3.2. Synthesis
3.2.1. Reaction of 6-(Hydroxymethyl)-2,2-dimethyl-1-azaspiro[4.4]nonan-1-oxyl 1a with Methanesulfonyl Chloride
Triethylamine (0.174 g, 1.71 mmol) was added to a solution of nitroxide
1a (0.20 g, 1.01 mmol) in dry CHCl
3 (5 mL) at 0 °C. Then MsCl (0.139 g, 1.21 mmol) was added dropwise to mixture under cooling on an ice bath. The mixture was stirred at room temperature for 6 h. The reaction was monitored by TLC (SiO
2, hexane-diethyl ether 1:1 mixture; stained with Dragendorff’s reagent, R
f(
2) = 0.45). The reaction mixture was washed with a saturated solution of NaCl (2×10 mL), the organic layer was dried with anhydrous Na
2SO
4. After evaporation of the solvent under reduced pressure the crude residue was purified by column chromatography (SiO
2, hexane-diethyl ether 1:1 mixture as an eluent) to give
3,3-dimethyloctahydrocyclopenta[c]pyrrolo[1,2-b]isoxazole (
2): 0.119 g, yield 65%. Physical properties and spectral characteristics coincide to the literature data [
4].
3.2.2. Reaction of 6-(Hydroxymethyl)-2,2-dimethyl-1-azaspiro[4.4]nonan-1-oxyl 1a with PPh3-CBr4
Carbon tetrabromide (0.84 g, 2.66 mmol) and PPh
3 (0.70 g, 2.66 mmol) were added to a solution of nitroxide
1a (0.25 g, 1.26 mmol) in dry CH
2Cl
2 (6 mL) and the reaction mixture was stirred at room temperature for 12 h to complete the reaction. The progress of reaction was monitored by TLC (SiO
2, hexane-diethyl ether 1:1 mixture; stained with Dragendorff’s reagent). After evaporation of the solvent under reduced pressure, the crude residue was purified by column chromatography (SiO
2, hexane-diethyl ether 1:1 mixture as an eluent) to give
2, 0.091 g, yield 40%. Physical properties and spectral characteristics coincide to the literature data [
4].
3.2.3. Reaction of (2,2-Dimethyl-1-azaspiro[4.4]nonan-6-yl)methanol 1b with Methanesulfonyl Chloride
Approximately three-fold excess of cold liquid NMe3 (1.53 mL, 17.34 mmol), prepared from aqueous solution of NMe3 and excess of solid NaOH, was added to the solution of 1b (1.058 g, 5.78 mmol) in dry CHCl3 at 0 °C. Then MsCl (1.06 g, 9.25 mmol) was added dropwise to the mixture under cooling on an ice bath and the reaction mass was stirred at room temperature for 12 h. The progress of the reaction was monitored by TLC (SiO2, methanol-ethyl acetate 1:4 mixture as an eluent; stained with iodine vapor, Rf(3salt) = 0.15). The reaction mixture was evaporated under reduced pressure. The residue was dissolved in excess of aqueous solution of NaOH and extracted with diethyl ether (3 × 10 mL). The combined organic layers were dried with anhydrous Na2CO3. After removal of the solvent under atmosphere pressure, the crude residue was purified by vacuum sublimation (P = 14 mm Hg, T = 110–120 °C) to give 3.
(5aS(R),8aR(S))-3,3-Dimethyloctahydrocyclopenta[2,3]azeto[1,2-a]pyrrole (3): 0.73 g, yield 77%. Colorless liquid. IR (neat) νmax: 2943, 2860, 2829, 1460, 1444, 1429, 1379, 1363, 1333, 1302, 1290, 1280, 1257, 1246, 1232, 1223, 1207, 1196, 1184, 1147, 1134, 1101, 1084, 1070, 1037, 980, 970, 951, 926, 904, 897, 872, 769, 717, 627 cm−1; 1H NMR (600 MHz; CDCl3, δ): 0.91 (s, 3H, CH3), 1.08 (s, 3H, CH3), 1.37 (ddd, Jd1 = 6.5 Hz, Jd2 = 12.9 Hz, Jd3 = 13.0 Hz, 1H, C(8)H2), 1.51 (dddd, Jd1 = 7.2 Hz, Jd2 = 8.1 Hz, Jd3 = 12.5 Hz, Jd4 = 12.08 Hz, 1H, C(6)H2), 1.59 (dd, Jd1 = 6.5 Hz, Jd2 = 11.5 Hz, 1H, C(2)H2), 1.61 (dd, Jd1 = 7.0 Hz, Jd2 = 12.8 Hz, 1H, C(6)H2), 1.65 (dd, Jd1 = 6.4 Hz, Jd2 = 13.0 Hz, 1H, C(8)H2), 1.74 (dd, Jd1 = 7.5 Hz, Jd2 = 12.9 Hz, 1H, C(1)H2), 1.81 (ddd, Jd1 = 6.5 Hz, Jd2 = 7.2 Hz, Jd3 = 12.5 Hz, 1H, C(7)H2), 1.88 (ddd, Jd1 = 6.5 Hz, Jd2 = 12.7 Hz, Jd3 = 12.9 Hz, 1H, C(1)H2), 1.96 (ddd, Jd1 = 7.5 Hz, Jd2 = 11.5 Hz, Jd3 = 12.7 Hz, 1H, C(2)H2), 2.15 (dddt, Jd1 = 6.4 Hz, Jd2 = 7.0 Hz, Jd3 = 12.7 Hz, Jt = 12.5 Hz, 1H, C(7)H2), 2.29 (ddd, Jd1 = 4.4 Hz, Jd2 = 8.1 Hz, Jd3 = 9.3 Hz, 1H, C(5a)H), 2.76 (dd, Jd1 = 4.4 Hz, Jd2 = 9.8 Hz, 1H, C(5)H2), 3.30 (dd, Jd1 = 9.3 Hz, Jd2 = 9.8 Hz, 1H, C(5)H2); 13C{1H} NMR (150 MHz; CDCl3, δ): 22.16 (CH3), 25.89 C(7), 27.85 (CH3), 31.14 C(6), 33.83 C(1), 36.95 C(2), 38.94 C(5a), 39.37 C(8), 48.96 C(5), 61.95 C(3), 82.21 C(8a).
