
Bacterial Diversity Correlates with Overall Survival in Cancers of the Head and Neck, Liver, and Stomach


Abstract
:1. Introduction
2. Results
2.1. Population Characteristics and Microbial Diversity Profiles
2.2. Microbial Profiles Differed across Institutions
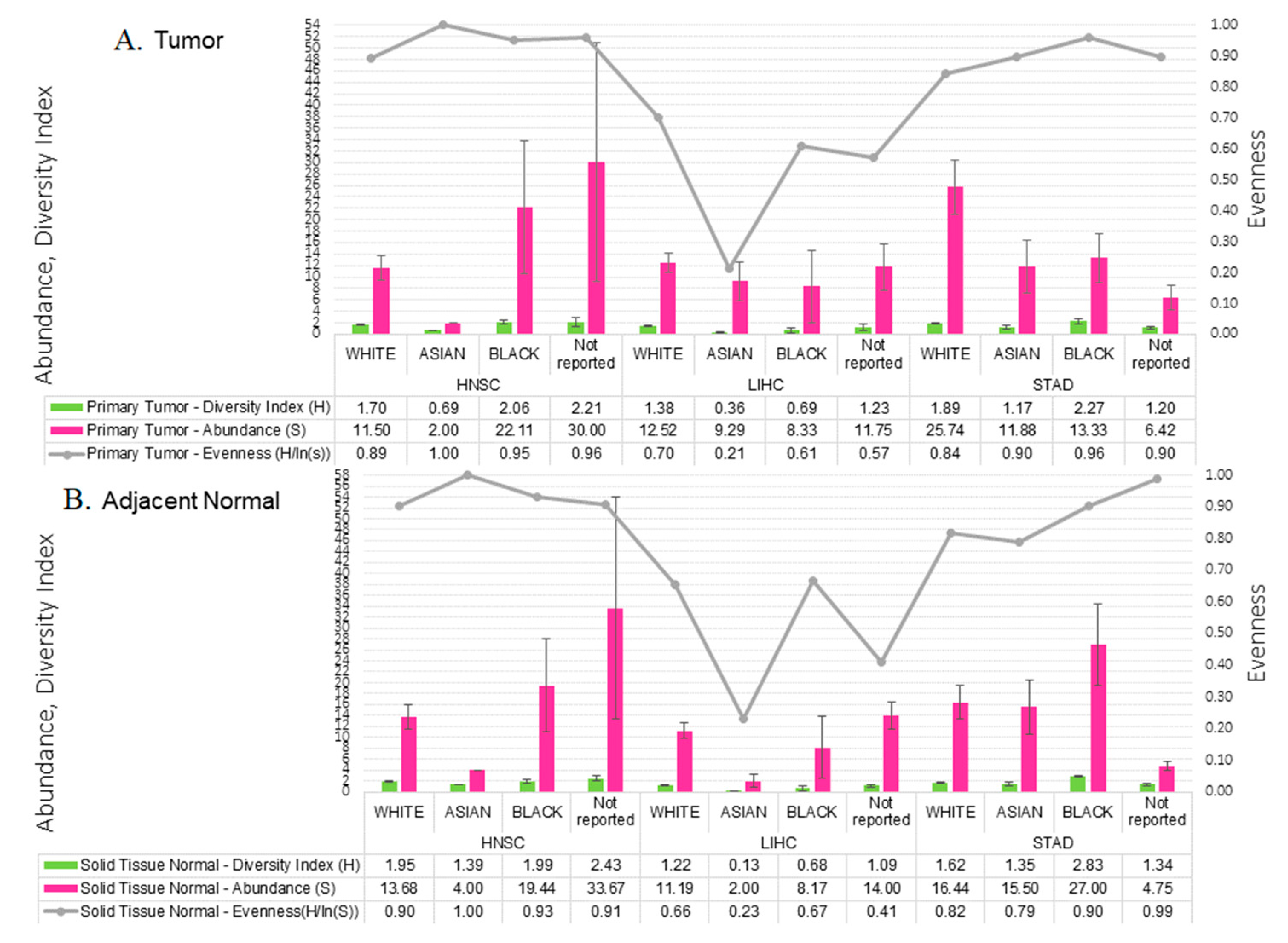
2.3. Microbial Relative Abundance Differed by Race
2.4. Tumor Microbial Diversity Differs by Clinicopathological Presentation
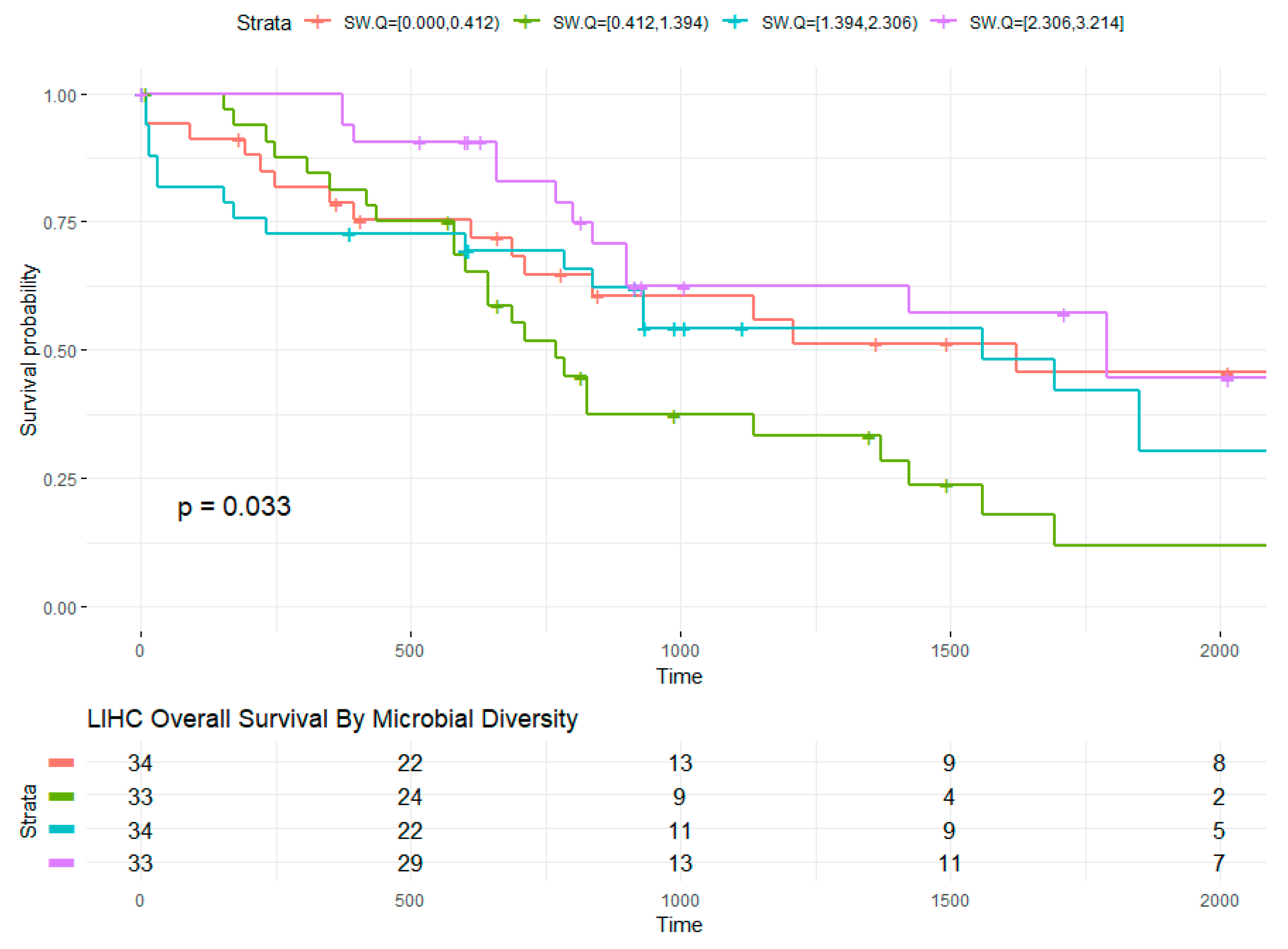
2.5. Microbial Within-Sample Diversity Is Associated with Overall Survival
3. Discussion
4. Materials and Methods
4.1. TCGA Exome Data and Bioinformatics Pipeline
4.2. Statistical Analyses
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Rodriguez, R.M.; Khadka, V.S.; Menor, M.; Hernandez, B.Y.; Deng, Y. Tissue-Associated Microbial Detection in Cancer Using Human Sequencing Data. BMC Bioinform. 2020, 21, 523. [Google Scholar] [CrossRef]
- van Elsland, D.; Neefjes, J. Bacterial Infections and Cancer. EMBO Rep. 2018, 19, e46632. [Google Scholar] [CrossRef]
- Torre, L.A.; Sauer, A.M.G.; Chen, M.S.; Kagawa-Singer, M.; Jemal, A.; Siegel, R.L. Cancer Statistics for Asian Americans, Native Hawaiians, and Pacific Islanders, 2016: Converging Incidence in Males and Females. CA Cancer J. Clin. 2016, 66, 182–202. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2015. CA Cancer J. Clin. 2015, 65, 5–29. [Google Scholar] [CrossRef] [PubMed]
- The Biology of Cancer Health Disparities-National Cancer Institute. Available online: https://www.cancer.gov/research/progress/discovery/biology-cancer-health-disparities (accessed on 9 July 2021).
- Gourin, C.G.; Podolsky, R.H. Racial Disparities in Patients with Head and Neck Squamous Cell Carcinoma. Laryngoscope 2006, 116, 1093–1106. [Google Scholar] [CrossRef]
