
Isotretinoin and Thalidomide Down-Regulate c-MYC Gene Expression and Modify Proteins Associated with Cancer in Hepatic Cells

 ,
,  , , and
, , and {kind=link}
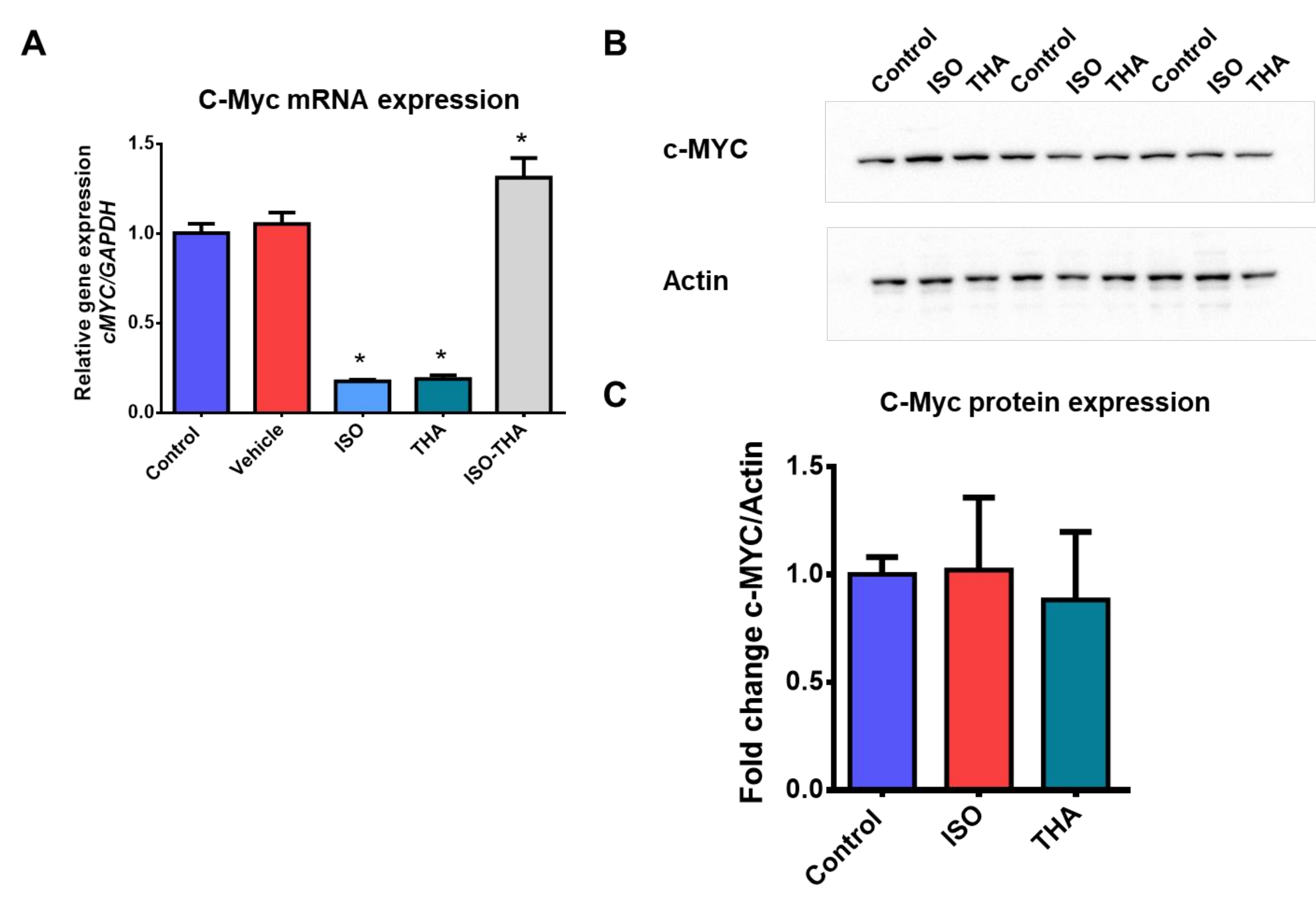{kind=link}
{kind=link}
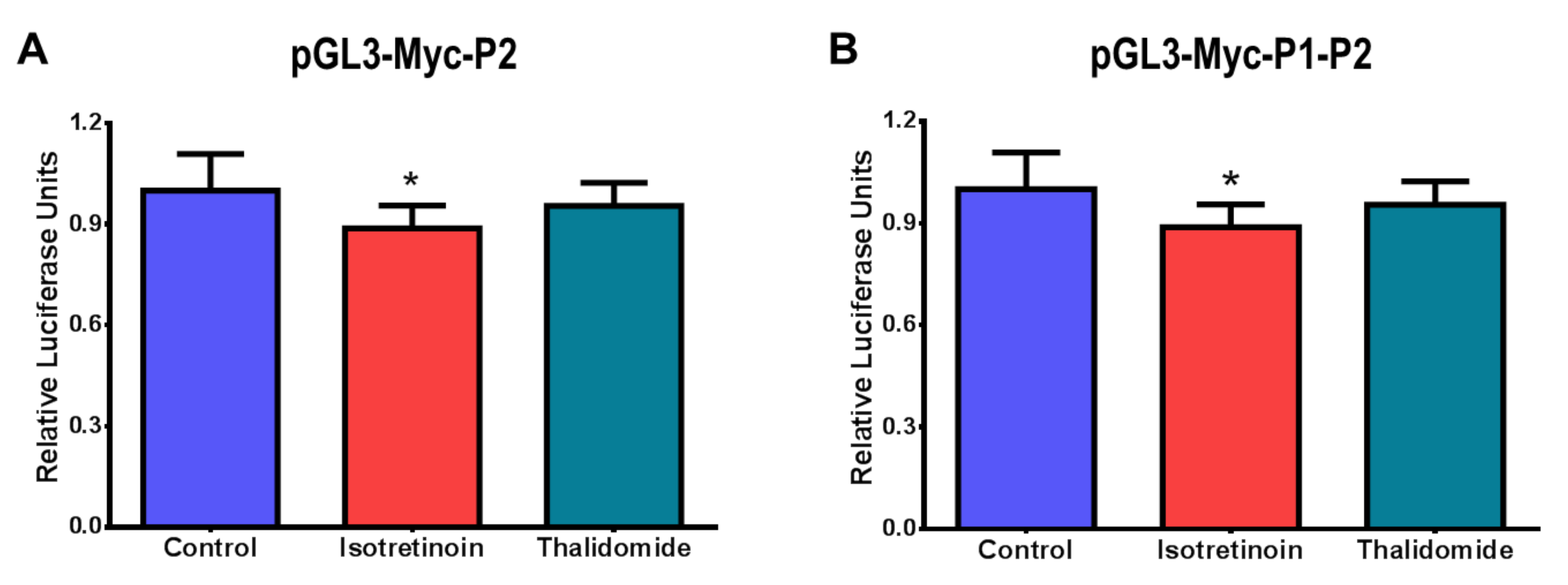{kind=link}
{kind=link}
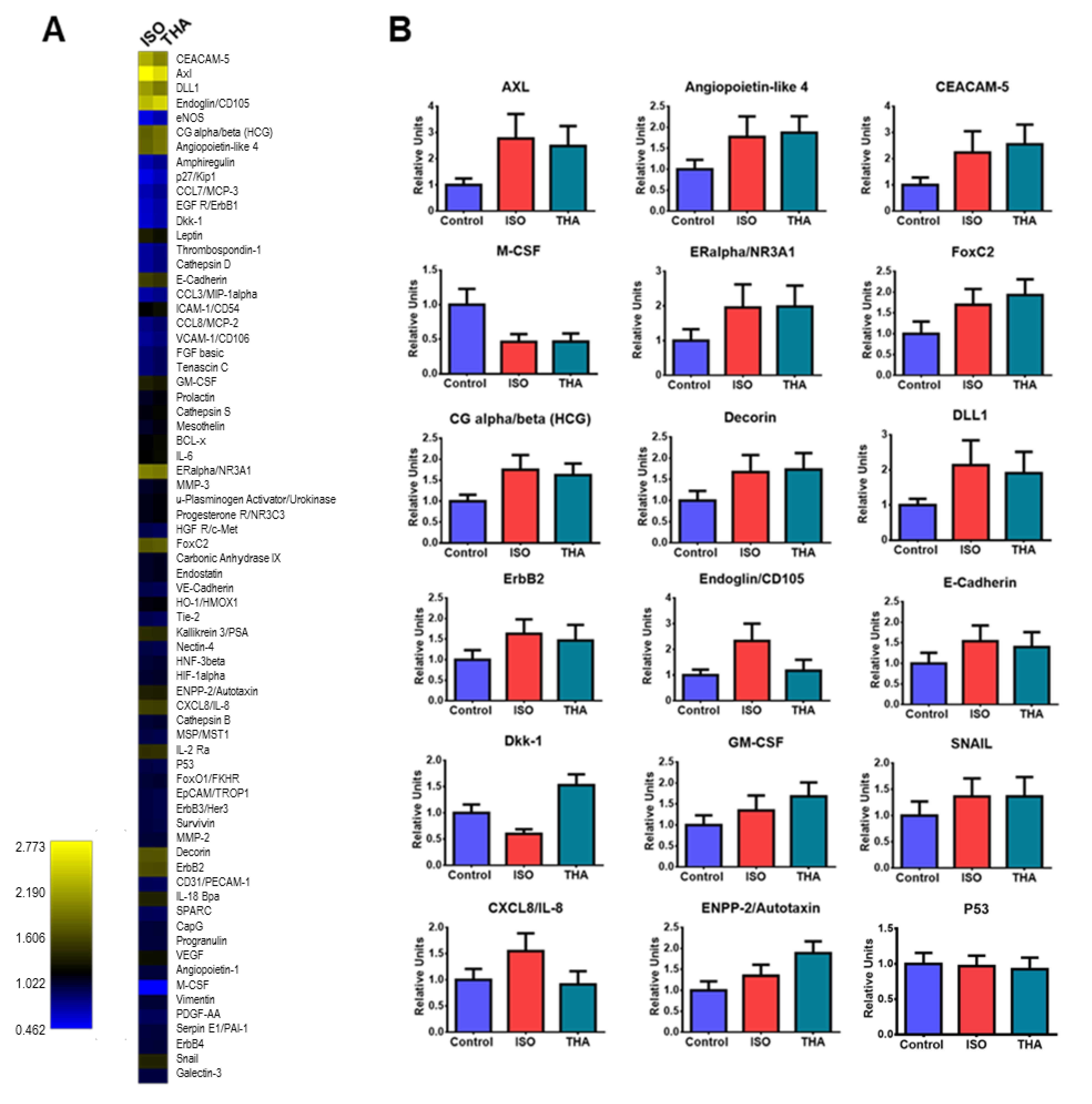{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Cell Culture and Treatments
4.3. Viability Assays
4.4. RNA Extraction
4.5. Reverse Transcription and Quantitative PCR Assays
4.6. Constructs, Transient Transfections and Promoter Activity Assays
4.7. Proteome Array and Densitometric Analysis
4.8. Western Blotting
4.9. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Mohammadian, M.; Allah Bakeshei, K.; Mohammadian-Hafshejani, A. International epidemiology of liver cancer: Geographical distribution, secular trends and predicting the future. J. Prev. Med. Hyg. 2020, 61, 259–289. [Google Scholar]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA. Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Maucort-Boulch, D.; de Martel, C.; Franceschi, S.; Plummer, M. Fraction and incidence of liver cancer attributable to hepatitis B and C viruses worldwide. Int. J. Cancer 2018, 142, 2471–2477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlaeger, C.; Longerich, T.; Schiller, C.; Bewerunge, P.; Mehrabi, A.; Toedt, G.; Kleeff, J.; Ehemann, V.; Eils, R.; Lichter, P.; et al. Etiology-dependent molecular mechanisms in human hepatocarcinogenesis. Hepatology 2008, 47, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Tsai, W.C.; Kung, P.T.; Wang, Y.H.; Kuo, W.Y.; Li, Y.H. Influence of the time interval from diagnosis to treatment on survival for early-stage liver cancer. PLoS. ONE. 2018, 13, 0199532. [Google Scholar] [CrossRef] [PubMed]
- Okuda, K.; Ohtsuki, T.; Obata, H.; Tomimatsu, M.; Okazaki, N.; Hasegawa, H.; Nakajima, Y.; Ohnishi, K. Natural history of hepatocellular carcinoma and prognosis in relation to treatment. Study of 850 patients. Cancer 1985, 56, 918–928. [Google Scholar] [CrossRef]