3.2.4. Reaction of (5aS,8aR)-3,3-Dimethyloctahydrocyclopenta[2,3]azeto[1,2-a]pyrrole 3 with HBr
An aqueous solution of HBr was added dropwise to a solution of amine 3 (0.27 g, 1.64 mmol) in diethyl ether (3 mL) with stirring to pH = 2–3. Then the organic and aqueous phases were separated, the aqueous one was evaporated to dryness under reduced pressure. The solid residue was crystallized from iso-propanol to give 3×HBr.
(5aS(R),8aR(S))-3,3-Dimethyloctahydro-1H-cyclopenta[2,3]azeto[1,2-a]pyrrol-4-ium bromide (3×HBr): 0.35 g, yield 87%. Colorless crystals, m.p. 194.5 °C with decomposition (iso-propanol). Elemental analysis: found: C, 53.66; H, 8.11; N, 5.70; Br, 32.49; calcd. for C11H20BrN: C, 53.67; H, 8.19; N, 5.69; Br, 32.46%; IR (KBr) νmax: 3014, 2960, 2928, 2868, 2800, 2754, 2733, 2673, 2950, 2632, 2596, 2571, 2546, 2521, 2496, 2482, 2442, 2415, 2363, 2334, 1470, 1446, 1425, 1406, 1387, 1381, 1350, 1331, 1317, 1304, 1273, 1242, 1213, 1192, 1171, 1146, 1117, 1090, 1065, 1034, 997, 985, 964, 947, 927, 893, 862, 845, 804, 787, 650, 627, 588, 486 cm−1; 1H NMR (600 MHz; CDCl3, δ): 1.39 (s, 3H, CH3), 1.46 (s, 3H, CH3), 1.82 (dddd, Jd1 = 6.5 Hz, Jd2 = 7.1 Hz, Jd3 = 13.0 Hz, Jd4 = 13.7 Hz, 1H, C(6)H2), 1.89 (dd, Jd1 = 6.6 Hz, Jd2 = 13.7 Hz, 1H, C(6)H2), 1.90 (ddd, Jd1 = 6.9 Hz, Jd2 = 13.1 Hz, Jd3 = 14.8 Hz, 1H, C(8)H2), 2.11 (dd, Jd1 = 6.4 Hz, Jd2 = 13.4 Hz, 1H, C(2)H2), 2.13 (ddd, Jd1 = 6.5 Hz, Jd2 = 6.9 Hz, Jd3 = 13.3 Hz, 1H, C(7)H2), 2.19 (dd, Jd1 = 7.1 Hz, Jd2 = 13.7 Hz, 1H, C(1)H2), 2.22 (dd, Jd1 = 6.1 Hz, Jd2 = 14.8 Hz, 1H, C(8)H2), 2.30 (ddddd, Jd1 = 6.1 Hz, Jd2 = 6.5 Hz, Jd3 = 13.0 Hz, Jd4 = 13.1 Hz, Jd5 = 13.3 Hz, 1H, C(7)H2), 2.36 (ddd, Jd1 = 6.4 Hz, Jd2 = 13.3 Hz, Jd3 = 13.7 Hz, 1H, C(1)H2), 2.53 (ddd, Jd1 = 7.1 Hz, Jd2 = 13.3 Hz, Jd3 = 13.3 Hz, 1H, C(2)H2), 2.80 (ddd, Jd1 = 5.6 Hz, Jd2 = 7.1 Hz, Jd3 = 9.4 Hz, 1H, C(5a)H), 3.51 (dd, Jd1 = 5.6 Hz, Jd2 = 12.3 Hz, 1H, C(5)H2), 4.19 (dd, Jd1 = 9.4 Hz, Jd2 = 12.3 Hz, 1H, C(5)H2); 13C{1H} NMR (150 MHz; CDCl3, δ): 21.31 (CH3), 24.80 (CH3), 25.79 C(7), 31.34 C(6), 32.53 C(1), 37.38 C(8), 37.42 C(2), 39.48 C(5a), 49.85 C(5), 67.74 C(3), 91.79 C(8a).