- Singh, G.K.; Siahpush, M.; Altekruse, S.F. Time Trends in Liver Cancer Mortality, Incidence, and Risk Factors by Unemployment Level and Race/Ethnicity, United States, 1969–2011. J. Community Health 2013, 38, 926–940. [Google Scholar] [CrossRef] [PubMed]
- Merchant, S.J.; Li, L.; Kim, J. Racial and Ethnic Disparities in Gastric Cancer Outcomes: More Important than Surgical Technique? World J. Gastroenterol. 2014, 20, 11546–11551. [Google Scholar] [CrossRef]
- Brooks, A.W.; Priya, S.; Blekhman, R.; Bordenstein, S.R. Gut Microbiota Diversity across Ethnicities in the United States. PLoS Biol. 2018, 16, e2006842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mager, D. Bacteria and Cancer: Cause, Coincidence or Cure? A Review. J. Transl. Med. 2006, 4, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paulos, C.M.; Wrzesinski, C.; Kaiser, A.; Hinrichs, C.S.; Chieppa, M.; Cassard, L.; Palmer, D.C.; Boni, A.; Muranski, P.; Yu, Z.; et al. Microbial Translocation Augments the Function of Adoptively Transferred Self/Tumor-Specific CD8+ T Cells via TLR4 Signaling. J. Clin. Investig. 2007, 117, 2197–2204. [Google Scholar] [CrossRef] [PubMed]
- Pandya, D.; Mariani, M.; He, S.; Andreoli, M.; Spennato, M.; Dowell-Martino, C.; Fiedler, P.; Ferlini, C. Epstein–Barr Virus MicroRNA Expression Increases Aggressiveness of Solid Malignancies. PLoS ONE 2015, 10, e0136058. [Google Scholar] [CrossRef]
- De Flora, S.; Bonanni, P. The Prevention of Infection-Associated Cancers. Carcinogenesis 2011, 32, 787–795. [Google Scholar] [CrossRef] [PubMed]
- De Flora, S.; La Maestra, S. Epidemiology of Cancers of Infectious Origin and Prevention Strategies. J. Prev. Med. Hyg. 2015, 56, E15–E20. [Google Scholar] [PubMed]