- Lin, S.; Hoffmann, K.; Schemmer, P. Treatment of hepatocellular carcinoma: A systematic review. Liver Cancer 2012, 1, 144–158. [Google Scholar] [CrossRef] [PubMed]
- Bahman, A.A.; Abaza, M.S.I.; Khoushiash, S.I.; Al-Attiyah, R.J. Sequencedependent effect of sorafenib in combination with natural phenolic compounds on hepatic cancer cells and the possible mechanism of action. Int. J. Mol. Med. 2018, 42, 1695–1715. [Google Scholar]
- Feng, J.; Dai, W.; Mao, Y.; Wu, L.; Li, J.; Chen, K.; Yu, Q.; Kong, R.; Li, S.; Zhang, J.; et al. Simvastatin re-sensitizes hepatocellular carcinoma cells to sorafenib by inhibiting HIF-1alpha/PPAR-gamma/PKM2-mediated glycolysis. J. Exp. Clin. Cancer Res. 2020, 39, 24. [Google Scholar] [CrossRef] [Green Version]
- Jiang, S.; Wang, R.; Zhang, X.; Wu, F.; Li, S.; Yuan, Y. Combination treatment of gemcitabine and sorafenib exerts a synergistic inhibitory effect on non-small cell lung cancer in vitro and in vivo via the epithelial-to-mesenchymal transition process. Oncol. Lett. 2020, 20, 346–356. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Cho, Y.Y.; Cho, E.J.; Yu, S.J.; Lee, J.H.; Yoon, J.H.; Kim, Y.J. Synergistic effect of ursodeoxycholic acid on the antitumor activity of sorafenib in hepatocellular carcinoma cells via modulation of STAT3 and ERK. Int. J. Mol. Med. 2018, 42, 2551–2559. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Zhan, Y.; Wu, Z.; Lin, M.; Jin, X.; Jiang, L.; Qiu, Y. A novel multitarget kinase inhibitor BZG with potent anticancer activity in vitro and vivo enhances efficacy of sorafenib through PI3K pathways in hepatocellular carcinoma cells. Biomed Pharmacother 2020, 125, 110033. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Liang, Q.; Yang, X.; Yu, Y.; Shen, X.; Sun, G. Combination of sorafenib and Valproic acid synergistically induces cell apoptosis and inhibits hepatocellular carcinoma growth via down-regulating Notch3 and pAkt. Am. J. Cancer Res. 2017, 7, 2503–2514. [Google Scholar]
