
Enabling Efficient Folding and High-Resolution Crystallographic Analysis of Bracelet Cyclotides

 , , and
, , and
Abstract
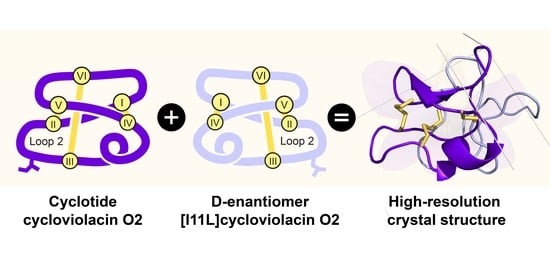
:
1. Introduction
2. Results and Discussion
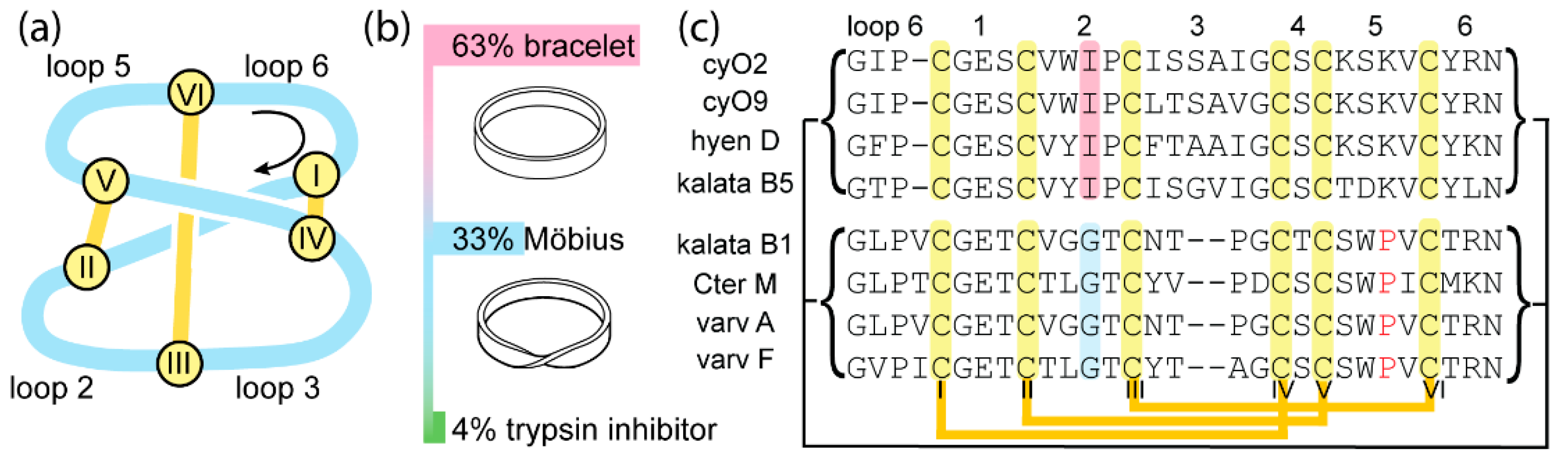
2.1. Substitution of Ile-11 to Gly or Leu Increases Folding Yield of Bracelet Cyclotides
2.2. Leu Mutants Are as Active as the Wild-Type Bracelet Cyclotides
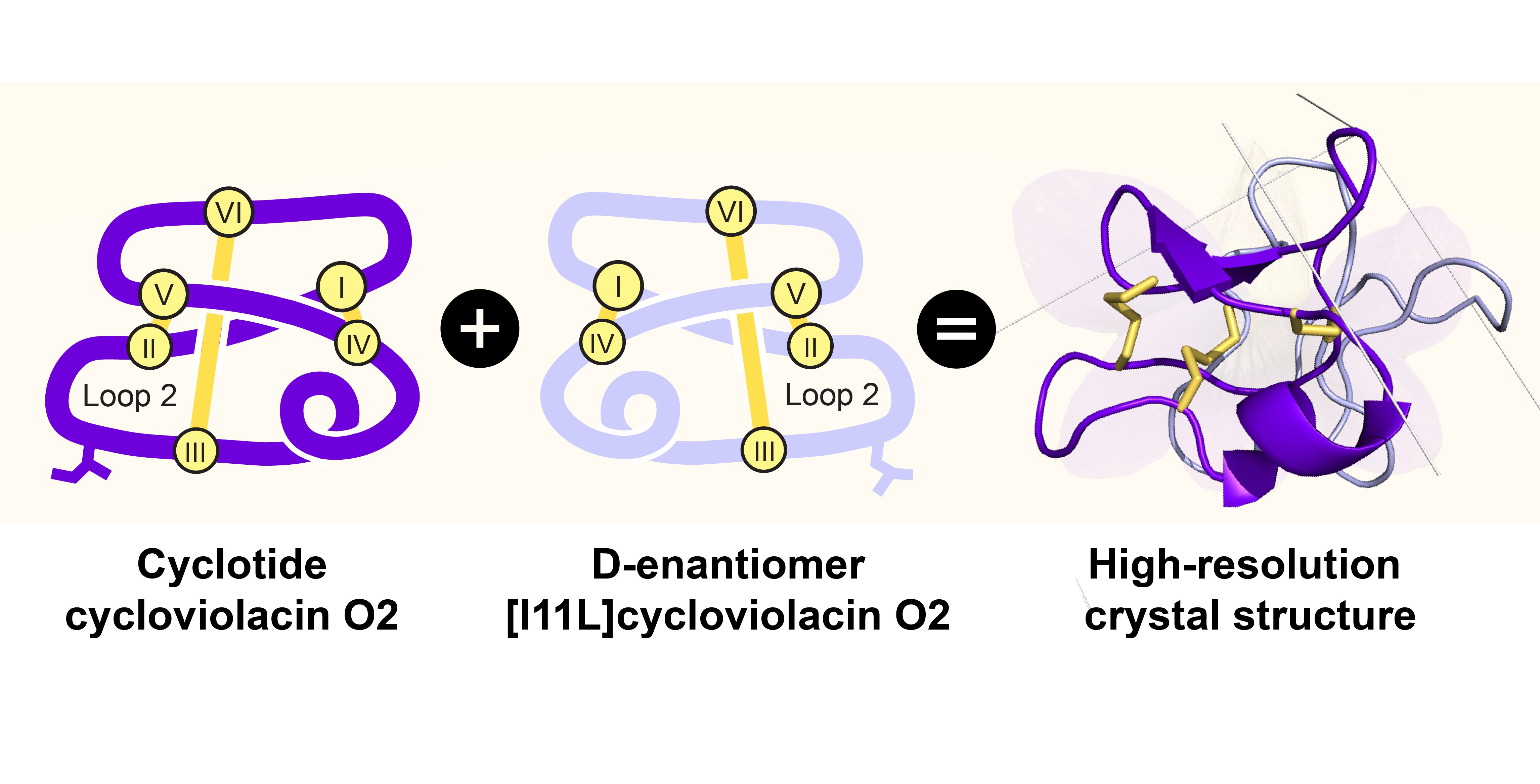
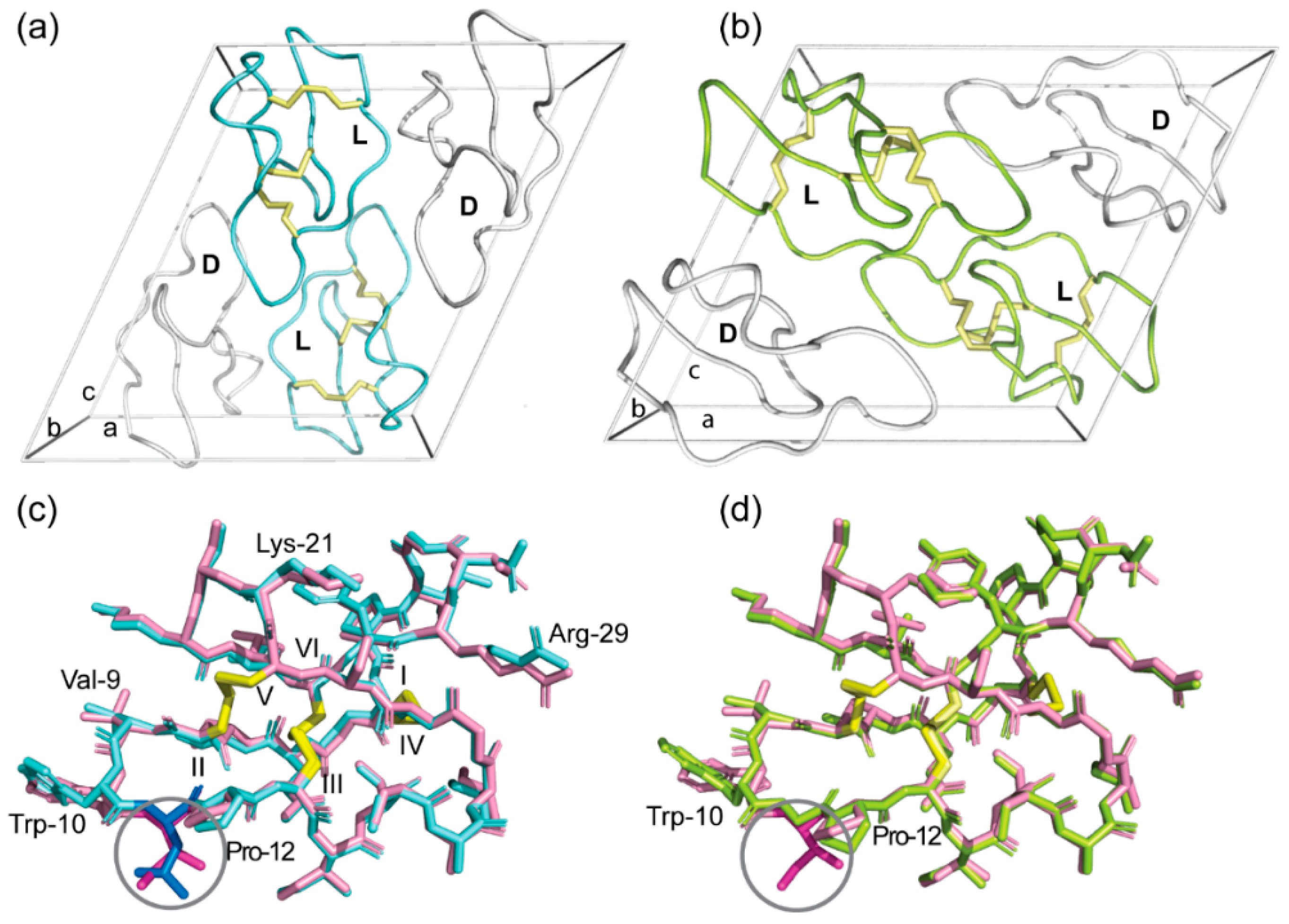
2.3. High-Resolution Structures of Bracelet Cyclotides Can Be Determined by Quasi-Racemic Crystallography
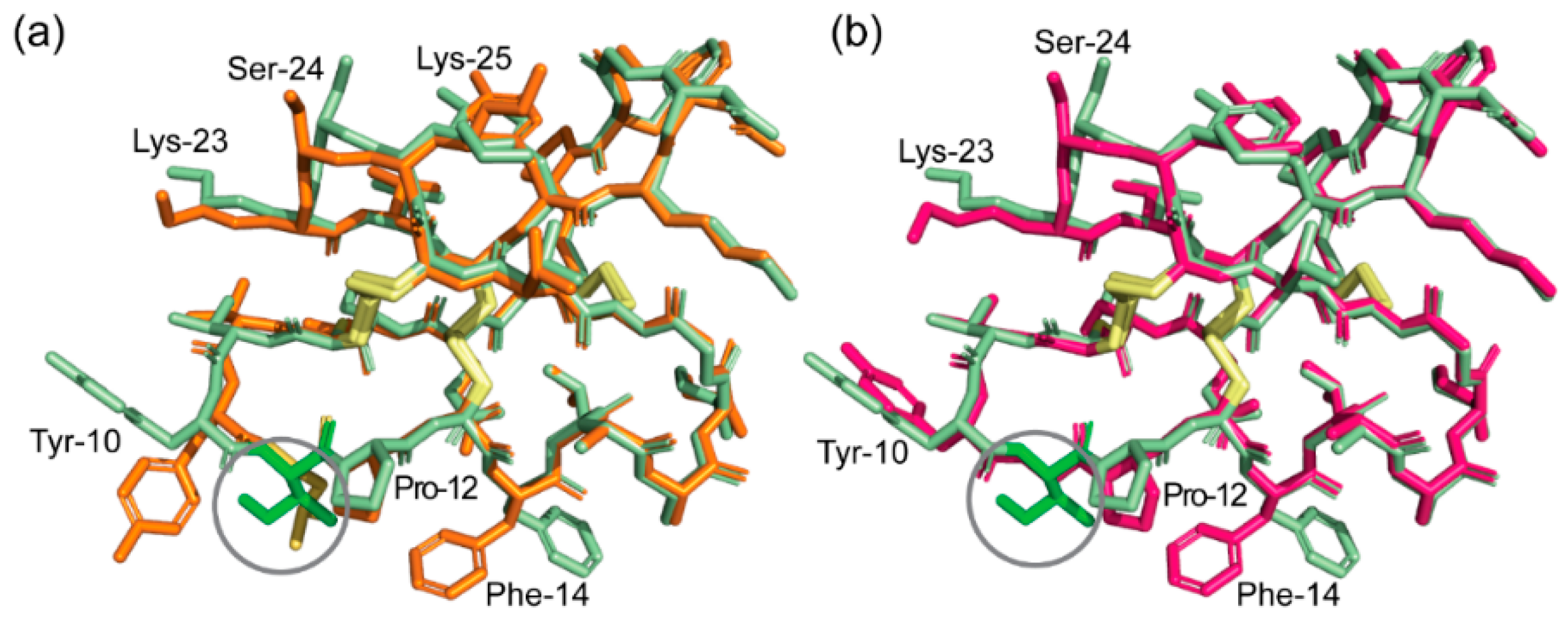
2.4. Racemic and Quasi-Racemic Crystal Structures of Ile-11 Mutant Bracelet Cyclotides
3. Materials and Methods
3.1. Plant Materials and Cyclotide Isolation
3.2. Selected Bracelet Cyclotides’ Synthesis and Folding
3.3. Cytotoxicity Study
3.4. Membrane-Binding Evaluation Using Surface Plasmon Resonance
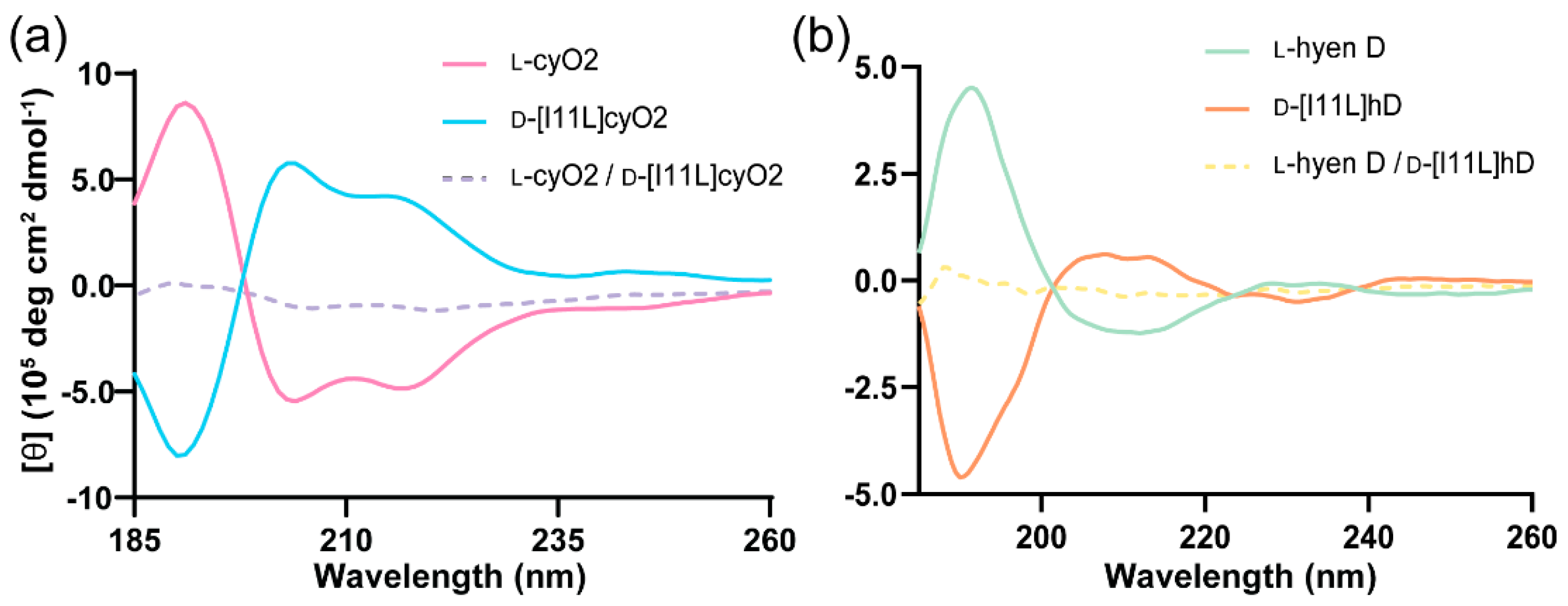
3.5. Circular Dichroism (CD)
3.6. Peptide Crystallization
3.7. Crystal Structure Determination
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- de Veer, S.J.; Kan, M.-W.; Craik, D.J. Cyclotides: From structure to function. Chem. Rev. 2019, 119, 12375–12421. [Google Scholar] [CrossRef]
- Huang, Y.-H.; Du, Q.; Craik, D.J. Cyclotides: Disulfide-rich peptide toxins in plants. Toxicon 2019, 172, 33–44. [Google Scholar] [CrossRef]
- Colgrave, M.L.; Craik, D.J. Thermal, chemical, and enzymatic stability of the cyclotide kalata B1: The importance of the cyclic cystine knot. Biochemistry 2004, 43, 5965–5975. [Google Scholar] [CrossRef]