- Farhana, L.; Antaki, F.; Murshed, F.; Mahmud, H.; Judd, S.L.; Nangia-Makker, P.; Levi, E.; Yu, Y.; Majumdar, A.P. Gut Microbiome Profiling and Colorectal Cancer in African Americans and Caucasian Americans. World J. Gastrointest. Pathophysiol. 2018, 9, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut Microbiome Modulates Response to Anti-PD-1 Immunotherapy in Melanoma Patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, R.M.; Hernandez, B.Y.; Menor, M.; Deng, Y.; Khadka, V.S. The Landscape of Bacterial Presence in Tumor and Adjacent Normal Tissue across 9 Major Cancer Types Using TCGA Exome Sequencing. Comput. Struct. Biotechnol. J. 2020, 18, 631–641. [Google Scholar] [CrossRef]
- Tae, H.; Karunasena, E.; Bavarva, J.H.; McIver, L.J.; Garner, H.R. Large Scale Comparison of Non-Human Sequences in Human Sequencing Data. Genomics 2014, 104, 453–458. [Google Scholar] [CrossRef]
- Gupta, V.K.; Paul, S.; Dutta, C. Geography, Ethnicity or Subsistence-Specific Variations in Human Microbiome Composition and Diversity. Front. Microbiol. 2017, 8, 1162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffman, K.L.; Hutchinson, D.S.; Fowler, J.; Smith, D.P.; Ajami, N.J.; Zhao, H.; Scheet, P.; Chow, W.-H.; Petrosino, J.F.; Daniel, C.R. Oral Microbiota Reveals Signs of Acculturation in Mexican American Women. PLoS ONE 2018, 13, e0194100. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Edwards, A.; Flemington, E.K.; Zhang, K. Racial Disparities in Patient Survival and Tumor Mutation Burden, and the Association between Tumor Mutation Burden and Cancer Incidence Rate. Sci. Rep. 2017, 7, 13639. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Cleveland, K.; Schnoll-Sussman, F.; McClure, B.; Bigg, M.; Thakkar, P.; Schultz, N.; Shah, M.A.; Betel, D. Identification of Low Abundance Microbiome in Clinical Samples Using Whole Genome Sequencing. Genome Biol. 2015, 16, 265. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HNSC (n = 69) | LIHC (n = 81) | STAD (n = 85) | |
|---|---|---|---|
| Sex | |||
| Female | 21 (30.4%) | 35 (43.2%) | 37 (43.5%) |
