
Role of 5-HT1A Receptor in Vilazodone-Mediated Suppression of L-DOPA-Induced Dyskinesia and Increased Responsiveness to Cortical Input in Striatal Medium Spiny Neurons in an Animal Model of Parkinson’s Disease


 ,
,  , and
, and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
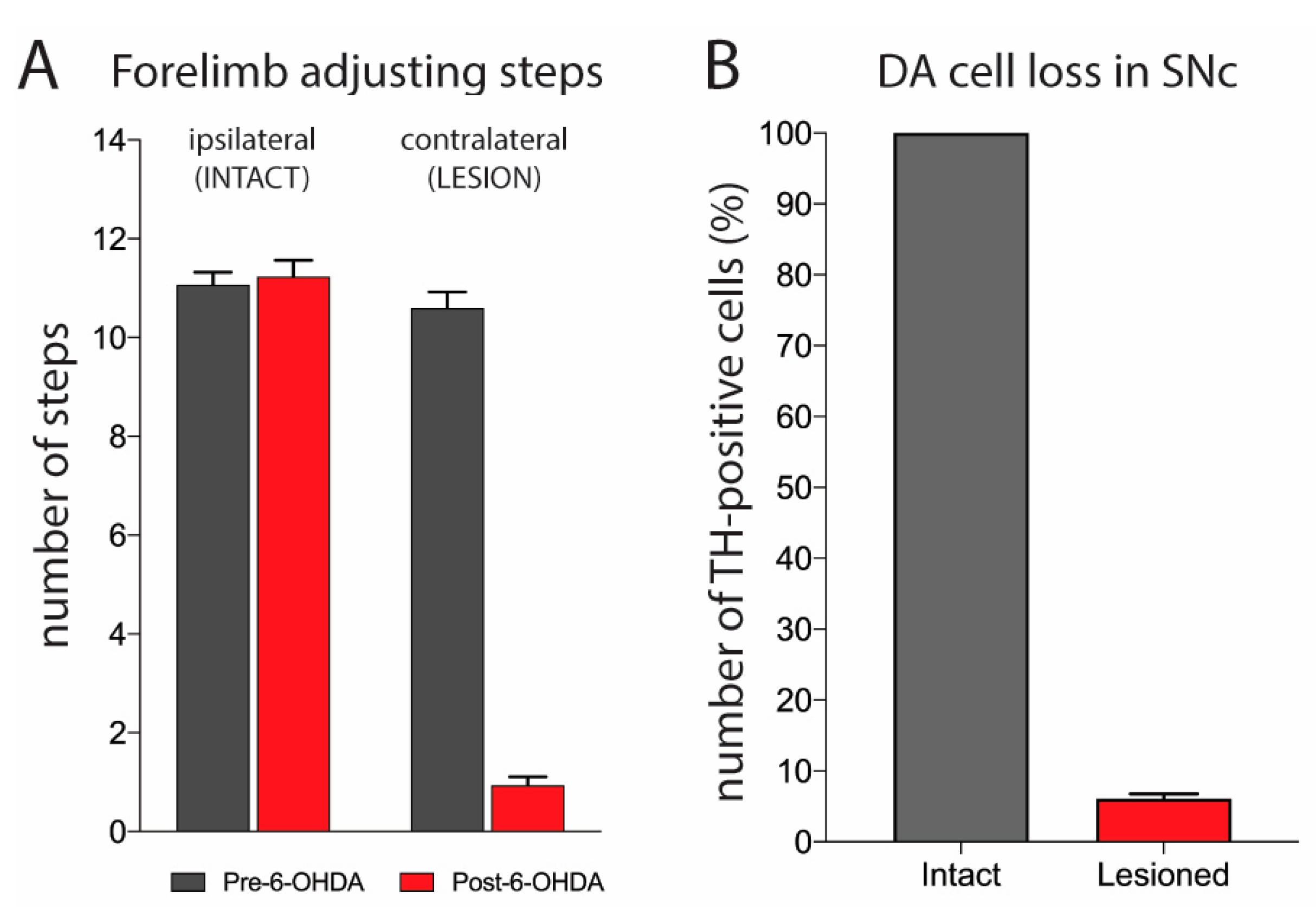
2.1. Evaluation of 6-OHDA Lesion
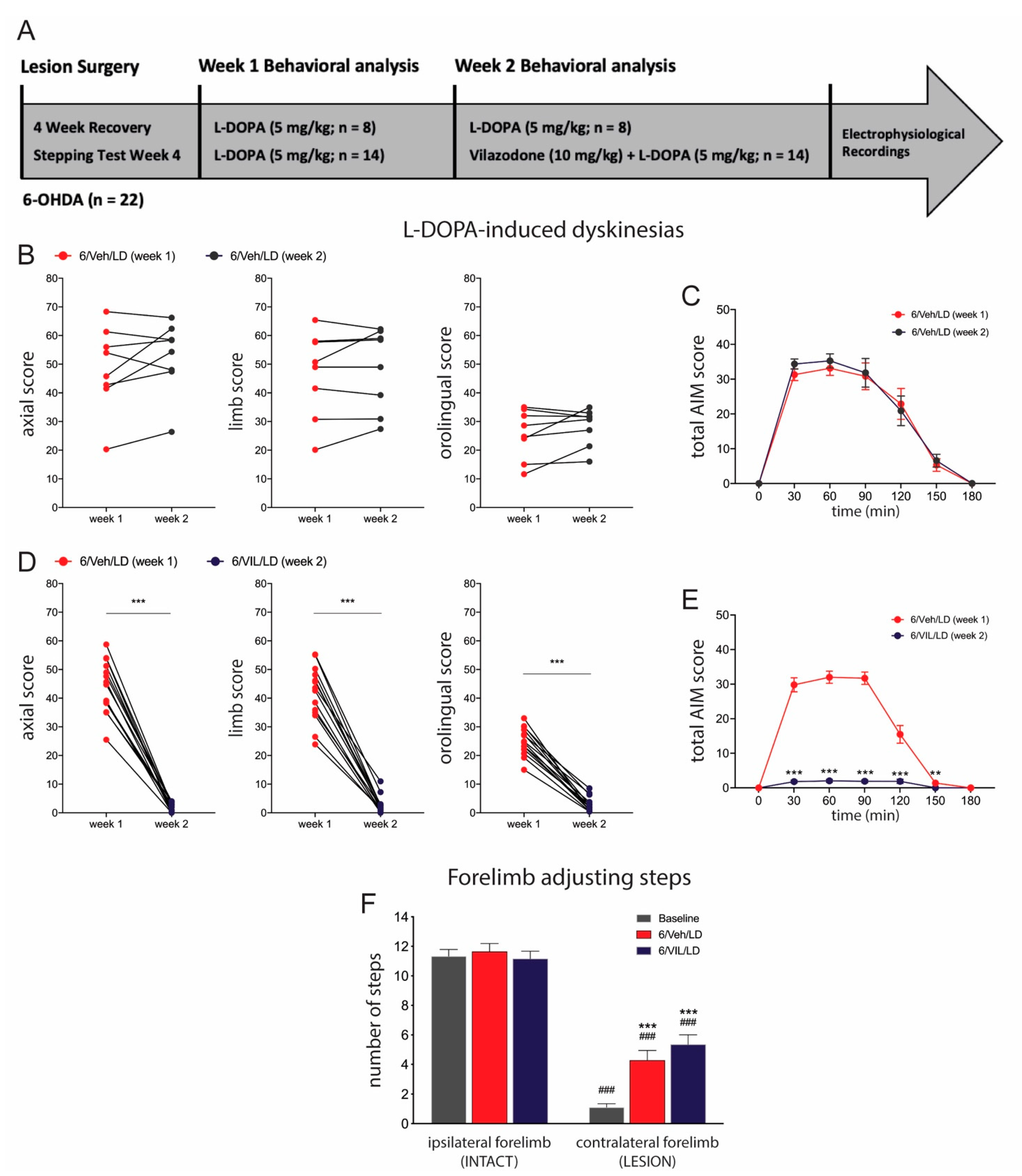
2.2. Vilazodone Suppresses Established AIMs, but Does Not Affect Improved Stepping Performance, after L-DOPA Treatment
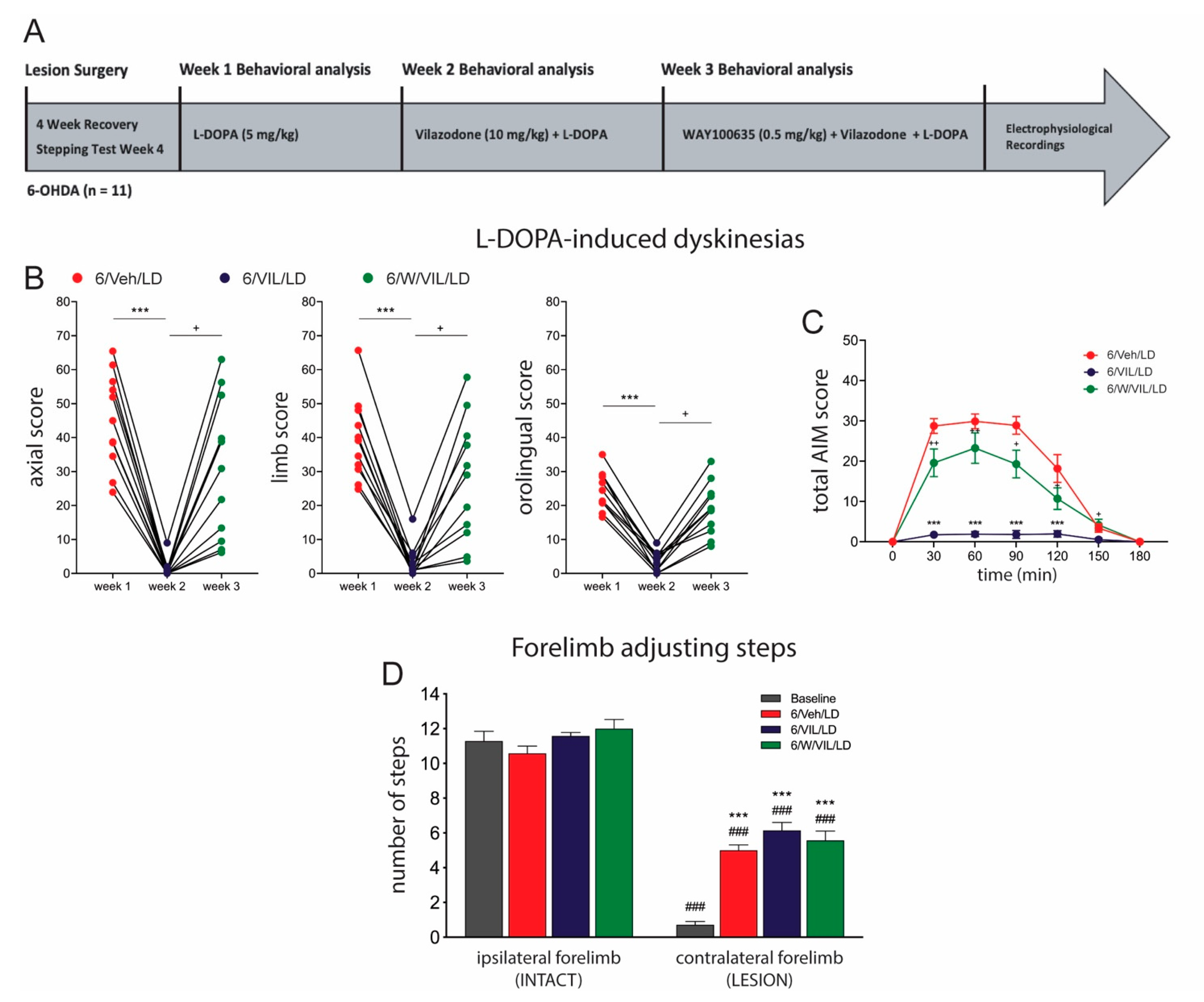
2.3. 5-HTr1A Agonism Mediates Impact of Vilazodone on AIMs
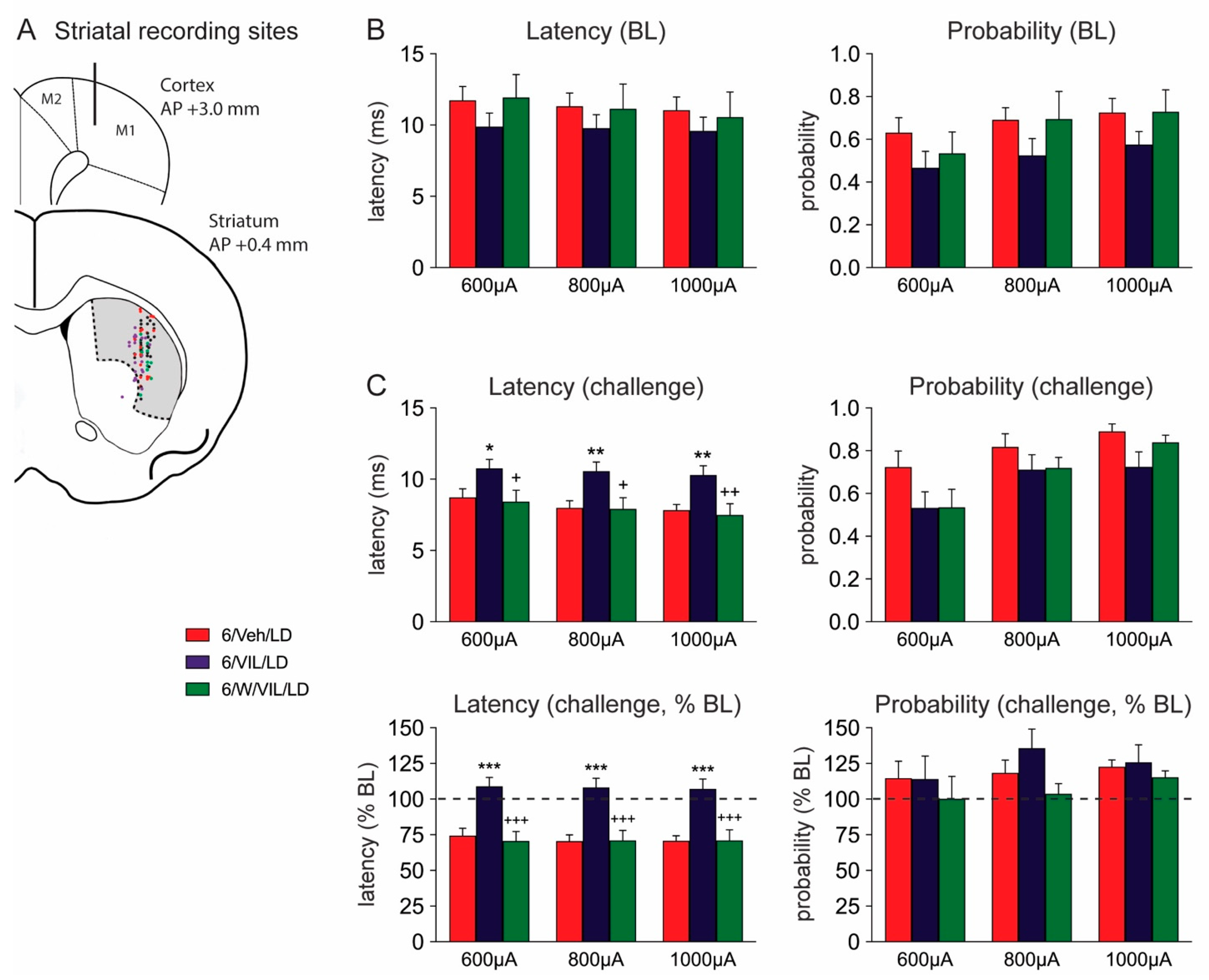
2.4. Effects of Vilazodone and 5-HTr1A Blockade on Striatal MSN Activity in Dyskinetic DA-Depleted Animals
3. Discussion
3.1. Characterization of Dopamine Lesion
3.2. Vilazodone Attenuates L-DOPA-Induced AIMs, but Does Not Block Prokinetic Effects of L-DOPA
3.3. Vilazodone Attenuates L-DOPA-Induced Facilitation of Corticostriatal Transmission in Dyskinetic Parkinsonian Rats
3.4. The Effects of Vilazodone Are Mediated by 5-HTr1A
4. Conclusions
5. Materials and Methods
5.1. Animals
5.2. 6-OHDA-Induced Dopaminergic Lesions and Stepping Test
5.3. Drug Treatments
5.4. Behavioral Analysis
5.5. In Vivo Single-Unit Electrophysiological Recordings
5.6. Tyrosine Hydroxylase Immunohistochemistry
5.7. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Birkmayer, W.; Hornykiewicz, O. The L-3,4-dioxyphenylalanine (DOPA)-effect in Parkinson-akinesia. Wien. Klin. Wochenschr. 1961, 73, 787–788. [Google Scholar] [PubMed]
- De la Fuente-Fernández, R.; Sossi, V.; Huang, Z.; Furtado, S.; Lu, J.Q.; Calne, D.B.; Ruth, T.J.; Stoessl, A.J. Levodopa-induced changes in synaptic dopamine levels increase with progression of Parkinson’s disease: Implications for dyskinesias. Brain 2004, 127, 2747–2754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercuri, N.B.; Bernardi, G. The ‘magic’ of L-Dopa: Why is it the gold standard Parkinson’s disease therapy? Trends Pharm. Sci. 2005, 26, 341–344. [Google Scholar] [CrossRef] [PubMed]
- Cotzias, G.C.; Papavasiliou, P.S.; Gellene, R. L-Dopa in parkinson’s syndrome. N. Engl. J. Med. 1969, 281, 272. [Google Scholar]
- Mones, R.J.; Elizan, T.S.; Siegel, G. L-Dopa induced dyskinesias in 152 patients with Parkinson’s disease. Trans. Am. Neurol. Assoc. 1970, 95, 286–287. [Google Scholar]
- Lane, E.L. L-DOPA for Parkinson’s disease-a bittersweet pill. Eur. J. Neurosci. 2019, 49, 384–398. [Google Scholar] [CrossRef]
- Ahlskog, J.E.; Muenter, M.D. Frequency of levodopa-related dyskinesias and motor fluctuations as estimated from the cumulative literature. Mov. Disord. 2001, 16, 448–458. [Google Scholar] [CrossRef]
- Hely, M.A.; Morris, J.G.; Reid, W.G.; Trafficante, R. Sydney multicenter study of Parkinson’s disease: Non-L-Dopa-responsive problems dominate at 15 years. Mov. Disord. 2005, 20, 190–199. [Google Scholar] [CrossRef]