- Jennings, C.; West, J.; Waine, C.; Craik, D.; Anderson, M. Biosynthesis and insecticidal properties of plant cyclotides: The cyclic knotted proteins from Oldenlandia affinis. Proc. Natl. Acad. Sci. USA 2001, 98, 10614–10619. [Google Scholar] [CrossRef] [Green Version]
- Slazak, B.; Kapusta, M.; Strömstedt, A.A.; Słomka, A.; Krychowiak, M.; Shariatgorji, M.; Andrén, P.E.; Bohdanowicz, J.; Kuta, E.; Göransson, U. How does the sweet violet (Viola odorata L.) fight pathogens and pests—Cyclotides as a comprehensive plant host defense system. Front. Plant Sci. 2018, 9, 1296. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.K.; Craik, D.J. Designing macrocyclic disulfide-rich peptides for biotechnological applications. Nat. Chem. Biol. 2018, 14, 417–427. [Google Scholar] [CrossRef]
- Camarero, J.A.; Campbell, M.J. The potential of the cyclotide scaffold for drug development. Biomedicines 2019, 7, 31. [Google Scholar] [CrossRef] [Green Version]
- Wong, C.T.T.; Rowlands, D.K.; Wong, C.-H.; Lo, T.W.C.; Nguyen, G.K.T.; Li, H.-Y.; Tam, J.P. Orally active peptidic bradykinin B1 receptor antagonists engineered from a cyclotide scaffold for inflammatory pain treatment. Angew. Chem. Int. Ed. 2012, 51, 5620–5624. [Google Scholar] [CrossRef]
- Simonsen, S.M.; Sando, L.; Rosengren, K.J.; Wang, C.K.; Colgrave, M.L.; Daly, N.L.; Craik, D.J. Alanine scanning mutagenesis of the prototypic cyclotide reveals a cluster of residues essential for bioactivity. J. Biol. Chem. 2008, 283, 9805–9813. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.-H.; Colgrave, M.L.; Clark, R.J.; Kotze, A.C.; Craik, D.J. Lysine-scanning mutagenesis reveals an amendable face of the cyclotide kalata B1 for the optimization of nematocidal activity. J. Biol. Chem. 2010, 285, 10797–10805. [Google Scholar] [CrossRef] [Green Version]
- Craik, D.J.; Daly, N.L.; Bond, T.; Waine, C. Plant cyclotides: A unique family of cyclic and knotted proteins that defines the cyclic cystine knot structural motif. J. Mol. Biol. 1999, 294, 1327–1336. [Google Scholar] [CrossRef]
- Gunasekera, S.; Daly, N.L.; Clark, R.J.; Craik, D.J. Dissecting the oxidative folding of circular cystine knot miniproteins. Antioxid. Redox Signal. 2009, 11, 971–980. [Google Scholar] [CrossRef] [Green Version]
- Aboye, T.L.; Clark, R.J.; Burman, R.; Roig, M.B.; Craik, D.J.; Göransson, U. Interlocking disulfides in circular proteins: Toward efficient oxidative folding of cyclotides. Antioxid. Redox Signal. 2011, 14, 77–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aboye, T.L.; Clark, R.J.; Craik, D.J.; Göransson, U. Ultra stable peptide scaffolds for protein engineering—Synthesis and folding of the circular cystine knotted cyclotide cycloviolacin O2. Chembiochem 2008, 9, 103–113. [Google Scholar] [CrossRef]
- Daly, N.L.; Clark, R.J.; Göransson, U.; Craik, D.J. Diversity in the disulfide folding pathways of cystine knot peptides. Lett. Pept. Sci. 2003, 10, 523–531. [Google Scholar] [CrossRef]