| Male | 48 (69.6%) | 46 (56.8%) | 48 (56.5%) |
| Race | |||
| Asian | 1 (1.4%) | 7(8.6%) | 16 (18.8%) |
| Black | 9 (13.0%) | 6 (7.4%) | 3 (3.5%) |
| Not reported | 3 (4.3%) | 4 (4.9%) | 12 (14.1%) |
| White | 56 (81.2%) | 64 (79.0%) | 54 (63.5%) |
| Age | |||
| Mean (SD) | 62.7 (12.2) | 64.3 (14.7) | 67.7 (10.5) |
| Missing | 0 (0%) | 2 (2.5%) | 0 (0%) |
| Vital Status | |||
| Deceased | 49 (71.0%) | 48 (59.3%) | 34 (40.0%) |
| Living | 20 (29.0%) | 33 (40.7%) | 51 (60.0%) |
| HNSC | LIHC | STAD | ||||
|---|---|---|---|---|---|---|
| Primary Tumor (n = 69) | Adjacent Normal (n = 69) | Primary Tumor (n = 81) | Adjacent Normal (n = 81) | Primary Tumor (n = 85) | Adjacent Normal (n = 85) | |
| Shannon | ||||||
| Mean (SD) | 1.76 (0.86) | 1.96 (0.87) | 1.23 (1.09) | 1.08 (1.04) | 1.67 (1.06) | 1.57 (1.12) |
| Richness | ||||||
| Mean (SD) | 13.6 (21.3) | 15.2 (19.8) | 11.9 (13.0) | 10.3 (11.2) | 20.0 (30.2) | 15.0 (20.3) |
| Evenness | ||||||
| Mean (SD) | 0.91 (0.13) | 0.91 (0.13) | 0.65 (0.31) | 0.62 (0.28) | 0.86 (0.18) | 0.84 (0.22) |
| Median [Min, Max] | 0.97 [0.41, 1] | 0.96 [0.31, 1] | 0.76 [0.07, 1] | 0.71 [0.08, 1] | 0.93 [0.33, 1] | 0.96 [0.05, 1] |
| Missing | 4 (5.8%) | 4 (5.8%) | 19 (23.5%) | 24 (29.6%) | 16 (18.8%) | 18 (21.2%) |
| HBV Status | ||||||
| Absent | 69 (100%) | 69 (100%) | 75 (92.6%) | 75 (92.6%) | 85 (100%) | 85 (100%) |
| Present | 0 (0%) | 0 (0%) | 6 (7.4%) | 6 (7.4%) | 0 (0%) | 0 (0%) |
| EBV Status | ||||||
| Absent | 63 (91.3%) | 66 (95.7%) | 80 (98.8%) | 80 (98.8%) | 60 (70.6%) | 59 (69.4%) |
| Present | 6 (8.7%) | 3 (4.3%) | 1 (1.2%) | 1 (1.2%) | 25 (29.4%) | 26 (30.6%) |
| HPV Status | ||||||
| Absent | 63 (91.3%) | 67 (97.1%) | 81 (100%) | 81 (100%) | 85 (100%) | 85 (100%) |
| Present | 6 (8.7%) | 2 (2.9%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) |
| Primary Tumor | Solid Tissue Normal | ||||