- Huot, P.; Johnston, T.H.; Koprich, J.B.; Fox, S.H.; Brotchie, J.M. The pharmacology of L-DOPA-induced dyskinesia in Parkinson’s disease. Pharmacol. Rev. 2013, 65, 171–222. [Google Scholar] [CrossRef] [Green Version]
- Damiano, A.M.; McGrath, M.M.; Willian, M.K.; Snyder, C.F.; LeWitt, P.A.; Reyes, P.F.; Richter, R.R.; Means, E.D. Evaluation of a measurement strategy for Parkinson’s disease: Assessing patient health-related quality of life. Qual. Life Res. 2000, 9, 87–100. [Google Scholar] [CrossRef]
- Chapuis, S.; Ouchchane, L.; Metz, O.; Gerbaud, L.; Durif, F. Impact of the motor complications of Parkinson’s disease on the quality of life. Mov. Disord. 2005, 20, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Smith, Y.; Wichmann, T.; Factor, S.A.; DeLong, M.R. Parkinson’s disease therapeutics: New developments and challenges since the introduction of levodopa. Neuropsychopharmacology 2012, 37, 213–246. [Google Scholar] [CrossRef] [PubMed]
- Marconi, R.; Lefebvre-Caparros, D.; Bonnet, A.M.; Vidailhet, M.; Dubois, B.; Agid, Y. Levodopa-induced dyskinesias in Parkinson’s disease phenomenology and pathophysiology. Mov. Disord. 1994, 9, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Cenci, M.A.; Konradi, C. Maladaptive striatal plasticity in L-DOPA-induced dyskinesia. Prog. Brain. Res. 2010, 183, 209–233. [Google Scholar]
- Murer, M.G.; Moratalla, R. Striatal signaling in L-DOPA-induced dyskinesia: Common mechanisms with drug abuse and long term memory involving D1 dopamine receptor stimulation. Front. Neuroanat. 2011, 5, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cenci, M.A. Molecular mechanisms of L-DOPA-induced dyskinesia. In Handbook of Basal Ganglia Structure and Function; Steiner, H., Tseng, K.Y., Eds.; Elsevier: London, UK, 2016; pp. 857–871. [Google Scholar]
- Klawans, H.L.; Goetz, C.; Nausieda, P.A.; Weiner, W.J. Levodopa-induced dopamine receptor hypersensitivity. Trans. Am. Neurol. Assoc. 1977, 102, 80–83. [Google Scholar] [CrossRef]
- Gerfen, C.R. D1 dopamine receptor supersensitivity in the dopamine-depleted striatum: Aberrant ERK1/2 signaling. In Handbook of Basal Ganglia Structure and Function; Steiner, H., Tseng, K.Y., Eds.; Elsevier: London, UK, 2010; pp. 491–500. [Google Scholar]
- Calabresi, P.; Pisani, A.; Rothwell, J.; Ghiglieri, V.; Obeso, J.A.; Picconi, B. Hyperkinetic disorders and loss of synaptic downscaling. Nat. Neurosci. 2016, 19, 868–875. [Google Scholar] [CrossRef]
- Abercrombie, E.D.; Bonatz, A.E.; Zigmond, M.J. Effects of ofl-Dopa on extracellular dopamine in striatum of normal and 6-hydroxydopamine-treated rats. Brain Res. 1990, 525, 36–44. [Google Scholar] [CrossRef]
- Lindgren, H.S.; Andersson, D.R.; Lagerkvist, S.; Nissbrandt, H.; Cenci, M.A. L-DOPA-induced dopamine efflux in the striatum and the substantia nigra in a rat model of Parkinson’s disease: Temporal and quantitative relationship to the expression of dyskinesia. J. Neurochem. 2010, 112, 1465–1476. [Google Scholar] [CrossRef]