- Croce, C.M. Oncogenes and cancer. N. Engl. J. Med. 2008, 358, 502–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, C.V.; O’Donnell, K.A.; Zeller, K.I.; Nguyen, T.; Osthus, R.C.; Li, F. The c-MYC target gene network. Semin Cancer Biol. 2006, 16, 253–264. [Google Scholar] [CrossRef]
- Wahlstrom, T.; Henriksson, M.A. Impact of MYC in regulation of tumor cell metabolism. Biochim. Biophys Acta. 2015, 1849, 563–569. [Google Scholar] [CrossRef]
- Tsujiuchi, T.; Tsutsumi, M.; Sasaki, Y.; Takahama, M.; Konishi, Y. Hypomethylation of CpG sites and c-myc gene overexpression in hepatocellular carcinomas, but not hyperplastic nodules, induced by a choline-deficient L-amino acid-defined diet in rats. Jpn. J. Cancer Res. 1999, 90, 909–913. [Google Scholar] [CrossRef]
- Bentley, D.L.; Groudine, M. Novel promoter upstream of the human c-myc gene and regulation of c-myc expression in B-cell lymphomas. Mol. Cell Biol. 1986, 6, 3481–3489. [Google Scholar] [CrossRef] [Green Version]
- Ashburn, T.T.; Thor, K.B. Drug repositioning: Identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef]
- Khalil, N.Y.; Darwish, I.A.; Al-Qahtani, A.A. Isotretinoin. Profiles Drug Subst. Excip. Relat. Methodol. 2020, 2020, 119–157. [Google Scholar]
- Matthay, K.K. Targeted isotretinoin in neuroblastoma: Kinetics, genetics, or absorption. Clin. Cancer Res. 2013, 19, 311–313. [Google Scholar] [CrossRef] [Green Version]
- Singhal, S.; Mehta, J. Thalidomide in cancer: Potential uses and limitations. Bio. Drugs 2001, 15, 163–172. [Google Scholar] [CrossRef]
- Caforio, M.; Sorino, C.; Iacovelli, S.; Fanciulli, M.; Locatelli, F.; Folgiero, V. Recent advances in searching c-MYC transcriptional cofactors during tumorigenesis. J. Exp. Clin. Cancer Res. 2018, 37, 239. [Google Scholar] [CrossRef] [Green Version]
- Wierstra, I.; Alves, J. The c-myc promoter: Still MysterY and challenge. Adv. Cancer Res. 2008, 99, 113–333. [Google Scholar]
- Bareket-Samish, A.; Cohen, I.; Haran, T.E. Signals for TBP/TATA box recognition. J. Mol. Biol. 2000, 299, 965–977. [Google Scholar] [CrossRef]
