M.; Brédas, J.L.; Kafafi, Z.H. Cooperative CH⋯π interactions in the crystal structure of 2,5-di(3-biphenyl)-1,1-dimethyl-3,4-diphenyl-silole and its effect on its electronic properties. J. Phys. Chem. C 2007, 111, 9543–9547. [Google Scholar] [CrossRef]
- Yao, Z.F.; Wang, J.Y.; Pei, J. Control of π-π stacking via crystal engineering in organic conjugated small molecule crystals. Cryst. Growth Des. 2018, 18, 7–15. [Google Scholar] [CrossRef]
- Mahmudov, K.T.; Kopylovich, M.N.; Guedes Da Silva, M.F.C.; Pombeiro, A.J.L. Chalcogen bonding in synthesis, catalysis and design of materials. Dalt. Trans. 2017, 46, 10121–10138. [Google Scholar] [CrossRef] [Green Version]
- Varadwaj, P.R.; Varadwaj, A.; Marques, H.M. Halogen bonding: A halogen-centered noncovalent interaction yet to be understood. Inorganics 2019, 7, 40. [Google Scholar] [CrossRef] [Green Version]
- Robertson, C.C.; Wright, J.S.; Carrington, E.J.; Perutz, R.N.; Hunter, C.A.; Brammer, L. Hydrogen bonding: Vs. halogen bonding: The solvent decides. Chem. Sci. 2017, 8, 5392–5398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.Z.; Jing, B.; Li, R.; Liu, Z.B.; Li, W.Z.; Luan, F.; Cheng, J.B.; Gong, B.A.; Sun, J.Z. Some measures for making halogen bonds stronger than hydrogen bonds in H2CS-HOX (X = F, Cl, and Br) complexes. Phys. Chem. Chem. Phys. 2011, 13, 2266–2271. [Google Scholar] [CrossRef]
- Domagała, M.; Lutyńska, A.; Palusiak, M. Extremely strong halogen bond. the case of a double-charge-assisted halogen bridge. J. Phys. Chem. A 2018, 122, 5484–5492. [Google Scholar] [CrossRef] [PubMed]
- Bankiewicz, B.; Palusiak, M. Cooperation/Competition between halogen bonds and hydrogen bonds in complexes of 2,6-diaminopyridines and X-CY3 (X = Cl, Br; Y = H, F). Symmetry 2021, 13, 766. [Google Scholar] [CrossRef]
- Uran, E.; Fotović, L.; Bedeković, N.; Stilinović, V.; Cinčić, D. The amine group as halogen bond acceptor in cocrystals of aromatic diamines and perfluorinated iodobenzenes. Crystals 2021, 11, 529. [Google Scholar] [CrossRef]
- Ateş, Ö.D.; Zorlu, Y.; Kanmazalp, S.D.; Chumakov, Y.; Gürek, A.G.; Ayhan, M.M. Halogen bonding driven crystal engineering of iodophthalonitrile derivatives. CrystEngComm 2018, 20, 3858–3867. [Google Scholar] [CrossRef]
- Mukherjee, A.; Sanz-Matias, A.; Velpula, G.; Waghray, D.; Ivasenko, O.; Bilbao, N.; Harvey, J.N.; Mali, K.S.; De Feyter, S. Halogenated building blocks for 2D crystal engineering on solid surfaces: Lessons from hydrogen bonding. Chem. Sci. 2019, 10, 3881–3891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, A.; Tothadi, S.; Desiraju, G.R. Halogen bonds in crystal engineering: Like hydrogen bonds yet different. Acc. Chem. Res. 2014, 47, 2514–2524. [Google Scholar] [CrossRef] [PubMed]
- Desiraju, G.R. A smorgasbord of halogen bonds. Acta Crystallogr. Sect. C Struct. Chem. 2019, 75, 1188–1189. [Google Scholar] [CrossRef]