- Politis, M.; Wu, K.; Loane, C.; Brooks, D.J.; Kiferle, L.; Turkheimer, F.E.; Bain, P.; Molloy, S.; Piccini, P. Serotonergic mechanisms responsible for levodopa-induced dyskinesias in Parkinson’s disease patients. J. Clin. Investig. 2014, 124, 1340–1349. [Google Scholar] [CrossRef]
- Larsen, M.B.; Sonders, M.S.; Mortensen, O.V.; Larson, G.A.; Zahniser, N.R.; Amara, S.G. Dopamine transport by the serotonin transporter: A mechanistically distinct mode of substrate translocation. J. Neurosci. 2011, 31, 6605–6615. [Google Scholar] [CrossRef] [Green Version]
- Nishijima, H.; Tomiyama, M. What mechanisms are responsible for the reuptake of levodopa-derived dopamine in Parkinsonian striatum? Front. Neurosci. 2016, 10, 575. [Google Scholar] [CrossRef] [Green Version]
- Rylander, D.; Parent, M.; O’Sullivan, S.S.; Dovero, S.; Lees, A.J.; Bezard, E.; Descarries, L.; Cenci, M.A. Maladaptive plasticity of serotonin axon terminals in levodopa-induced dyskinesia. Ann. Neurol. 2010, 68, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Zeng, B.Y.; Iravani, M.M.; Jackson, M.J.; Rose, S.; Parent, A.; Jenner, P. Morphological changes in serotoninergic neurites in the striatum and globus pallidus in levodopa primed MPTP treated common marmosets with dyskinesia. Neurobiol. Dis. 2010, 40, 599–607. [Google Scholar] [CrossRef]
- Bédard, C.; Wallman, M.J.; Pourcher, E.; Gould, P.V.; Parent, A.; Parent, M. Serotonin and dopamine striatal innervation in Parkinson’s disease and Huntington’s chorea. Parkinsonism Relat. Disord. 2011, 17, 593–598. [Google Scholar] [CrossRef]
- Arai, R.; Karasawa, N.; Geffard, M.; Nagatsu, I. L-DOPA is converted to dopamine in serotonergic fibers of the striatum of the rat: A double-labeling immunofluorescence study. Neurosci. Lett. 1995, 195, 195–198. [Google Scholar] [CrossRef]
- Tanaka, H.; Kannari, K.; Maeda, T.; Tomiyama, M.; Suda, T.; Matsunaga, M. Role of serotonergic neurons in L-DOPA-derived extracellular dopamine in the striatum of 6-OHDA-lesioned rats. NeuroReport 1999, 10, 631–634. [Google Scholar] [CrossRef]
- Carta, M.; Carlsson, T.; Kirik, D.; Björklund, A. Dopamine released from 5-HT terminals is the cause of L-DOPA-induced dyskinesia in parkinsonian rats. Brain 2007, 130, 1819–1833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navailles, S.; Bioulac, B.; Gross, C.; De Deurwaerdère, P. Serotonergic neurons mediate ectopic release of dopamine induced by L-DOPA in a rat model of Parkinson’s disease. Neurobiol. Dis. 2010, 38, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Carta, M.; Björklund, A. The serotonergic system in L-DOPA-induced dyskinesia: Pre-clinical evidence and clinical perspective. J. Neural. Transm. 2018, 125, 1195–1202. [Google Scholar] [CrossRef] [PubMed]
- Lanza, K.; Bishop, C. Serotonergic targets for the treatment of L-DOPA-induced dyskinesia. J. Neural. Transm. 2018, 125, 1203–1216. [Google Scholar] [CrossRef]