- Goransson, U.; Craik, D.J. Disulfide mapping of the cyclotide kalata B1. Chemical proof of the cystic cystine knot motif. J. Biol. Chem. 2003, 278, 48188–48196. [Google Scholar] [CrossRef] [Green Version]
- Rosengren, K.J.; Daly, N.L.; Plan, M.R.; Waine, C.; Craik, D.J. Twists, knots, and rings in proteins structural definition of the cyclotide framework. J. Biol. Chem. 2003, 278, 8606–8616. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.K.; Hu, S.H.; Martin, J.L.; Sjögren, T.; Hajdu, J.; Bohlin, L.; Claeson, P.; Göransson, U.; Rosengren, K.J.; Tang, J.; et al. Combined X-ray and NMR analysis of the stability of the cyclotide cystine knot fold that underpins its insecticidal activity and potential use as a drug scaffold. J. Biol. Chem. 2009, 284, 10672–10683. [Google Scholar] [CrossRef] [Green Version]
- Yeates, T.O.; Kent, S.B.H. Racemic protein crystallography. Annu. Rev. Biophys. 2012, 41, 41–61. [Google Scholar] [CrossRef]
- Pentelute, B.L.; Gates, Z.P.; Tereshko, V.; Dashnau, J.L.; Vanderkooi, J.M.; Kossiakoff, A.A.; Kent, S.B.H. X-ray structure of snow flea antifreeze protein determined by racemic crystallization of synthetic protein enantiomers. J. Am. Chem. Soc. 2008, 130, 9695–9701. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.K.; King, G.J.; Northfield, S.E.; Ojeda, P.G.; Craik, D.J. Racemic and quasi-racemic X-ray structures of cyclic disulfide-rich peptide drug scaffolds. Angew. Chem. Int. Ed. 2014, 53, 11236–11241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, Q.; Chan, L.Y.; Gilding, E.K.; Henriques, S.T.; Condon, N.D.; Ravipati, A.S.; Kaas, Q.; Huang, Y.-H.; Craik, D.J. Discovery and mechanistic studies of cytotoxic cyclotides from the medicinal herb Hybanthus enneaspermus. J. Biol. Chem. 2020, 295, 10911–10925. [Google Scholar] [CrossRef]