- Zurita, J.; Rodriguez, V.; Zambrano, C.; Ramón Mora, J.; Rincón, L.; Javier Torres, F. Theoretical description of R–X· · · NH3 halogen bond complexes: Effect of the R group on the complex stability and sigma-hole electron depletion. Molecules 2020, 25, 530. [Google Scholar] [CrossRef] [Green Version]
- Wolters, L.P.; Schyman, P.; Pavan, M.J.; Jorgensen, W.L.; Bickelhaupt, F.M.; Kozuch, S. The many faces of halogen bonding: A review of theoretical models and methods. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2014, 4, 523–540. [Google Scholar] [CrossRef]
- Bauzá, A.; Frontera, A. Halogen and chalcogen bond energies evaluated using electron density properties. ChemPhysChem 2020, 21, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Janjić, G.V.; Jelić, S.T.; Trišović, N.P.; Popović, D.M.; Dordević, I.S.; Milčić, M.K. New Theoretical insight into fluorination and fluorine-fluorine interactions as a driving force in crystal structures. Cryst. Growth Des. 2020, 20, 2943–2951. [Google Scholar] [CrossRef]
- Bolotin, D.S.; Il’in, M.V.; Suslonov, V.V.; Novikov, A.S. Symmetrical noncovalent interactions Br... Br observed in crystal structure of exotic primary peroxide. Symmetry 2020, 12, 637. [Google Scholar] [CrossRef] [Green Version]
- Cheng, N.; Bi, F.; Liu, Y.; Zhang, C.; Liu, C. The structures and properties of halogen bonds involving polyvalent halogen in complexes of FXOn (X = Cl, Br; N = 0–3)-CH3CN. New J. Chem. 2014, 38, 1256–1263. [Google Scholar] [CrossRef]
- Grabowski, S.J. Halogen bond with the multivalent halogen acting as the Lewis acid center. Chem. Phys. Lett. 2014, 605–606, 131–136. [Google Scholar] [CrossRef]
- Bauzá, A.; Onero, D.Q.; Frontera, A. Substituent effects in multivalent halogen bonding complexes: A combined theoretical and crystallographic study. Molecules 2018, 23, 18. [Google Scholar] [CrossRef] [Green Version]
- Gurbanov, A.V.; Mertsalov, D.F.; Zubkov, F.I.; Nadirova, M.A.; Nikitina, E.V.; Truong, H.H.; Grigoriev, M.S.; Zaytsev, V.P.; Mahmudov, K.T.; Pombeiro, A.J.L. Role of halogen substituents on halogen bonding in 4,5-dibromohexahydro-3a,6-epoxyisoindol-1(4h)-ones. Crystals 2021, 11, 112. [Google Scholar] [CrossRef]
- Suslonov, V.V.; Soldatova, N.S.; Ivanov, D.M.; Galmés, B.; Frontera, A.; Resnati, G.; Postnikov, P.S.; Kukushkin, V.Y.; Bokach, N.A. Diaryliodonium tetrachloroplatinates(II): Recognition of a trifurcated metal-involving μ 3 -I···(Cl,Cl,Pt) Halogen Bond. Cryst. Growth Des. 2021, 21, 5360–5372. [Google Scholar] [CrossRef]
- Novikov, A.S.; Ivanov, D.M.; Bikbaeva, Z.M.; Bokach, N.A.; Kukushkin, V.Y. Noncovalent interactions involving iodofluorobenzenes: The interplay of halogen bonding and weak lp(O)···π-holearene interactions. Cryst. Growth Des. 2018, 18, 7641–7654. [Google Scholar] [CrossRef]
- Khetmalis, Y.M.; Shivani, M.; Murugesan, S.; Chandra Sekhar, K.V.G. Oxindole and its derivatives: A review on recent progress in biological activities. Biomed. Pharmacother. 2021, 141, 111842. [Google Scholar] [CrossRef]