3.2.5. Reaction of ((5R(S),6R(S))-2,2-Dimethyl-1-azaspiro[4.4]nonan-6-yl)methanol 1b with PPh3-CBr4
Carbon tetrabromide (1.80 g, 5.53 mmol) and PPh3 (1.45 g, 5.53 mmol) were added to a solution of 1b (0.40 g, 2.21 mmol) in dry CH2Cl2 (8 mL) and the reaction mixture was stirred at room temperature for 12 h to complete the reaction. The progress of reaction was monitored by TLC (SiO2, methanol-ethyl acetate 1:6 mixture; stained with iodine vapor, Rf(3×HBr) = 0.2). After evaporation of the solvent under reduced pressure, the crude residue was purified by column chromatography (SiO2, methanol-ethyl acetate 1:5 mixture) and recrystallized from iso-propanol to give 3×HBr, 0.35 g, yield 65%.
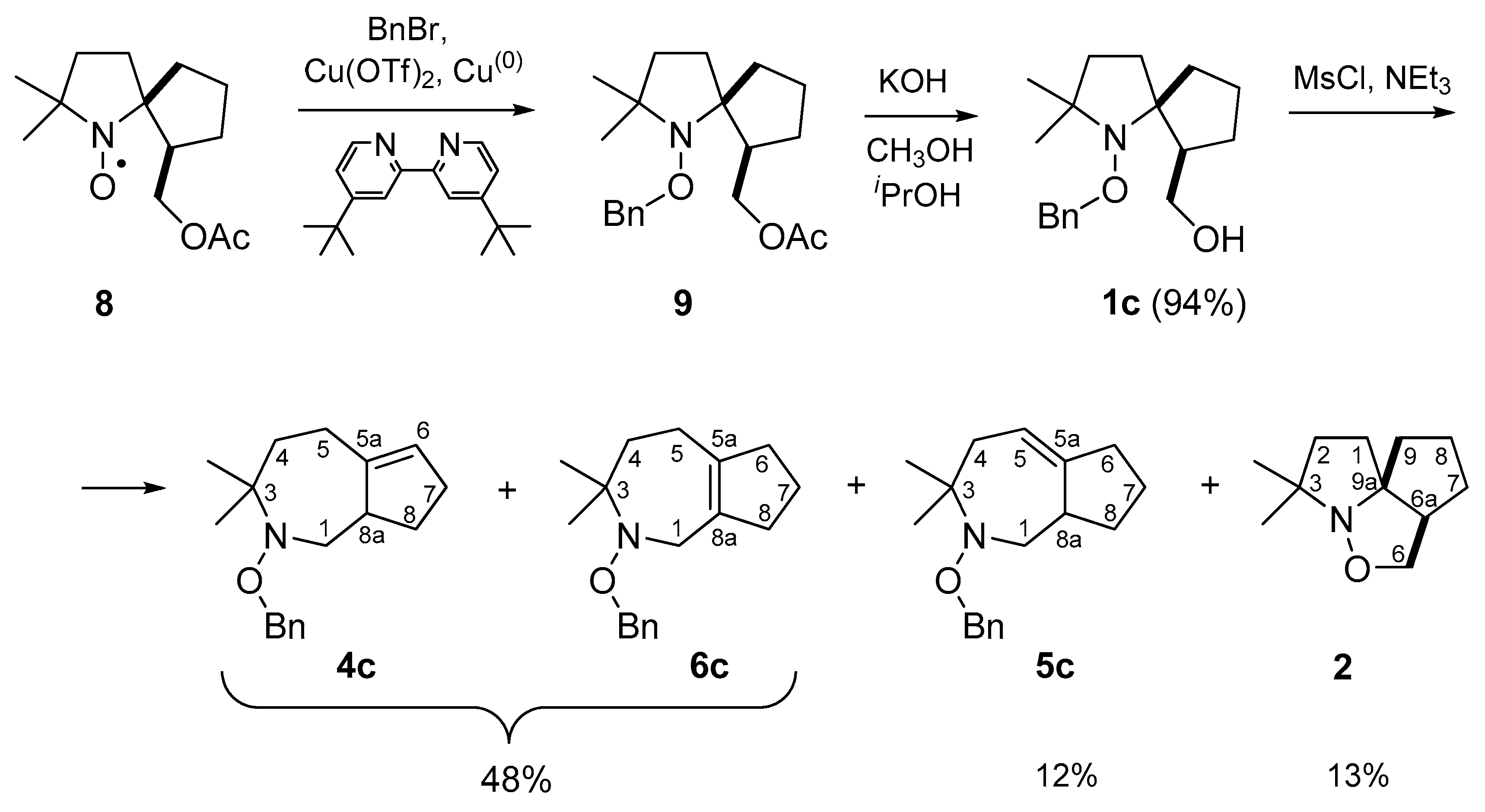
3.2.6. (1-(Benzyloxy)-2,2-dimethyl-1-azaspiro[4.4]nonan-6-yl)-methanol (1c)
An excess of KOH suspension in methanol-
iso-propanol 1:3 mixture (8 mL) was added to a solution of alkoxyamine
9 [
4] (0.342 g, 1.03 mmol) in iso-propanol (25 mL). The reaction mixture was left at room temperature for 12 h to complete the reaction. The progress of reaction was monitored by TLC (SiO
2, hexane-diethyl ether 1:1 mixture; stained with Dragendorff’s reagent, R
f(
1c) = 0.45). Methanol and
iso-propanol were evaporated under reduced pressure. A saturated solution of NaCl (15 mL) was added to the residue, and the resulting mixture was extracted with CHCl
3 (3 × 15 mL). The combined organic extracts were dried with anhydrous Na
2SO
4. After evaporation of the solvent under reduced pressure, the crude residue was purified by column chromatography (SiO
2, hexane-diethyl ether 1:1 mixture) to give
1c.