- Du, Q.; Huang, Y.-H.; Bajpai, A.; Frosig- Jorgensen, M.; Zhao, G.; Craik, D.J. Evaluation of the in vivo aphrodisiac activity of a cyclotide extract from Hybanthus enneaspermus. J. Nat. Prod. 2020, 83, 3736–3743. [Google Scholar] [CrossRef]
- Herrmann, A.; Svangård, E.; Claeson, P.; Gullbo, J.; Bohlin, L.; Göransson, U. Key role of glutamic acid for the cytotoxic activity of the cyclotide cycloviolacin O2. Cell. Mol. Life Sci. 2006, 63, 235–245. [Google Scholar] [CrossRef]
- Svangård, E.; Burman, R.; Gunasekera, S.; Lövborg, H.; Gullbo, J.; Göransson, U. Mechanism of action of cytotoxic cyclotides: Cycloviolacin O2 disrupts lipid membranes. J. Nat. Prod. 2007, 70, 643–647. [Google Scholar] [CrossRef] [PubMed]
- Göransson, U.; Herrmann, A.; Burman, R.; Haugaard-Jönsson, L.M.; Rosengren, K.J. The conserved Glu in the cyclotide cycloviolacin O2 has a key structural role. Chembiochem 2009, 10, 2354–2360. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, A.; Burman, R.; Mylne, J.S.; Karlsson, G.; Gullbo, J.; Craik, D.J.; Clark, R.J.; Göransson, U. The alpine violet, Viola biflora, is a rich source of cyclotides with potent cytotoxicity. Phytochemistry 2008, 69, 939–952. [Google Scholar] [CrossRef]
- Tang, J.; Wang, C.K.; Pan, X.; Yan, H.; Zeng, G.; Xu, W.; He, W.; Daly, N.L.; Craik, D.J.; Tan, N. Isolation and characterization of cytotoxic cyclotides from Viola tricolor. Peptides 2010, 31, 1434–1440. [Google Scholar] [CrossRef]