- Yamato, H.; Kannari, K.; Shen, H.; Suda, T.; Matsunaga, M. Fluoxetine reduces L-DOPA-derived extracellular DA in the 6-OHDA-lesioned rat striatum. NeuroReport 2001, 12, 1123–1126. [Google Scholar] [CrossRef] [PubMed]
- Bishop, C.; George, J.A.; Buchta, W.; Goldenberg, A.A.; Mohamed, M.; Dickinson, S.O.; Eissa, S.; Eskow Jaunarajs, K.L. Serotonin transporter inhibition attenuates l-DOPA-induced dyskinesia without compromising I-DOPA efficacy in hemi-parkinsonian rats. Eur. J. Neurosci. 2012, 36, 2839–2848. [Google Scholar] [CrossRef] [Green Version]
- Conti, M.M.; Ostock, C.Y.; Lindenbach, D.; Goldenberg, A.A.; Kampton, E.; Dell’isola, R.; Katzman, A.C.; Bishop, C. Effects of prolonged selective serotonin reuptake inhibition on the development and expression of L-DOPA-induced dyskinesia in hemi-parkinsonian rats. Neuropharmacology 2014, 77, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fidalgo, C.; Ko, W.K.; Tronci, E.; Li, Q.; Stancampiano, R.; Chuan, Q.; Bezard, E.; Carta, M. Effect of serotonin transporter blockade on L-DOPA-induced dyskinesia in animal models of Parkinson’s disease. Neuroscience 2015, 298, 389–396. [Google Scholar] [CrossRef]
- Kannari, K.; Kurahashi, K.; Tomiyama, M.; Maeda, T.; Arai, A.; Baba, M.; Suda, T.; Matsunaga, M. Tandospirone citrate, a selective 5-HT1A agonist, alleviates L-DOPA-induced dyskinesia in patients with Parkinson’s disease. No Shinkei 2002, 54, 133–137. [Google Scholar]
- Tomiyama, M.; Kimura, T.; Maeda, T.; Kannari, K.; Matsunaga, M.; Baba, M. A serotonin 5-HT1A receptor agonist prevents behavioral sensitization to L-DOPA in a rodent model of Parkinson’s disease. Neurosci. Res. 2005, 52, 185–194. [Google Scholar] [CrossRef]
- Bishop, C.; Taylor, J.L.; Kuhn, D.M.; Eskow, K.L.; Park, J.Y.; Walker, P.D. MDMA and fenfluramine reduce L-DOPA-induced dyskinesia via indirect 5-HT1A receptor stimulation. Eur. J. Neurosci. 2006, 23, 2669–2676. [Google Scholar] [CrossRef] [PubMed]
- Eskow, K.L.; Gupta, V.; Alam, S.; Park, J.Y.; Bishop, C. The partial 5-HT1A agonist buspirone reduces the expression and development of L-DOPA-induced dyskinesia in rats and improves L-DOPA efficacy. Pharmacol. Biochem. Behav. 2007, 87, 306–314. [Google Scholar] [CrossRef]
- Muñoz, A.; Li, Q.; Gardoni, F.; Marcello, E.; Qin, C.; Carlsson, T.; Kirik, D.; Di Luca, M.; Björklund, A.; Bezard, E.; et al. Study of the antidyskinetic effect of eltoprazine in animal models of levodopa-induced dyskinesia. Mov. Disord. 2013, 28, 1088–1096. [Google Scholar]
- Bézard, E.; Muñoz, A.; Tronci, E.; Pioli, E.Y.; Li, Q.; Porras, G.; Björklund, A.; Carta, M. Anti-dyskinetic effect of anpirtoline in animal models of L-DOPA-induced dyskinesia. Neurosci. Res. 2013, 77, 242–246. [Google Scholar] [CrossRef]
- Ghiglieri, V.; Mineo, D.; Vannelli, A.; Cacace, F.; Mancini, M.; Pendolino, V.; Napolitano, F.; di Maio, A.; Mellone, M.; Stanic, J.; et al. Modulation of serotonergic transmission by eltoprazine in L-DOPA-induced dyskinesia: Behavioral, molecular, and synaptic mechanisms. Neurobiol. Dis. 2016, 86, 140–153. [Google Scholar] [CrossRef]