- Marcu, K.B.; Patel, A.J.; Yang, Y. Differential regulation of the c-MYC P1 and P2 promoters in the absence of functional tumor suppressors: Implications for mechanisms of deregulated MYC transcription. Curr. Top Microbiol Immunol 1997, 224, 47–56. [Google Scholar] [PubMed]
- Kaur, M.; Cole, M.D. MYC acts via the PTEN tumor suppressor to elicit autoregulation and genome-wide gene repression by activation of the Ezh2 methyltransferase. Cancer Res. 2013, 73, 695–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reigada, C.; Valera-Vera, E.A.; Sayé, M.; Errasti, A.E.; Avila, C.C.; Miranda, M.R.; Pereira, C.A. Trypanocidal Effect of Isotretinoin through the Inhibition of Polyamine and Amino Acid Transporters in Trypanosoma cruzi. PLoS. Negl. Trop. Dis. 2017, 11, e0005472. [Google Scholar] [CrossRef] [PubMed]
- Becker, E.; Schmidt, T.S.B.; Bengs, S.; Poveda, L.; Opitz, L.; Atrott, K.; Stanzel, C.; Biedermann, L.; Rehman, A.; Jonas, D.; et al. Effects of oral antibiotics and isotretinoin on the murine gut microbiota. Int. J. Antimicrob Agents 2017, 50, 342–351. [Google Scholar] [CrossRef] [Green Version]
- Csoka, A.B.; Szyf, M. Epigenetic side-effects of common pharmaceuticals: A potential new field in medicine and pharmacology. Med. Hypotheses 2009, 73, 770–780. [Google Scholar] [CrossRef]
- Vargesson, N. The teratogenic effects of thalidomide on limbs. J. Hand Surg. Eur. Vol. 2019, 44, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Leivo, T.; Arjomaa, P.; Oivula, J.; Vesterinen, M.; Kiistala, U.; Autio, P.; Oikarinen, A. Differential modulation of transforming growth factor-beta by betamethasone-17- valerate and isotretinoin: Corticosteroid decreases and isotretinoin increases the level of transforming growth factor-beta in suction blister fluid. Skin Pharmacol. Appl. Skin Physiol. 2000, 13, 150–156. [Google Scholar] [CrossRef]
- Morath, C.; Dechow, C.; Lehrke, I.; Haxsen, V.; Waldherr, R.; Floege, J.; Ritz, E.; Wagner, J. Effects of retinoids on the TGF-beta system and extracellular matrix in experimental glomerulonephritis. J. Am. Soc. Nephrol. 2001, 12, 2300–2309. [Google Scholar] [CrossRef]