(1-(Benzyloxy)-2,2-dimethyl-1-azaspiro[4.4]nonan-6-yl)-methanol (1c): 0.28 g, yield 94%. Colorless oil. Elemental analysis: found: C, 74.85; H, 9.46; N, 4.91; calcd. for C18H27NO2: C, 74.70; H, 9.40; N, 4.73%; IR (neat) νmax: 3427, 3089, 3064, 3032, 2958, 2870, 1608, 1497, 1454, 1363, 1317, 1257, 1155, 1082, 1028, 908, 845, 752, 735, 696, 613 cm−1; 1H NMR (500 MHz; CDCl3, δ): 1.21 (s, 3H), 1.31 (s, 3H), 1.40–1.57 (m, 3H), 1.60–1.80 (m, 5H), 1.80–1.90 (m, 2H), 2.30–2.38 (m, 1H), 3.78–3.82 (m, 2H), 4.76 (d, Jd = 10 Hz, 1H), 4.94 (d, Jd = 10 Hz, 1H), 5.56 (br.s, 1H), 7.25–7.36 (m, 5H); 13C{1H} NMR (125 MHz; CDCl3, δ): 22.55, 22.81, 27.15, 29.96, 34.44, 35.99, 36.09, 50.41, 64.47, 66.57, 76.46, 78.81, 127.83, 128.17, 128.39, 137.12.
3.2.7. Reaction of (1-(Benzyloxy)-2,2-dimethyl-1-azaspiro[4.4]nonan-6-yl)-methanol 1c with Methanesulfonyl Chloride
Triethylamine (0.62 g, 6.16 mmol) was added to a solution of 1c (1.018 g, 3.52 mmol) in dry CHCl3 (15 mL) at 0 °C. Then MsCl (0.605 g, 5.29 mmol) was added dropwise to the mixture upon stirring and cooling on an ice bath, and the reaction mass was left at room temperature for 72 h. The reaction was monitored by TLC (SiO2, hexane-diethyl ether 1:2 mixture, stained with Dragendorff’s reagent; Rf(1c) = 0.55). The mixture was concentrated under reduced pressure, the crude residue was subjected to column chromatography (SiO2, hexane-diethyl ether 20:1 mixture, stained with Dragendorff’s reagent; Rf(4c) = 0.4, Rf(5c) = 0.3, and then: hexane-diethyl ether 1:2 mixture, Rf(2) = 0.45) to give 2, 5c, and a mixture of 4c and 6c 10:1 with preparative yields 13, 12, and 48%, respectively. Pure 4c was isolated using preparative HPLC (Zorbax C8 (250 mm × 4.6 mm, i.d., 5 µm); mobile phase: acetonitrile/water (8:2 v/v).
2-(Benzyloxy)-3,3-dimethyl-1,2,3,4,5,7,8,8a-octahydrocyclopenta[c]azepine (4c): Colorless oil. Elemental analysis: C, 79.86; H, 9.21; N, 5.17; calcd. for C18H25NO: C, 79.66; H, 9.28; N, 5.16%; HRMS (EI/DFS) m/z [M]+ calcd. for (C18H25NO)+: 271.1931; found: 271.1932. IR (neat) νmax: 3109, 3090, 3066, 3034, 2931, 2850, 1944, 1647, 1606, 1497, 1454, 1433, 1379, 1362, 1307, 1288, 1265, 1242, 1209, 1176, 1155, 1140, 1126, 1084, 1041, 1030, 997, 958, 931, 910, 870, 847, 733, 696, 665, 648, 617, 600 cm−1; 1H NMR (400 MHz; CDCl3, δ): 1.07 (s, 3H, CH3), 1.25 (s, 3H, CH3), 1.32 (ddd, Jd1 = 3.1 Hz, Jd2 = 7.1 Hz, Jd3 = 14.6 Hz, 1H, C(4)H2), 1.34 (tdd, Jt = 7.5 Hz, Jd1 = 9.0 Hz, Jd2 = 12.6 Hz, 1H, C(8)H2), 1.85–1.95 (m, 1H, C(4)H2), 2.01 (dtd, Jd1 = 4.1 Hz, Jt = 8.0 Hz, Jd2 = 12.6 Hz, 1H, C(8)H2), 2.17–2.26 (m, 2H, C(7)H2), 2.26–2.38 (m, 2H, C(5)H2), 2.82 (dd, Jd1 = 10.7 Hz, Jd2 = 13.2 Hz, 1H, C(1)H2), 2.90–2.98 (m, 1H, C(8a)H), 3.07 (dd, Jd1 = 3.4 Hz, Jd2 = 13.2 Hz, 1H, C(1)H2), 4.64 (d, Jd = 10.8 Hz, 1H, CH2Ph), 4.67 (d, Jd = 10.8 Hz, 1H, CH2Ph), 5.29–5.32 (m, 1H, C(6)H), 7.25–7.29 (m, 1H, Ph), 7.31–7.37 (m, 4H, Ph); 13C{1H} NMR (100 MHz; CDCl3, δ): 19.81 (CH3), 24.27 C(5), 28.87 (CH3), 29.53 C(8), 30.87 C(7), 36.75 C(4), 45.41 C(8a), 57.66 C(1), 61.35 C(3), 75.44 (CH2Ph), 122.95 C(6), 127.48 (Ph), 128.10 (Ph), 128.22 (Ph), 137.73 (Ph), 147.92 C(5a).