- Grégoire, L.; Samadi, P.; Graham, J.; Bédard, P.J.; Bartoszyk, G.D.; Di Paolo, T. Low doses of sarizotan reduce dyskinesias and maintain antiparkinsonian efficacy of l-Dopa in parkinsonian monkeys. Parkinsonism Relat. Disord. 2009, 15, 445–452. [Google Scholar] [CrossRef]
- Rosengarten, H.; Bartoszyk, G.D.; Quartermain, D.; Lin, Y. The effect of chronic administration of sarizotan, 5-HT1A agonist/D3/D4 ligand, on haloperidol-induced repetitive jaw movements in rat model of tardive dyskinesia. Prog. Neuropsychopharmacol. Biol. Psychiatry 2006, 30, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Goetz, C.G.; Damier, P.; Hicking, C.; Laska, E.; Müller, T.; Olanow, C.W.; Rascol, O.; Russ, H. Sarizotan as a treatment for dyskinesias in Parkinson’s disease: A double-blind placebo-controlled trial. Mov. Disord. 2007, 22, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Goetz, C.G.; Laska, E.; Hicking, C.; Damier, P.; Müller, T.; Nutt, J.; Olanow, W.C.; Rascol, O.; Russ, H. Placebo influences on dyskinesia in Parkinson’s disease. Mov. Disord. 2008, 23, 700–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahli, Z.T.; Banerjee, P.; Tarazi, F.I. The preclinical and clinical effects of vilazodone for the treatment of major depressive disorder. Expert Opin. Drug Discov. 2016, 11, 515–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, Z.A.; Starr, K.R.; Langmead, C.J.; Hill, M.; Bartoszyk, G.D.; Hagan, J.J.; Middlemiss, D.N.; Dawson, L.A. Neurochemical evaluation of the novel 5-HT1A receptor partial agonist/serotonin reuptake inhibitor, vilazodone. Eur. J. Pharmacol. 2005, 510, 49–57. [Google Scholar] [CrossRef]
- Owen, R.T. Vilazodone: A new treatment option for major depressive disorder. Drugs Today 2011, 47, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Cruz, M.P. Vilazodone HCl (Viibryd): A serotonin partial agonist and reuptake inhibitor for the treatment of major depressive disorder. Pharm. Ther. 2012, 37, 28–31. [Google Scholar]
- Meadows, S.M.; Conti, M.M.; Gross, L.; Chambers, N.E.; Avnor, Y.; Ostock, C.Y.; Lanza, K.; Bishop, C. Diverse serotonin actions of Vilazodone reduce L-3,4-dihydroxyphenylalanine-induced dyskinesia in hemi-parkinsonian rats. Mov. Disord. 2018, 33, 1740–1749. [Google Scholar] [CrossRef]
- Altwal, F.; Moon, C.; West, A.R.; Steiner, H. The multimodal serotonergic agent vilazodone inhibits L-DOPA-induced gene regulation in striatal projection neurons and associated dyskinesia in an animal model of Parkinson’s disease. Cells 2020, 9, 2265. [Google Scholar] [CrossRef]
- Tseng, K.Y.; Caballero, A.; Dec, A.; Cass, D.K.; Simak, N.; Sunu, E.; Park, M.J.; Blume, S.R.; Sammut, S.; Park, D.J. Inhibition of striatal soluble guanylyl cyclase-cGMP signaling reverses basal ganglia dysfunction and akinesia in experimental parkinsonism. PLoS ONE 2011, 6, e27187. [Google Scholar] [CrossRef]