- Xu, Q.; Kopp, J.B. Retinoid and TGF-beta families: Crosstalk in development, neoplasia, immunity, and tissue repair. Semin. Nephrol. 2012, 32, 287–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yagi, K.; Furuhashi, M.; Aoki, H.; Goto, D.; Kuwano, H.; Sugamura, K.; Miyazono, K.; Kato, M. c-myc is a downstream target of the Smad pathway. J. Biol. Chem. 2002, 277, 854–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luscher, B.; Mitchell, P.J.; Williams, T.; Tjian, R. Regulation of transcription factor AP-2 by the morphogen retinoic acid and by second messengers. Genes Dev. 1989, 3, 1507–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaubatz, S.; Imhof, A.; Dosch, R.; Werner, O.; Mitchell, P.; Buettner, R.; Eilers, M. Transcriptional activation by Myc is under negative control by the transcription factor AP-2. EMBO. J. 1995, 14, 1508–1519. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Hitchler, M.J.; Sun, W.; Sarsour, E.H.; Goswami, P.C.; Klingelhutz, A.J.; Domann, F.E. AP-2alpha Inhibits c-MYC Induced Oxidative Stress and Apoptosis in HaCaT Human Keratinocytes. J. Oncol. 2009, 2009, 780874. [Google Scholar] [CrossRef] [Green Version]
- Layton, A.M.; Dreno, B.; Gollnick, H.; Mobaken, H.; Shear, N. Isotretinoin therapy and the incidence of acne relapse: A nested case-control study. Br. J. Dermatol 2009, 160, 217–218. [Google Scholar] [CrossRef]
- Keifer, J.A.; Guttridge, D.C.; Ashburner, B.P.; Baldwin, A.S., Jr. Inhibition of NF-kappa B activity by thalidomide through suppression of IkappaB kinase activity. J. Biol. Chem. 2001, 276, 22382–22387. [Google Scholar] [CrossRef] [Green Version]
- Moreira, A.L.; Friedlander, D.R.; Shif, B.; Kaplan, G.; Zagzag, D. Thalidomide and a thalidomide analogue inhibit endothelial cell proliferation in vitro. J. Neurooncol. 1999, 43, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Zheng, Y.; Zhang, Y.; Zhang, J.; Xie, F.; Guo, S.; Gu, J.; Yang, J.; Zheng, P.; Lai, J.; et al. Methylation-mediated LINC00261 suppresses pancreatic cancer progression by epigenetically inhibiting c-MYC transcription. Theranostics 2020, 10, 10634–10651. [Google Scholar] [CrossRef] [PubMed]