- Taylor, S.J.; Padyana, A.K.; Abeywardane, A.; Liang, S.; Hao, M.H.; De Lombaert, S.; Proudfoot, J.; Farmer, B.S.; Li, X.; Collins, B.; et al. Discovery of potent, selective chymase inhibitors via fragment linking strategies. J. Med. Chem. 2013, 56, 4465–4481. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Chen, J.; Huang, H.; Li, S.; Wu, H.; Hu, C.; Tang, J.; Zhang, Q. (Z)-(thienylmethylene)oxindole-based polymers for high-performance solar cells. Macromolecules 2016, 49, 2145–2152. [Google Scholar] [CrossRef]
- Tingare, Y.S.; Su, C.; Shen, M.T.; Tsai, S.H.; Ho, S.Y.; Li, W.R. New oxindole-bridged acceptors for organic sensitizers: Substitution and performance studies in dye-sensitized solar cells. Molecules 2020, 25, 2159. [Google Scholar] [CrossRef] [PubMed]
- Wilcken, R.; Zimmermann, M.O.; Lange, A.; Joerger, A.C.; Boeckler, F.M. Principles and applications of halogen bonding in medicinal chemistry and chemical biology. J. Med. Chem. 2013, 56, 1363–1388. [Google Scholar] [CrossRef]
- Hachuła, B.; Zerzucha, P.; Zubko, M.; Kusz, J. 6-chloro-2-oxindole: X-ray and DFT-calculated study. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 2011, 67, o413–o416. [Google Scholar] [CrossRef]
- Bader, R.F.W. A quantum theory of molecular structure and its applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Bader, R.F.W.; Essén, H. The characterization of atomic interactions. J. Chem. Phys. 1984, 80, 1943. [Google Scholar] [CrossRef]
- Lipkowski, P.; Grabowski, S.J.; Robinson, T.L.; Leszczynski, J. Properties of the C-H⋯H dihydrogen bond: An ab initio and topological analysis. J. Phys. Chem. A 2004, 108, 10865–10872. [Google Scholar] [CrossRef]
- Bianchi, R.; Gervasio, G.; Marabello, D. Experimental electron density analysis of Mn2(CO)10: Metal-metal and metal-ligand bond characterization. Inorg. Chem. 2000, 39, 2360–2366. [Google Scholar] [CrossRef] [PubMed]
- Usoltsev, A.N.; Adonin, S.A.; Abramov, P.A.; Novikov, A.S.; Shayapov, V.R.; Plyusnin, P.E.; Korolkov, I.V.; Sokolov, M.N.; Fedin, V.P. 1D and 2D polybromotellurates(IV): Structural studies and thermal stability. Eur. J. Inorg. Chem. 2018, 2018, 3264–3269. [Google Scholar] [CrossRef]
- Capdevila-Cortada, M.; Novoa, J.J. The nature of the C-Br⋯Br-C intermolecular interactions found in molecular crystals: A general theoretical-database study. CrystEngComm 2015, 17, 3354–3365. [Google Scholar] [CrossRef]
- Contreras-García, J.; Johnson, E.R.; Keinan, S.; Chaudret, R.; Piquemal, J.; Beratan, D.N.; Yang, W. NCIPLOT: A program for plotting noncovalent interaction regions. J. Chem. Theory Comput. 2011, 7, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Gassoumi, B.; Chaabene, M.; Ghalla, H.; Chaabane, R. Ben Role of hydrogen bonding interactions within of the conformational preferences of calix[n = 4,6,8]arene: DFT and QTAIM analysis. J. Mol. Model. 2020, 26, 12. [Google Scholar] [CrossRef]