- Jayasinghe, V.R.; Flores-Barrera, E.; West, A.R.; Tseng, K.Y. Frequency-dependent corticostriatal disinhibition resulting from chronic dopamine depletion: Role of local striatal cGMP and GABA-AR signaling. Cereb. Cortex 2017, 27, 625–634. [Google Scholar] [CrossRef] [Green Version]
- Padovan-Neto, F.E.; Patterson, S.; Voelkner, N.M.; Altwal, F.; Beverley, J.A.; West, A.R.; Steiner, H. Selective regulation of 5-HT1B serotonin receptor expression in the striatum by dopamine depletion and repeated L-DOPA treatment: Relationship to L-DOPA-induced dyskinesias. Mol. Neurobiol. 2020, 57, 736–751. [Google Scholar] [CrossRef] [PubMed]
- Mallet, N.; Le Moine, C.; Charpier, S.; Gonon, F. Feedforward inhibition of projection neurons by fast-spiking GABA interneurons in the rat striatum in vivo. J. Neurosci. 2005, 25, 3857–3869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padovan-Neto, F.E.; Cavalcanti-Kiwiatkoviski, R.; Carolino, R.O.; Anselmo-Franci, J.; Del Bel, E. Effects of prolonged neuronal nitric oxide synthase inhibition on the development and expression of L-DOPA-induced dyskinesia in 6-OHDA-lesioned rats. Neuropharmacology 2015, 89, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.; DeLong, M.R.; Papa, S.M. Inversion of dopamine responses in striatal medium spiny neurons and involuntary movements. J. Neurosci. 2008, 28, 7537–7547. [Google Scholar] [CrossRef]
- Rangel-Barajas, C.; Silva, I.; Lopéz-Santiago, L.M.; Aceves, J.; Erlij, D.; Florán, B. L-DOPA-induced dyskinesia in hemiparkinsonian rats is associated with up-regulation of adenylyl cyclase type V/VI and increased GABA release in the substantia nigra reticulata. Neurobiol. Dis. 2011, 41, 51–61. [Google Scholar] [CrossRef]
- Ryan, M.B.; Bair-Marshall, C.; Nelson, A.B. Aberrant striatal activity in parkinsonism and levodopa-induced dyskinesia. Cell Rep. 2018, 23, 3438–3446. [Google Scholar] [CrossRef]
- Chang, J.W.; Wachtel, S.R.; Young, D.; Kang, U.J. Biochemical and anatomical characterization of forepaw adjusting steps in rat models of Parkinson’s disease: Studies on medial forebrain bundle and striatal lesions. Neuroscience 1999, 88, 617–628. [Google Scholar] [CrossRef]
- Winkler, C.; Kirik, D.; Björklund, A.; Cenci, M.A. L-DOPA-induced dyskinesia in the intrastriatal 6-hydroxydopamine model of parkinson’s disease: Relation to motor and cellular parameters of nigrostriatal function. Neurobiol. Dis. 2002, 10, 165–186. [Google Scholar] [CrossRef]
- Cenci, M.A.; Lundblad, M. Post- versus presynaptic plasticity in L-DOPA-induced dyskinesia. J. Neurochem. 2006, 99, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Lindenbach, D.; Palumbo, N.; Ostock, C.Y.; Vilceus, N.; Conti, M.M.; Bishop, C. Side effect profile of 5-HT treatments for Parkinson’s disease and L-DOPA-induced dyskinesia in rats. Br. J. Pharmacol. 2015, 172, 119–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Page, M.E.; Cryan, J.F.; Sullivan, A.; Dalvi, A.; Saucy, B.; Manning, D.R.; Lucki, I. Behavioral and neurochemical effects of 5-(4-[4-(5-Cyano-3-indolyl)-butyl)-butyl]-1-piperazinyl)-benzofuran-2-carboxamide (EMD 68843): A combined selective inhibitor of serotonin reuptake and 5-hydroxytryptamine(1A) receptor partial agonist. J. Pharmacol. Exp. Ther. 2002, 302, 1220–1227. [Google Scholar] [CrossRef] [Green Version]