- Lv, P.; Meng, Q.; Liu, J.; Wang, C. Thalidomide Accelerates the Degradation of Extracellular Matrix in Rat Hepatic Cirrhosis via Down-Regulation of Transforming Growth Factor-beta1. Yonsei. Med. J. 2015, 56, 1572–1581. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.L.; Xu, P.; Chen, H.H.; Zhao, Y.; Shen, J.; Jiang, C.; Jiang, S.; Ni, S.Z.; Xu, B.; Li, L. Thalidomide Inhibits TGF-beta1-induced Epithelial to Mesenchymal Transition in Alveolar Epithelial Cells via Smad-Dependent and Smad-Independent Signaling Pathways. Sci. Rep. 2017, 7, 14727. [Google Scholar] [CrossRef] [Green Version]
- Reszegi, A.; Horváth, Z.; Fehér, H.; Wichmann, B.; Tátrai, P.; Kovalszky, I.; Baghy, K. Protective Role of Decorin in Primary Hepatocellular Carcinoma. Front. Oncol. 2020, 10, 645. [Google Scholar] [CrossRef] [PubMed]
- Akazawa, A.; Kono, H.; Hara, M.; Furuya, S.; Nakata, Y.; Wakana, H.; Fukushima, H.; Sun, C.; Fujii, H.; Ichikawa, D. M-CSF Receptor Antagonists Inhibit the Initiation and Progression of Hepatocellular Carcinoma in Mice. Anticancer Res. 2019, 39, 4787–4794. [Google Scholar] [CrossRef]
- Xiu, M.X.; Liu, Y.M.; Kuang, B.H. The Role of DLLs in Cancer: A Novel Therapeutic Target. Onco. Targets. Ther. 2020, 13, 3881–3901. [Google Scholar] [CrossRef]
- Liu, Y.; Ma, H.; Yao, J. ERα, a key target for cancer therapy: A review. Onco. Targets. Ther. 2020, 13, 2183–2191. [Google Scholar] [CrossRef] [Green Version]
- Rankin, E.B.; Giaccia, A.J. The Receptor Tyrosine Kinase AXL in Cancer Progression. Cancers 2016, 8, 103. [Google Scholar] [CrossRef]
- Powell, E.; Shao, J.; Picon, H.M.; Bristow, C.; Ge, Z.; Peoples, M.; Robinson, F.; Jeter-Jones, S.L.; Schlosberg, C.; Grzeskowiak, C.L.; et al. A functional genomic screen in vivo identifies CEACAM5 as a clinically relevant driver of breast cancer metastasis. NPJ. Breast. Cancer 2018, 4, 9. [Google Scholar] [CrossRef]
- Wang, Y.; Shi, J.; Chai, K.; Ying, X.; Zhou, B.P. The Role of Snail in EMT and Tumorigenesis. Curr. Cancer Drug Targets 2013, 13, 963–972. [Google Scholar] [CrossRef]