- He, W.; Chan, L.Y.; Zeng, G.; Daly, N.L.; Craik, D.J.; Tan, N. Isolation and characterization of cytotoxic cyclotides from Viola philippica. Peptides 2011, 32, 1719–1723. [Google Scholar] [CrossRef]
- Wong, C.T.; Taichi, M.; Nishio, H.; Nishiuchi, Y.; Tam, J.P. Optimal oxidative folding of the novel antimicrobial cyclotide from Hedyotis biflora requires high alcohol concentrations. Biochemistry 2011, 50, 7275–7283. [Google Scholar] [CrossRef]
- Henriques, S.T.; Huang, Y.-H.; Castanho, M.A.; Bagatolli, L.A.; Sonza, S.; Tachedjian, G.; Daly, N.L.; Craik, D.J. Phosphatidylethanolamine-binding is a conserved feature of cyclotide-membrane interactions. J. Biol. Chem. 2012, 287, 33629–33643. [Google Scholar] [CrossRef] [Green Version]
- Henriques, S.T.; Craik, D.J. Cyclotide structure and function: The role of membrane binding and permeation. Biochemistry 2017, 56, 669–682. [Google Scholar] [CrossRef]
- Wang, C.K.; Colgrave, M.L.; Gustafson, K.R.; Ireland, D.C.; Göransson, U.; Craik, D.J. Anti-HIV cyclotides from the Chinese medicinal herb Viola yedoensis. J. Nat. Prod. 2008, 71, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.K.; Wacklin, H.P.; Craik, D.J. Cyclotides insert into lipid bilayers to form membrane pores and destabilize the membrane through hydrophobic and phosphoethanolamine-specific interactions. J. Biol. Chem. 2012, 287, 43884–43898. [Google Scholar] [CrossRef] [Green Version]
- Colgrave, M.L.; Kotze, A.C.; Ireland, D.C.; Wang, C.K.; Craik, D.J. The anthelmintic activity of the cyclotides: Natural variants with enhanced activity. Chembiochem 2008, 9, 1939–1945. [Google Scholar] [CrossRef]