2-(Benzyloxy)-3,3-dimethyl-1,2,3,4,6,7,8,8a-octahydrocyclopenta[c]azepine (5c): 0.11 g, yield 12%. Colorless oil. HRMS (EI/DFS) m/z [M]+ calcd. for (C18H25NO)+: 271.1931; found: 271.1927. IR (neat) νmax: 3088, 3063, 3030, 2947, 2897, 2864, 1606, 1587, 1497, 1452, 1433, 1377, 1360, 1331, 1306, 1275, 1221, 1194, 1176, 1155, 1132, 1105, 1082, 1040, 1005, 968, 935, 912, 876, 852, 825, 798, 748, 733, 696, 677, 654, 634, 609, 600, 565, 542, 463; 1H NMR (500 MHz; DMSO-d6, 333 K, δ): 0.98 (s, 3H, CH3), 1.20 (s, 3H, CH3), 1.20–1.28 (m, 1H, C(8)H2), 1.36–1.46 (m, 1H, C(7)H2), 1.60–1.68 (m, 1H, C(7)H2), 1.72–1.82 (m, 1H, C(4)H2), 1.83–1.93 (m, 1H, C(8)H2), 2.23 (br.s, 2H, C(6)H2), 2.44 (br.s, 1H, C(4)H2), 2.68 (m, 1H, C(1)H2), 2.78 (br.s, 1H, C(8a)H), 3.04 (dd, Jd1 = 12.4 Hz, Jd2 = 2.4 Hz, 1H, C(1)H2), 4.58–4.66 (m, 2H, CH2Ph), 5.36–5.42 (m, 1H, C(5)H), 7.24–7.36 (m, 5H, Ph); 13C{1H} NMR (125 MHz; DMSO-d6, 333 K, δ): 18.75 (CH3), 25.12 C(7), 29.57 (CH3), 32.04 C(8), 33.31 C(6), 36.91 (C4), 39.00 C(8a), 53.87 C(1), 59.20 C(3), 74.40 (CH2Ph), 116.72 C(5), 127.34 (Ph), 127.96 (Ph), 128.08 (Ph), 137.57 (Ph), 149.34 C(5a); 15N NMR (51 MHz, HCONH2, DMSO-d6, δ): 185.7.
2-(Benzyloxy)-3,3-dimethyl-1,2,3,4,5,6,7,8-octahydrocyclopenta[c]azepine (6c): (the data from the NMR spectrum of the mixture): 1H NMR (500 MHz; DMSO-d6, 333 K, δ): 1.18 (s, 6H), 1.65–1.69 (m, 2H), 1.75–1.83 (m, 2H), 2.05–2.10 (m, 2H), 2.15–2.40 (m, 4H), 3.50 (s, 2H), 4.63 (br.s., 2H), 7.30–7.42 (m. 5H); 13C{1H} NMR (125 MHz; DMSO-d6, 333 K, δ): 22.45, 24.33, 24.90, 37.66, 37.80, 38.89, 51.87, 61.38, 75.71, 128.09, 128.65, 129.04, 133.04, 138.17, 138.27.
3.2.8. Reaction of (1-(Benzyloxy)-2,2-dimethyl-1-azaspiro[4.4]nonan-6-yl)-methanol 1c with PPh3-CBr4
Carbon tetrabromide (1.21 g, 3.63 mmol) and PPh3 (0.98 g, 3.75 mmol) were added to a solution of 1c (0.35 g, 1.21 mmol) in dry CHCl3 (7 mL) and the reaction mixture was stirred at room temperature for 48 h. The reaction was monitored by TLC (SiO2, hexane-diethyl ether 1:2 mixture). After evaporation of the solvent under reduced pressure, the crude residue was subjected to column chromatography (SiO2, hexane-diethyl ether 20:1 mixture as an eluent) to give a mixture of 4c, 5c, and 6c (0.111 g, yield 34%).
3.2.9. 6-(Hydroxymethyl)-2,2-dimethyl-1-azaspiro[4.4]nonan-1-yl benzoate (1d)
Manganese dioxide (7.47 g, 85 mmol) was added to a solution of nitroxide 1а (0.85 g, 4.29 mmol) in diethyl ether (20 mL). A solution of benzhydrazide (1.16 g, 8.58 mmol) in methanol was added dropwise to the resulting mixture, and the reaction mass was stirred for 30 min. Then manganese dioxide was filtered off through celite, the filtrate was evaporated, and the residue was purified by column chromatography (SiO2, hexane-diethyl ether 1:1 mixture as an eluent, detected under UV lamp and stained with Dragendorff’s reagent, Rf(1d) = 0.45).