- Kuznetsov, M.L. Relationships between interaction energy and electron density properties for homo halogen bonds of the [(A)NY–X···X–Z(b)M] type (X = Cl, Br, I). Molecules 2019, 24, 2733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartashevich, E.V.; Tsirelson, V.G. Interplay between non-covalent interactions in complexes and crystals with halogen bonds. Russ. Chem. Rev. 2014, 83, 1181–1203. [Google Scholar] [CrossRef]
- Mata, I.; Alkorta, I.; Espinosa, E.; Molins, E. Relationships between interaction energy, intermolecular distance and electron density properties in hydrogen bonded complexes under external electric fields. Chem. Phys. Lett. 2011, 507, 185–189. [Google Scholar] [CrossRef]
- Javan, M.B.; Ganji, M.D. Theoretical investigation on the encapsulation of atomic hydrogen into heterofullerene nanocages. Curr. Appl. Phys. 2013, 13, 1525–1531. [Google Scholar] [CrossRef]
- Shariatinia, Z.; Shahidi, S. A DFT study on the physical adsorption of cyclophosphamide derivatives on the surface of fullerene C60 nanocage. J. Mol. Graph. Model. 2014, 52, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Silva, R.A.L.; de Brito, S.F.; Machado, D.F.S.; Carvalho-Silva, V.H.; de Oliveira, H.C.B.; Ribeiro, L. The influence of the configuration of the (C70)2 dimer on its rovibrational spectroscopic properties: A theoretical survey. J. Mol. Model. 2018, 24, 235. [Google Scholar] [CrossRef] [PubMed]
- Saccone, M.; Spengler, M.; Pfletscher, M.; Kuntze, K.; Virkki, M.; Wölper, C.; Gehrke, R.; Jansen, G.; Metrangolo, P.; Priimagi, A.; et al. Photoresponsive halogen-bonded liquid crystals: The role of aromatic fluorine substitution. Chem. Mater. 2019, 31, 462–470. [Google Scholar] [CrossRef]
- Berger, R.; Resnati, G.; Metrangolo, P.; Weber, E.; Hulliger, J. Organic fluorine compounds: A great opportunity for enhanced materials properties. Chem. Soc. Rev. 2011, 40, 3496. [Google Scholar] [CrossRef]
- Kharandiuk, T.; Hussien, E.J.; Cameron, J.; Petrina, R.; Findlay, N.J.; Naumov, R.; Klooster, W.T.; Coles, S.J.; Ai, Q.; Goodlett, S.; et al. Noncovalent close contacts in fluorinated thiophene-phenylene-thiophene conjugated units: Understanding the nature and dominance of O···H versus S···F and O···F interactions with respect to the control of polymer conformation. Chem. Mater. 2019, 31, 7070–7079. [Google Scholar] [CrossRef]
- REQAB; Rigaku Corporation: Tokyo, Japan, 1998.
- CrystalClear v 2.0; Rigaku Americas: The Woodlands, TX, USA; Rigaku Corporation: Tokyo, Japan, 2011.
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef] [Green Version]
- CrystalStructure v 4.1; Rigaku Americas and Rigaku; Rigaku Americas: The Woodlands, TX, USA; Rigaku Corporation: Tokyo, Japan, 2010.
- SCALE3 ABSPACK, CrysAlisPro, Version 1.171.40.80a; Rigaku Corporation: Tokyo, Japan, 2020.
- CrysAlisPro, v.171.40.80a; Rigaku Corporation: Tokyo, Japan, 2020.