- Sellnow, R.C.; Newman, J.H.; Chambers, N.; West, A.R.; Steece-Collier, K.; Sandoval, I.M.; Benskey, M.J.; Bishop, C.; Manfredsson, F.P. Regulation of dopamine neurotransmission from serotonergic neurons by ectopic expression of the dopamine D2 autoreceptor blocks levodopa-induced dyskinesia. Acta Neuropathol. Commun. 2019, 7, 8. [Google Scholar] [CrossRef] [PubMed]
- Ashby, C.R.J.; Kehne, J.H.; Bartoszyk, G.D.; Renda, M.J.; Athanasiou, M.; Pierz, K.A.; Seyfried, C.A. Electrophysiological evidence for rapid 5-HT₁A autoreceptor inhibition by vilazodone, a 5-HT₁A receptor partial agonist and 5-HT reuptake inhibitor. Eur. J. Pharmacol. 2013, 714, 359–365. [Google Scholar] [CrossRef] [Green Version]
- El Mansari, M.; Crnic, A.; Oosterhof, C.; Blier, P. Long-term administration of the antidepressant vilazodone modulates rat brain monoaminergic systems. Neuropharmacology 2015, 99, 696–704. [Google Scholar] [CrossRef]
- Picconi, B.; Centonze, D.; Håkansson, K.; Bernardi, G.; Greengard, P.; Fisone, G.; Cenci, M.A.; Calabresi, P. Loss of bidirectional striatal synaptic plasticity in L-DOPA–induced dyskinesia. Nat. Neurosci. 2003, 6, 501–506. [Google Scholar] [CrossRef]
- Aubert, I.; Guigoni, C.; Håkansson, K.; Li, Q.; Dovero, S.; Barthe, N.; Bioulac, B.H.; Gross, C.E.; Fisone, G.; Bloch, B.; et al. Increased D1 dopamine receptor signaling in levodopa-induced dyskinesia. Ann. Neurol. 2004, 57, 17–26. [Google Scholar] [CrossRef]
- Cenci, M.A.; Lee, C.S.; Bjorklund, A. L-DOPA-induced dyskinesia in the rat is associated with striatal overexpression of prodynorphin- and glutamic acid decarboxylase mRNA. Eur. J. Neurosci. 1998, 10, 2694–2706. [Google Scholar] [CrossRef] [PubMed]
- Girasole, A.E.; Lum, M.Y.; Nathaniel, D.; Bair-Marshall, C.J.; Guenthner, C.J.; Luo, L.; Kreitzer, A.C.; Nelson, A.B. A subpopulation of striatal neurons mediates levodopa-induced dyskinesia. Neuron 2018, 97, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Fienberg, A.A.; Hiroi, N.; Mermelstein, P.G.; Song, W.J.; Snyder, G.L.; Nishi, A.; Cheramy, A.; O’Callaghan, J.P.; Miller, D.B.; Cole, D.G.; et al. DARPP-32: Regulator of the efficacy of dopaminergic neurotransmission. Science 1998, 281, 838–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santini, E.; Valjent, E.; Usiello, A.; Carta, M.; Borgkvist, A.; Girault, J.A.; Hervé, D.; Greengard, P.; Fisone, G. Critical involvement of cAMP/DARPP-32 and extracellular signal-regulated protein kinase signaling in L-DOPA-induced dyskinesia. J. Neurosci. 2007, 27, 6995–7005. [Google Scholar] [CrossRef] [PubMed]