6-(Hydroxymethyl)-2,2-dimethyl-1-azaspiro[4.4]nonan-1-yl benzoate (1d): 1.06 g, yield 81%. Yellowish oil. Elemental analysis: C, 70.98; H, 8.41; N, 4.86; calcd. for C18H25NO3: C, 71.26; H, 8.31; N, 4.62%; HRMS (EI/DFS) m/z [M]+ calcd. for (C18H25NO3)+: 303.1829; found: 303.1826. IR (neat) νmax: 3452, 3088, 3064, 3032, 2958, 2875, 1740, 1601, 1583, 1491, 1452, 1410, 1385, 1367, 1315, 1261, 1244, 1176, 1113, 1082, 1063, 1026, 1001, 976, 947, 924, 897, 885, 854, 802, 710, 687, 667, 646, 617 cm−1; NMR (400 MHz; CDCl3, δ): 1.19 (s, 3H), 1.25 (s, 3H), 1.38–1.56 (m, 2H), 1.56–1.69 (m, 3H), 1.70–1.92 (m, 4H), 2.04 (dt, Jd = 12.4 Hz, Jt = 8.5 Hz, 1H), 2.32–2.41 (m, 1H), 3.61 (dd, Jd1 = 6.8 Hz, Jd2 = 12.1 Hz, 1H), 3.80 (dd, Jd1 = 2.5 Hz, Jd2 = 12.1 Hz, 1H), 5.29 (br.s, 1H), 7.39–7.46 (m, 2H), 7.52–7.59 (m, 1H), 7.97–8.03 (m, 2H); 13C{1H} NMR (100 MHz; CDCl3, δ): 21.88, 22.35, 26.05, 27.93, 35.08, 35.41, 35.46, 51.50, 62.32, 67.59, 77.79, 128.51, 128.40, 129.31, 133.17, 166.01.
3.2.10. Reaction of (1-(Benzoyloxy)-2,2-dimethyl-1-azaspiro[4.4]nonan-6-yl)-methanol 1d with PPh3-CBr4
Carbon tetrabromide (0.638 g, 1.92 mmol) and PPh3 (0.50 g, 1.92 mmol) were added to a solution of 1d (0.194 g, 0.64 mmol) in dry CHCl3 (6 mL) and the reaction mixture was stirred at room temperature for 12 h. The reaction was monitored by TLC (SiO2, hexane-diethyl ether 2:1 mixture; detected under UV lamp and stained with Dragendorff’s reagent). After evaporation of the solvent under reduced pressure, the crude residue was subjected to column chromatography (SiO2, from hexane-ethyl acetate 50:1 to hexane-ethyl acetate 25:1 mixture as an eluent) to give 4d, 5d, and 13.
3,3-Dimethyl-1,4,5,7,8,8a-hexahydrocyclopenta[c]azepin-2(3H)-yl benzoate (4d): 0.055 g, yield 30%. Colorless oil. Elemental analysis: C, 75.88; H, 8.25; N, 4.80; calcd. for C18H23NO2: C, 75.76; H, 8.12; N, 4.62%; HRMS (EI/DFS) m/z [M]+ calcd. for (C18H23NO2)+: 285.1723; found: 285.1720. IR (neat) νmax: 3088, 3061, 3037, 2976, 2931, 2850, 1741, 1645, 1601, 1583, 1491, 1450, 1383, 1363, 1313, 1259, 1238, 1176, 1157, 1126, 1084, 1063, 1024, 999, 960, 935, 906, 889, 874, 862, 802, 708, 687, 671, 646 cm−1; 1H NMR (600 MHz; CDCl3, δ): 1.19 (s, 3H, CH3), 1.22 (s, 3H, CH3), 1.34 (dddd, Jd1 = 6.6 Hz, Jd2 = 6.9 Hz, Jd3 = 8.7 Hz, Jd4 = 13.0 Hz, 1H, C(8)H2), 1.46 (ddd, Jd1 = 4.1 Hz, Jd2 = 5.2 Hz, Jd3 = 15.0 Hz, 1H, C(4)H2), 2.04 (dddd, Jd1 = 4.7 Hz, Jd2 = 8.4 Hz, Jd3 = 8.7 Hz, Jd4 = 12.7 Hz, 1H, C(8)H2), 2.07–2.18 (m, 1H, C(4)H2), 2.17–2.29 (m, 2H, C(7)H2), 2.37–2.48 (m, 2H, C(5)H2), 3.06 (dd, Jd1 = 11.0 Hz, Jd2 = 13.0 Hz, 1H, C(1)H2), 3.15–3.29 (m, 2H, C(8a)H + C(1)H2), 5.30–5.32 (m, 1H, C(6)H), 7.39–7.43 (m, 2H, Ph), 7.51–7.55 (m, 1H, Ph), 7.98–8.02 (m, 2H, Ph); 13C{1H} NMR (150 MHz; CDCl3, δ): 20.30 (CH3), 24.23 C(5), 29.29 (CH3), 29.41 C(8), 30.77 C(7), 35.55 C(4), 44.02 C(8a), 59.15 C(1), 62.27 C(3), 123.80 C(6), 128.27 (Ph), 129.23 (Ph), 129.58 (Ph), 132.74 (Ph), 146.74 C(5a), 164.97 (C = O).