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Capdevila-Cortada, M.; Castelló, J.; Novoa, J.J. The nature of the C-Cl⋯Cl-C intermolecular interactions found in molecular crystals: A general theoretical-database study covering the 2.75–4.0 Å range. CrystEngComm 2014, 16, 8232–8242. [Google Scholar] [CrossRef]
- Chai, J.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Balmohammadi, Y.; Khavasi, H.R.; Naghavi, S.S. Existence of untypical halogen-involving interactions in crystal packings: A statistical and first-principles study. CrystEngComm 2020, 22, 2756–2765. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Boys, S.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Weinhold, F.; Landis, C.R. Natural bond orbitals and extensions of localized bonding concepts. Chem. Educ. Res. Pr. 2001, 2, 91–104. [Google Scholar] [CrossRef]
- Oliveira, B.G.; Araújo, R.C.M.U.; Ramos, M.N. A topologia molecular QTAIM e a descrição mecânico-quântica de ligações de hidrogênio e ligações de di-hidrogênio. Quim. Nova 2010, 33, 1155–1162. [Google Scholar] [CrossRef]
- Kumar, P.S.V.; Raghavendra, V.; Subramanian, V. Bader’s Theory of atoms in molecules (AIM) and its applications to chemical bonding. J. Chem. Sci. 2016, 128, 1527–1536. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Clarendon Press: Oxford, UK, 1990; ISBN 9780198558651. [Google Scholar]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing noncovalent interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Paredes, J.; Carrillo-Torres, R.C.; Hernández-Negrete, O.; Sotelo-Mundo, R.R.; Glossman-Mitnik, D.; Esparza-Ponce, H.E.; Alvarez-Ramos, M.E. Experimental and theoretical study on the molecular structure, covalent and non-covalent interactions of 2,4-dinitrodiphenylamine: X-ray diffraction and QTAIM approach. J. Mol. Struct. 2017, 1141, 53–63. [Google Scholar] [CrossRef]
- Thamotharan, S.; Kothandapani, J.; Selva Ganesan, S.; Venkataramanan, N.S.; Madan Kumar, S.; Byrappa, K.; Percino, J.; Robles, F. Quantitative analysis of intermolecular interactions in 2,2’-((4-bromophenyl)methylene)bis(3-hydroxy-5,5-dimethylcyclohex-2-en-1-one): Insights from crystal structure, PIXEL, Hirshfeld surfaces and QTAIM analysis. J. Chem. Sci. 2018, 130, 20. [Google Scholar] [CrossRef] [Green Version]
- Gao, H.; Feng, W.; Li, X.; Li, N.; Du, Y.; Wu, Y.; Bai, H.; Qiao, W. Insights into the non-covalent interaction between modified nucleobases and graphene nanoflake from first-principles. Phys. E Low-Dimens. Syst. Nanostruct. 2019, 107, 73–79. [Google Scholar] [CrossRef]
- Gatti, C.; Macetti, G.; Boyd, R.J.; Matta, C.F. An electron density source-function study of dna base pairs in their neutral and ionized ground states. J. Comput. Chem. 2018, 39, 1112–1128. [Google Scholar] [CrossRef] [PubMed]
- Usoltsev, A.N.; Adonin, S.A.; Novikov, A.S.; Abramov, P.A.; Sokolov, M.N.; Fedin, V.P. Chlorotellurate(iv) supramolecular associates with “trapped” Br2: Features of non-covalent halogen⋯halogen interactions in crystalline phases. CrystEngComm 2020, 22, 1985–1990. [Google Scholar] [CrossRef]
- Borissova, A.O.; Korlyukov, A.A.; Antipin, M.Y.; Lyssenko, K.A. Estimation of dissociation energy in donor-acceptor complex AuCl·PPh3 via topological analysis of the experimental electron density distribution function. J. Phys. Chem. A 2008, 112, 11519–11522. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Williams, T.; Kelley, C. Gnuplot 5.2.8: An Interactive Plotting Program. 2019. Available online: http://www.gnuplot.info/ (accessed on 5 September 2021).