- Guthy, D.A.; Voshol, H. Antibody-based arrays in disease proteomics. Antib. Technol. J. 2015, 15–25. [Google Scholar]
- Sanchez-Carbayo, M. Antibody arrays: Technical considerations and clinical applications in cancer. Clin. Chem. 2006, 52, 1651–1659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Becerril-Esquivel, C.; Penuelas-Urquides, K.; Blancas-Sanchez, E.; Zapata-Benavides, P.; Silva-Ramirez, B.; Chavez-Reyes, A.; Castorena-Torres, F.; Cisneros, B.; Bermudez de Leon, M. The polyaromatic hydrocarbon beta-naphthoflavone alters binding of YY1, Sp1, and Sp3 transcription factors to the Dp71 promoter in hepatic cells. Mol. Med. Rep. 2018, 17, 6150–6155. [Google Scholar] [PubMed] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramírez-Flores, P.N.; Barraza-Reyna, P.J.; Aguirre-Vázquez, A.; Camacho-Moll, M.E.; Guerrero-Beltrán, C.E.; Resendez-Pérez, D.; González-Villasana, V.; Garza-González, J.N.; Silva-Ramírez, B.; Castorena-Torres, F.; et al. Isotretinoin and Thalidomide Down-Regulate c-MYC Gene Expression and Modify Proteins Associated with Cancer in Hepatic Cells. Molecules 2021, 26, 5742. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26195742
Ramírez-Flores PN, Barraza-Reyna PJ, Aguirre-Vázquez A, Camacho-Moll ME, Guerrero-Beltrán CE, Resendez-Pérez D, González-Villasana V, Garza-González JN, Silva-Ramírez B, Castorena-Torres F, et al. Isotretinoin and Thalidomide Down-Regulate c-MYC Gene Expression and Modify Proteins Associated with Cancer in Hepatic Cells. Molecules. 2021; 26(19):5742. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26195742
Chicago/Turabian StyleRamírez-Flores, Patricia Nefertari, Paulina J. Barraza-Reyna, Alain Aguirre-Vázquez, María E. Camacho-Moll, Carlos Enrique Guerrero-Beltrán, Diana Resendez-Pérez, Vianey González-Villasana, Jesús Norberto Garza-González, Beatriz Silva-Ramírez, Fabiola Castorena-Torres, and et al. 2021. "Isotretinoin and Thalidomide Down-Regulate c-MYC Gene Expression and Modify Proteins Associated with Cancer in Hepatic Cells" Molecules 26, no. 19: 5742. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26195742