|---|---|---|---|---|---|
| N = 235 | N = 235 | ||||
| Cohort | Female | Male | Female | Male | |
| N = 93 | N = 142 | N = 93 | N = 142 | p-Value | |
| HNSC | |||||
| White | 1.75 (0.79) | 1.68 (0.75) | 2.05 (0.95) | 1.90 (0.82) | |
| Asian | -- | 0.69 (0.00) | -- | 1.39 (0.00) | |
| Black | 1.36 (0.03) | 2.26 (1.05) | 1.78 (0.17) | 2.05 (1.08) | |
| Not reported | 0.69 (0.00) | 2.97 (1.12) | 1.35 (0.00) | 2.96 (1.08) | 0.360 |
| LIHC | |||||
| White | 1.28 (1.01) | 1.47 (1.10) | 1.31 (1.02) | 1.13 (1.05) | |
| Asian | -- | 0.36 (0.48) | -- | 0.13 (0.26) | |
| Black | 0.05 (0.05) | 1.01 (1.24) | 0.00 (0.00) | 1.01 (1.07) | |
| Not reported | 2.02 (1.10) | 0.43 (0.43) | 0.92 (0.67) | 1.26 (0.52) | 0.001 |
| STAD | |||||
| White | 2.06 (0.87) | 1.71 (0.97) | 1.51 (1.09) | 1.73 (1.05) | |
| Asian | 1.28 (1.29) | 1.08 (1.33) | 1.56 (1.45) | 1.19 (1.42) | |
| Black | -- | 2.27 (0.68) | -- | 2.83 (0.25) | |
| Not reported | 1.35 (0.21) | 1.15 (0.87) | 1.19 (0.85) | 1.39 (0.76) | 0.007 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodriguez, R.M.; Menor, M.; Hernandez, B.Y.; Deng, Y.; Khadka, V.S. Bacterial Diversity Correlates with Overall Survival in Cancers of the Head and Neck, Liver, and Stomach. Molecules 2021, 26, 5659. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26185659
Rodriguez RM, Menor M, Hernandez BY, Deng Y, Khadka VS. Bacterial Diversity Correlates with Overall Survival in Cancers of the Head and Neck, Liver, and Stomach. Molecules. 2021; 26(18):5659. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26185659
Chicago/Turabian StyleRodriguez, Rebecca M., Mark Menor, Brenda Y. Hernandez, Youping Deng, and Vedbar S. Khadka. 2021. "Bacterial Diversity Correlates with Overall Survival in Cancers of the Head and Neck, Liver, and Stomach" Molecules 26, no. 18: 5659. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26185659