- Barnes, N.M.; Sharp, T. A review of central 5-HT receptors and their function. Neuropharmacology 1999, 38, 1083–1152. [Google Scholar] [CrossRef]
- Paxinos, G.; Watson, C. The Rat Brain in Stereotaxic Coordinates, 6th ed.; Academic Press: New York, NY, USA, 1998. [Google Scholar]
- Olsson, M.; Nikkhah, G.; Bentlage, C.; Björklund, A. Forelimb akinesia in the rat Parkinson model: Differential effects of dopamine agonists and nigral transplants as assessed by a new stepping test. J. Neurosci. 1995, 15, 3863–3875. [Google Scholar] [CrossRef] [PubMed]
- Threlfell, S.; Sammut, S.; Menniti, F.S.; Schmidt, C.J.; West, A.R. Inhibition of phosphodiesterase 10A increases the responsiveness of striatal projection neurons to cortical stimulation. J. Pharmacol. Exp. Ther. 2009, 328, 785–795. [Google Scholar] [CrossRef]
- Sammut, S.; Threlfell, S.; West, A.R. Nitric oxide-soluble guanylyl cyclase signaling regulates corticostriatal transmission and short-term synaptic plasticity of striatal projection neurons recorded in vivo. Neuropharmacology 2010, 58, 624–631. [Google Scholar] [CrossRef] [Green Version]
- Ondracek, J.M.; Dec, A.; Hoque, K.E.; Lim, S.A.; Rasouli, G.; Indorkar, R.P.; Linardakis, J.; Klika, B.; Mukherji, S.J.; Burnazi, M.; et al. Feed-forward excitation of striatal neuron activity by frontal cortical activation of nitric oxide signaling in vivo. Eur. J. Neurosci. 2008, 27, 1739–1754. [Google Scholar] [CrossRef]
- Zhang, Y.; Meredith, G.E.; Mendoza-Elias, N.; Rademacher, D.J.; Tseng, K.Y.; Steece-Collier, K. Aberrant restoration of spines and their synapses in L-DOPA-induced dyskinesia: Involvement of corticostriatal but not thalamostriatal synapses. J. Neurosci. 2013, 33, 11655–11667. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Altwal, F.; Padovan-Neto, F.E.; Ritger, A.; Steiner, H.; West, A.R. Role of 5-HT1A Receptor in Vilazodone-Mediated Suppression of L-DOPA-Induced Dyskinesia and Increased Responsiveness to Cortical Input in Striatal Medium Spiny Neurons in an Animal Model of Parkinson’s Disease. Molecules 2021, 26, 5790. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26195790
Altwal F, Padovan-Neto FE, Ritger A, Steiner H, West AR. Role of 5-HT1A Receptor in Vilazodone-Mediated Suppression of L-DOPA-Induced Dyskinesia and Increased Responsiveness to Cortical Input in Striatal Medium Spiny Neurons in an Animal Model of Parkinson’s Disease. Molecules. 2021; 26(19):5790. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26195790
Chicago/Turabian StyleAltwal, Feras, Fernando E. Padovan-Neto, Alexandra Ritger, Heinz Steiner, and Anthony R. West. 2021. "Role of 5-HT1A Receptor in Vilazodone-Mediated Suppression of L-DOPA-Induced Dyskinesia and Increased Responsiveness to Cortical Input in Striatal Medium Spiny Neurons in an Animal Model of Parkinson’s Disease" Molecules 26, no. 19: 5790. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26195790