3,3-Dimethyl-3,4,6,7,8,8a-hexahydrocyclopenta[c]azepin-2(1H)-yl benzoate (5d): 0.027 g, yield 15%. Colorless oil. HRMS (EI/DFS) m/z [M]+ calcd. for (C18H23NO2)+: 285.1723; found: 285.1725. IR (neat) νmax: 3088, 3061, 3032, 2949, 2931, 2899, 2866, 2854, 1743, 1601, 1583, 1491, 1450, 1435, 1381, 1362, 1331, 1313, 1257, 1248, 1192, 1176, 1130, 1084, 1063, 1024, 1007, 993, 970, 939, 893, 870, 851, 831, 802, 754, 710, 688, 671 cm−1; 1H NMR (500 MHz; DMSO-d6, δ): 1.10 (s, 3H, CH3), 1.16 (s, 3H, CH3), 1.21–1.28 (m, 1H, C(8)H2), 1.38–1.49 (m, 1H, C(7)H2), 1.61–1.71 (m, 1H, C(7)H2), 1.84–1.96 (m, 2H, C(4)H, C(8)H), 2.28 (br.s, 2H, C(6)H2), 2.62 (br.s, 1H, C(4)H), 2.91 (br.s, 1H, C(8a)H), 2.99 (t, Jt = 12.4 Hz, 1H, C(1)H2), 3.12 (dd, Jd1 = 12.4 Hz, Jd2 = 2.0 Hz, 1H, C(1)H2), 5.44–5.51 (m, 1H, C(5)H), 7.52 (t, Jt = 7.7 Hz, 2H, m-Ph), 7.64 (tt, Jt1 = 7.5 Hz, Jt2 = 1.1 Hz, 1H, p-Ph), 7.94 (dd, Jd1 = 8.2 Hz, Jd2 = 1.1 Hz, 2H, o-Ph); 13C{1H} NMR (125 MHz; DMSO-d6, δ): 19.25 (CH3), 25.05 C(7), 29.34 (CH3), 31.83 (C8), 33.36 C(6), 36.62 C(4), 38.61 C(8a), 55.36 C(1), 59.89 C(3), 116.70 C(5), 128.69 (m-Ph), 128.73 (o-Ph), 129.22 (i-Ph), 133.03 (p-Ph), 149.51(C(5a), 163.84 (C=O); 15N NMR (51 MHz, HCONH2, DMSO-d6, δ): 194.4.
6-((Benzoyloxy)methyl)-2,2-dimethyl-1-oxo-1-azaspiro[4.4]nonan-1-oxyl (13): 0.027 g, yield 14%. Yellow crystalline solid, m.p. 68.2 °C with decomposition (hexane). Elemental analysis: C, 71.77; H, 7.97; N, 4.68; calcd. for C18H24NO3: C, 71.50; H, 8.00; N, 4.63%; HRMS (EI/DFS) m/z [M]+ calcd. for (C18H24NO3)+: 302.1751; found: 302.1752. IR (KBr) νmax: 3070, 3063, 2968, 2928, 2873, 2854, 1716, 1601, 1583, 1491, 1454, 1406, 1371, 1360, 1350, 1313, 1282, 1273, 1250, 1203, 1180, 1163, 1130, 1115, 1084, 1072, 1022, 991, 968, 951, 928, 893, 849, 717, 688, 665, 590, 569, 447 cm−1; UV (EtOH) λmax (log ε): 229 (4.06).
3.2.11. Reduction of Nitroxide 13 with Zn in CF3COOH for NMR
A suspension of zinc dust (100 mg) in a solution of nitroxide 13 (0.015 g, 0.050 mmol) in CD3OD (0.4 mL) in a small glass vial was heated to reflux upon vigorous stirring and trifluoroacetic acid (0.1 mL) was added dropwise. The mixture was stirred for 10–15 min, and the solution was transferred into an NMR tube through a pipette tip with a tightly inserted paper filter. The vial was rinsed with a small portion of CDCl3–CD3OD mixture and this solution was filtered into the same NMR tube until the normal NMR sample volume was reached.
(5R(S),6R(S))-6-((Benzoyloxy)methyl)-2,2-dimethyl-1-azaspiro[4.4]nonan-1-ium 2,2,2-trifluoroacetate (14): 1H NMR (400 MHz; CDCl3–CD3OD mixture, δ): 1.47 (s, 3H), 1.50 (s, 3H), 1.71–1.87 (m, 2H), 1.88–2.07 (m, 3H), 2.08–2.24 (m, 2H), 2.44–2.56 (m, 1H), 4.36 (dd, Jd1 = 7.1 Hz, Jd2 = 11.8 Hz, 1H), 4.48 (dd, Jd1 = 6.3 Hz, Jd2 = 11.8 Hz, 1H), 7.40–7.50 (m, 2H), 7.55–7.63 (m, 1H), 7.93–8.03 (m, 2H).