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | 1 | 2 |
|---|---|---|
| Formula | C8H6BrNO | C8H5BrFNO |
| Formula mass (g mol−1) | 212.05 | 230.04 |
| Crystal System | Monoclinic | Monoclinic |
| Space Group | P 21/c | P 21/c |
| a (Å) | 4.286(2) | 7.4802(10) |
| b (Å) | 12.704(6) | 14.5825(16) |
| c (Å) | 14.187(7) | 14.0709(16) |
| β (°) | 93.458(7) | 95.616(11) |
| Z | 4 | 8 |
| V (Å3) | 771.2(7) | 1527.5(3) |
| calcd (g cm−3) | 1.826 | 2.001 |
| T (K) | 173 | 173 |
| μ (mm−1) | 5.279 | 5.341 |
| F (000) | 416 | 896 |
| Total Reflections | 6480 | 13,980 |
| Independent Reflections | 1406 | 2791 |
| Data/restraints/parameters | 1406/0/104 | 2791/0/217 |
| Rint | 0.0857 | 0.0599 |
| R1 (I ≥ 2σ(I)) | 0.0455 | 0.0404 |
| wR2 (F2) (I ≥ 2σ(I)) | 0.0978 | 0.1038 |
| R1 (all data) | 0.0635 | 0.0499 |
| wR2 (F2) (all data) | 0.1057 | 0.1123 |
| GOF | 1.038 | 1.081 |
| CCDC deposition number | 2101968 | 2101969 |
| Short Intermolecular Contact Type | Structure 1 Contact Atoms | Structure 1 Distance (Å) (Distance—vdW (Å)) | Structure 2 Contact Atoms | Structure 2 Distance (Å) (Distance—vdW(Å)) |
|---|---|---|---|---|
| Br···Br | Br1···Br11 | 3.525 −0.175) | NA | None < vdW |
| N-H···O | H1···O12 | 2.047 (−0.673) | H1···O13 | 1.917 (−0.803) |
| H2···O24 | 1.929 (−0.791) | |||
| C-H···F | NA | NA | H2A5···F2 | 2.555 (−0.115) |
| H10B···F16 | 2.646 (−0.024) | |||
| C-H···Br | None < vdW | NA | H2A···Br2 | 2.997 (−0.053) 2.979 (−0.071) 3.006 (−0.044) 2.917 (−0.133) |
| H2B···Br17 | ||||
| H15···Br18 | ||||
| H10A···Br29 | ||||
| C-H···O | H710···O1 H2B11···O1 | 2.492 (−0.228) 2.691 (−0.029) | H56···O2 | 2.419 (−0.301) 2.498 (−0.222) 2.690 (−0.030) |
| H13···O15 | ||||
| H2B3···O2 |
| Dimer | CP | |||||||
|---|---|---|---|---|---|---|---|---|
| 1-BrBr | 1 | 0.637 | 2.588 | 1.354 | 0.512 | −0.376 | 1.360 | −0.285 |
| 2-CHBrBr | 1 | 0.376 | 1.241 | 0.744 | 0.236 | −0.162 | 1.460 | −0.193 |
| 2 | 0.282 | 1.098 | 0.709 | 0.204 | −0.133 | 1.534 | 0.412 | |
| 3 | 0.481 | 1.642 | 0.837 | 0.327 | −0.243 | 1.344 | −0.333 | |
| 1-CHOCHBr | 1 | 0.285 | 0.980 | 0.592 | 0.186 | −0.127 | 1.467 | −0.182 |
| 2 | 0.124 | 0.471 | 0.327 | 0.085 | −0.052 | 1.623 | 0.432 | |
| 3 | 0.738 | 0.323 | 1.856 | 0.622 | −0.437 | 1.425 | −0.610 | |
| 2a-CHOCHF | 1 | 0.777 | 3.439 | 1.916 | 0.668 | −0.477 | 1.402 | −0.719 |
| 2 | 0.158 | 0.778 | 0.510 | 0.143 | −0.925 | 1.551 | 0.652 | |
| 3 | 0.595 | 2.909 | 1.701 | 0.557 | −0.387 | 1.440 | −0.551 | |
| 2b-CHOCHF | 1 | 0.898 | 3.970 | 2.248 | 0.768 | −0.543 | 1.414 | −0.893 |
| 2 | 0.136 | 0.719 | 0.510 | 0.129 | −0.776 | 1.657 | 0.306 | |
| 3 | 0.459 | 2.154 | 1.283 | 0.410 | −0.282 | 1.455 | −0.413 | |
| 1-NHONHO | 1 | 1.693 | 11.505 | 4.763 | 2.400 | −1.924 | 1.248 | −2.125 |