- Lindholm, P.; Göransson, U.; Johansson, S.; Claeson, P.; Gullbo, J.; Larsson, R.; Bohlin, L.; Backlund, A. Cyclotides: A novel type of cytotoxic agents. Mol. Cancer Ther. 2002, 1, 365–369. [Google Scholar] [PubMed]
- Cheneval, O.; Schroeder, C.I.; Durek, T.; Walsh, P.; Huang, Y.-H.; Liras, S.; Price, D.A.; Craik, D.J. Fmoc-based synthesis of disulfide-rich cyclic peptides. J. Org. Chem. 2014, 79, 5538–5544. [Google Scholar] [CrossRef]
- Henriques, S.T.; Huang, Y.H.; Chaousis, S.; Sani, M.A.; Poth, A.G.; Separovic, F.; Craik, D.J. The prototypic cyclotide kalata B1 has a unique mechanism of entering cells. Chem. Biol. 2015, 22, 1087–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide Name | Amino Acid Sequence | Folding Yield (%) |
|---|---|---|
| cyO2 | GIPCGESCVWIPCISSAIGCSCKSKVCYRN | negligible |
| [I11G]cyO2 | GIPCGESCVWGPCISSAIGCSCKSKVCYRN | 51.6 |
| [I11L]cyO2 | GIPCGESCVWLPCISSAIGCSCKSKVCYRN | 30.8 |
| cyO9 | GIPCGESCVWIPCLTSAVGCSCKSKVCYRN | 8.2 |
| [I11G]cyO9 | GIPCGESCVWGPCLTSAVGCSCKSKVCYRN | 59.2 |
| [I11L]cyO9 | GIPCGESCVWLPCLTSAVGCSCKSKVCYRN | 27.3 |
| hyen D | GFPCGESCVYIPCFTAAIGCSCKSKVCYKN | negligible |
| [I11G]hD 1 | GFPCGESCVYGPCFTAAIGCSCKSKVCYKN | 57.3 |
| [I11L]hD 2 | GFPCGESCVYLPCFTAAIGCSCKSKVCYKN | 44.7 |
| kB5 | GTPCGESCVYIPCISGVIGCSCTDKVCYLN | negligible |
| [I11G]kB5 | GTPCGESCVYGPCISGVIGCSCTDKVCYLN | 50.5 |
| [I11L]kB5 | GTPCGESCVYLPCISGVIGCSCTDKVCYLN | 13.3 |
| Peptide Names | POPC/POPE (80:20) | CC50 1 on HeLa (μM) 2 | |