3.2.12. 3,3,4-Trimethyloctahydro-1H-cyclopenta[2,3]azeto[1,2-a]pyrrol-4-ium iodide (15)
Iodomethane (0.23g, 1.64 mmol) was added to a solution of 3 (0.09 g, 0.545 mmol) in dry diethyl ether (2 mL) and the reaction mass was left over 12 h. Then the precipitate formed was filtered off and washed with diethyl ether.
3,3,4-Trimethyloctahydro-1H-cyclopenta[2,3]azeto[1,2-a]pyrrol-4-ium iodide (15): 0.133 g, yield 80%. White crystalline solid, m.p. 187.2–187.4 °C. Elemental analysis: C, 47.19; H, 7.23; N, 4.56; calcd. for C12H22IN: C, 46.91; H, 7.22; N, 4.59%. IR (KBr) νmax: 3007, 2956, 2874, 2833, 2816, 2756, 2600, 2580, 1470, 1443, 1433, 1396, 1383, 1358, 1335, 1315, 1304, 1294, 1275, 1240, 1217, 1196, 1178, 1149, 1138, 1099, 1086, 1068, 1034, 986, 953, 931, 916, 895, 881, 852, 839, 796, 779, 735, 642, 606, 571, 499, 411; 1H NMR (300 MHz; CDCl3, δ): 1.34 (s, 3H), 1.53 (s, 3H), 1.80–2.39 (m, 8H), 2.50 (dd, Jd1 = 5.9 Hz, Jd2 = 16.0 Hz, 1H), 2.60 (ddd, Jd1 = 6.2 Hz, Jd2 = 13.0 Hz, Jd3 = 13.1 Hz, 1H), 2.77–2.89 (m, 1H), 2.94 (c, 3H), 3.52 (dd, Jd1 = 6.2 Hz, Jd2 = 12.6 Hz, 1H), 4.65 (dd, Jd1 = 9.9 Hz, Jd2 = 12.6 Hz, 1H); 13C{1H} NMR (75 MHz; CDCl3, δ): 20.72, 24.74, 26.90, 30.09, 33.16, 33.44, 37.80, 38.72, 40.20, 62.65, 74.37, 99.18.
3.2.13. 2,3,3-Trimethyl-1,2,3,4,5,7,8,8a-octahydrocyclopenta[c]azepine (16)
Wet silver (I) oxide (5.94 mmol) and water (12 mL) were added to salt 15 (0.729 g, 2.37 mmol) and stirred for 12 h. Then the solid residue was filtered off, the filtrate was concentrated in vacuum. The residue was heated on a water bath under reflux until the disappearance of a solid insoluble in diethyl ether, and then was extracted with diethyl ether. After evaporation of the solvent under reduced pressure, the crude residue was purified by column chromatography (alumina, pentane-diethyl ether 6:1 as an eluent) to give 16.
2,3,3-Trimethyl-1,2,3,4,5,7,8,8a-octahydrocyclopenta[c]azepine (16): 0.296 g, yield 70%. Colorless oil. HRMS (EI/DFS) m/z [M]+ calcd. for (C12H21N)+: 179.1669; found: 179.1668. Elemental analysis: C, 80.36; H, 11.82; N, 7.60; calcd. for C12H21N: C, 80.38; H, 11.81; N, 7.81%. IR (neat) νmax: 3466, 3039, 2964, 2945, 2926, 2847, 2798, 2789, 2777, 1649, 1618, 1464, 1454, 1429, 1377, 1362, 1344, 1315, 1288, 1267, 1236, 1205, 1174, 1128, 1109, 1088, 1049, 1024, 1005, 974, 952.7 941, 918, 908, 852, 800, 694, 638, 577, 530, 488. 1H NMR (600 MHz; CDCl3, δ): 0.94 (s, 3H, CH3), 1.09 (dd, Jd1 = 0.4 Hz, Jd2 = 0.7 Hz, 3H, CH3), 1.22 (dddd, Jd1 = 8.8 Hz, Jd2 = 8.9 Hz, Jd3 = 9.2 Hz, Jd4 = 12.5 H, 1H, C(8)H2), 1.32–1.39 (m, 1H, C(4)H2), 1.65–1.71 (m, 1H, C(4)H2), 1.94 (dddd, Jd1 = 5.1 Hz, Jd2 = 5.5 Hz, Jd3 = 7.8 Hz, Jd4 = 12.6 Hz, 1H, C(8)H2), 2.15–2.21 (m, 2H, C(7)H2), 2.23–2.29 (m, 2H, C(5)H2), 2.32 (s, 3H, CH3), 2.35 (dd-quartet, Jd1 = 4.2 Hz, Jd2 = 13.9 Hz, Jquartet = 0.4 Hz, 1H, C(1)H2), 2.55 (dd, Jd1 = 10.7 Hz, Jd2 = 13.9 Hz, 1H, C(1)H2), 2.72–2.79 (m, 1H, C(8a)H), 5.20–5.22 (m, 1H, C(6)H); 13C{1H} NMR (150 MHz; CDCl3, δ): 20.27 (CH3), 24.70 C(5), 28.19 (CH3), 29.35 C(8), 30.83 C(7), 38.72 (NCH3), 39.95 C(4), 48.35 C(8a), 55.96 C(3), 57.34 C(1), 122.48 C(6), 148.90 C(5a).


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}