| 2 | 0.265 | 1.612 | 0.875 | 0.315 | −0.228 | 1.384 | 0.579 | |
| 3 | 1.693 | 11.505 | 4.763 | 2.400 | −1.924 | 1.248 | −2.125 | |
| 2a-NHONHO | 1 | 2.588 | 12.721 | 3.898 | 2.790 | −2.401 | 1.162 | −3.626 |
| 2 | 0.392 | 2.332 | 1.398 | 0.443 | −0.303 | 1.461 | 0.918 | |
| 3 | 2.588 | 12.721 | 3.898 | 2.790 | −2.401 | 1.162 | −3.626 | |
| 2b-NHONHO | 1 | 2.588 | 12.721 | 3.898 | 2.790 | −2.401 | 1.162 | −3.626 |
| 2 | 0.392 | 2.332 | 1.398 | 0.443 | −0.303 | 1.461 | 0.918 | |
| 3 | 2.588 | 12.721 | 3.898 | 2.790 | −2.401 | 1.162 | −3.626 |
| Complex | Donor | Acceptor | E(2) (kcal/mol) |
|---|---|---|---|
| 1-BrBr | LP (1) Br | BD*(1) Br-C | 0.79 |
| 2-CHBrBr | LP (2) Br | BD*(1) Br-C | 0.27 |
| LP (2) Br | BD*(1) C-H | 0.26 | |
| 1-CHOCHBr | LP (1) O | BD*(1) C-H | 0.37 |
| 2a-CHOCHF | LP (1) O | BD*(1) C-H | 0.25 |
| 2b-CHOCHF | LP (1) O | BD*(1) C-H | 0.65 |
| 1-NHONHO | LP (2) O | BD*(1) N-H | 3.07 |
| LP (2) O | BD*(1) N-H | 3.07 | |
| 2a-NHONHO | LP (1) O | BD*(1) N-H | 2.70 |
| LP (2) O | BD*(1) N-H | 5.97 | |
| LP (1) O | BD*(1) N-H | 2.70 | |
| LP (2) O | BD*(1) N-H | 5.97 | |
| 2b-NHONHO | LP (1) O | BD*(1) N-H | 2.68 |
| LP (2) O | BD*(1) N-H | 5.54 | |
| LP (1) O | BD*(1) N-H | 2.68 | |
| LP (2) O | BD*(1) N-H | 5.54 |
| Complex | ||||||
|---|---|---|---|---|---|---|
| 1-BrBr | −0.765 | — | −1.370 | −1.830 | −0.885 | −1.656 |
| 2-CHBrBr | −1.478 | — | −1.473 | −2.013 | −0.952 | −3.315 |
| 1-CHOCHBr | −3.219 | −1.768 | −2.051 | −2.891 | −1.326 | −3.029 |
| 2a-CHOCHF | −2.958 | −2.709 | −3.143 | −4.383 | −2.032 | −3.151 |
| 2b-CHOCHF | −2.999 | −2.587 | −3.001 | −4.212 | −1.940 | −3.359 |
| 1-NHONHO | −10.942 | −12.072 | — | — | −9.054 | — |
| 2a-NHONHO | −11.465 | −15.064 | — | — | −11.298 | — |
| 2b-NHONHO | −11.782 | −15.064 | — | — | −11.298 | — |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silva, R.A.L.; da Silva Filho, D.A.; Moberg, M.E.; Pappenfus, T.M.; Janzen, D.E. Halogen Interactions in Halogenated Oxindoles: Crystallographic and Computational Investigations of Intermolecular Interactions. Molecules 2021, 26, 5487. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26185487
Silva RAL, da Silva Filho DA, Moberg ME, Pappenfus TM, Janzen DE. Halogen Interactions in Halogenated Oxindoles: Crystallographic and Computational Investigations of Intermolecular Interactions. Molecules. 2021; 26(18):5487. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26185487
Chicago/Turabian StyleSilva, Rodrigo A. Lemos, Demetrio A. da Silva Filho, Megan E. Moberg, Ted M. Pappenfus, and Daron E. Janzen. 2021. "Halogen Interactions in Halogenated Oxindoles: Crystallographic and Computational Investigations of Intermolecular Interactions" Molecules 26, no. 18: 5487. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26185487