|---|---|---|---|
| P/L Max (mol/mol) | KD (μM) | ||
| cyO2 | 0.28 ± 0.00 | 3.90 ± 0.11 | 1.05 ± 0.06 |
| [I11G]cyO2 | n.b. 3 | n.a. 4 | >64 |
| [I11L]cyO2 | 0.31 ± 0.01 | 4.71 ± 0.17 | 0.95 ± 0.08 |
| cyO9 | 0.30 ± 0.01 | 4.83 ± 0.17 | 2.35 ± 0.13 |
| [I11G]cyO9 | n.b. | n.a. | >64 |
| [I11L]cyO9 | 0.31 ± 0.00 | 4.26 ± 0.06 | 1.06 ± 0.03 |
| hyen D | 0.37 ± 0.01 | 4.48 ± 0.17 | 0.85 ± 0.03 |
| [I11G]hD | 0.24 ± 0.01 | 81.0 ± 7.91 | 50.2 ± 4.61 |
| [I11L]hD | 0.36 ± 0.01 | 4.39 ± 0.50 | 0.70 ± 0.02 |
| kB5 | 0.22 ± 0.00 | 5.02 ± 0.17 | 3.11 ± 0.20 |
| [I11G]kB5 | n.b. | n.a. | >64 |
| [I11L]kB5 | 0.22 ± 0.00 | 4.95 ± 0.12 | 4.31 ± 0.48 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, Y.-H.; Du, Q.; Jiang, Z.; King, G.J.; Collins, B.M.; Wang, C.K.; Craik, D.J. Enabling Efficient Folding and High-Resolution Crystallographic Analysis of Bracelet Cyclotides. Molecules 2021, 26, 5554. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26185554
Huang Y-H, Du Q, Jiang Z, King GJ, Collins BM, Wang CK, Craik DJ. Enabling Efficient Folding and High-Resolution Crystallographic Analysis of Bracelet Cyclotides. Molecules. 2021; 26(18):5554. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26185554
Chicago/Turabian StyleHuang, Yen-Hua, Qingdan Du, Zhihao Jiang, Gordon J. King, Brett M. Collins, Conan K. Wang, and David J. Craik. 2021. "Enabling Efficient Folding and High-Resolution Crystallographic Analysis of Bracelet Cyclotides" Molecules 26, no. 18: 5554. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26185554