
Controversy of Peptide Cyclization from Tripeptide


1
Medical Imaging Research Center, Institute for Radiological Research, Chang Gung Memorial Hospital, Chang Gung University, Taoyuan 333423, Taiwan
2
Department of Nephrology and Clinical Poison Center, Chang Gung Memorial Hospital, Taoyuan 333423, Taiwan
3
Department of Chemistry, National Dong Hwa University, Hualien 974003, Taiwan
4
Department of Life Science, National Dong Hwa University, Hualien 974003, Taiwan
*
Authors to whom correspondence should be addressed.
Molecules 2021, 26(2), 389; https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26020389
Submission received: 16 November 2020
/
Revised: 5 January 2021
/
Accepted: 5 January 2021
/
Published: 13 January 2021
(This article belongs to the Special Issue Cyclic Peptide Analogues and Non-peptide Mimetics)
Abstract
:The present investigation reports an attempt to synthesize naturally occurring α-cyclic tripeptide cyclo(Gly-l-Pro-l-Glu) 1, [cyclo(GPE)], previously isolated from the Ruegeria strain of bacteria with marine sponge Suberites domuncula. Three linear precursors, Boc-GPE(OBn)2, Boc-PE(OBn)G and Boc-E(OBn)GP, were synthesized using a solution phase peptide coupling protocol. Although cyclo(GPE) 1 was our original target, all precursors were dimerized and cyclized at 0 °C with high dilution to form corresponding α-cyclic hexapeptide, cyclo(GPE(OBn))2 7, which was then converted to cyclic hexapeptide cyclo(GPE)2 2. Cyclization at higher temperature induced racemization and gave cyclic tripeptide cyclo(GPDE(OBn)) 9. Structure characteristics of the newly synthesized cyclopeptides were determined using 1H-NMR, 13C-NMR and high-resolution mass spectrometry. The chemical shift values of carbonyls of 2 and 7 are larger than 170 ppm, indicating the formation of a cyclic hexapeptide.
1. Introduction
Marine sponge-derived natural cyclic peptides have generated much interest in recent years due to their privileged structures and persuasive biological activities (therapeutic potential). The surfaces and internal spaces of sponges provide a specific environmental niche that contains a high number of bacteria to exceed those of seawater by two or three orders of magnitude [1]. Moreover, several studies have increased interest in sponge-associated bacteria recently by proving that these bioactive compounds, when isolated from marine sponges, are symbiotically produced microorganisms [2]. Among these cyclic peptides, naturally occurring α-cyclic tripeptides and tetrapeptides are highly constrained because they have a small ring size [3]. Diverse biological activities of natural cyclic peptides, such as antimicrobial, cytotoxic, anti-HIV, nematicidal and anti-inflammatory characteristics [4,5,6,7], increases their importance as targets for synthetic chemists. However, many biologically active cyclic peptides have been isolated [8,9,10,11,12], but not fully characterized [13,14]. Another hurdle is to meet the supply problem and target identification. Synthesis [15,16] is particularly useful for biological applications as well as to confirm the actual structure of natural products and target identification to overcome the hurdle.
It is extremely challenged for successful drug development of these often complex molecules. In many cases, total synthesis of natural products revealed structural incorrectness of the reported natural products [17,18,19]. Previously, we reported the synthesis and X-ray crystallographic characterization of marine cyclic tetrapeptide cyclo(Gly-l-Ser-l-Pro-l-Glu) [20]. Antitumor and antimicrobial activity of some cyclic tetrapeptides and tripeptides were examined [21]. In addition, some α-cyclic tripeptides also synthesized as the inhibitors for HMG-CoA reductase [22]. As a continuation of natural cyclopeptide synthesis, we attempted to synthesize a rare example of naturally occurring marine α-cyclic tripeptide, cyclo(Gly-l-Pro-l-Glu) 1 [8], for applications in vivo as it is more permeable and more resistant to degradation by digestive proteases [23]. When rendered into its linear form, Gly-l-Pro-l-Glu (GPE) is the N-terminal sequence of insulin-like growth factor (IGF-1) [24,25]. There is evidence that the peptide has neuroprotective properties and improves long-term function following brain injury or disease, including hypoxic-ischemic brain injury, chemical toxins and in animal models of Parkinson’s and Alzheimer’s disease [26,27,28]. Furthermore, GPE and GPE analogues are thus of interest as promising neuroprotective agents for the treatment of central nervous system (CNS) diseases [27,28]. Despite having therapeutic significance, several challenging features are still encountered in the synthesis, such as dimerization and racemization of linear natural amino acid sequence, cyclization only with all 3 amide linkages in cis configuration and less conformation to raise the accuracy of molecular docking due to its rigidity [22,29,30,31]. These factors can produce contamination to influence the in vivo ability of formulation/characterization for clinical applications.
To address these problems, this paper explores an attempt to synthesize the reported natural cyclic tripeptide 1 since it possesses a side-chain carbonyl group that may enable further functionalization after cyclization. Although the literature reported natural cyclic tripeptide, the characterization data of our synthesized compound turn out to be cyclic hexapeptide 2. In addition, the cyclization of linear precursor at room temperature (RT) was involved with racemization to give a cyclic d-tripeptide 9.
2. Results and Discussion
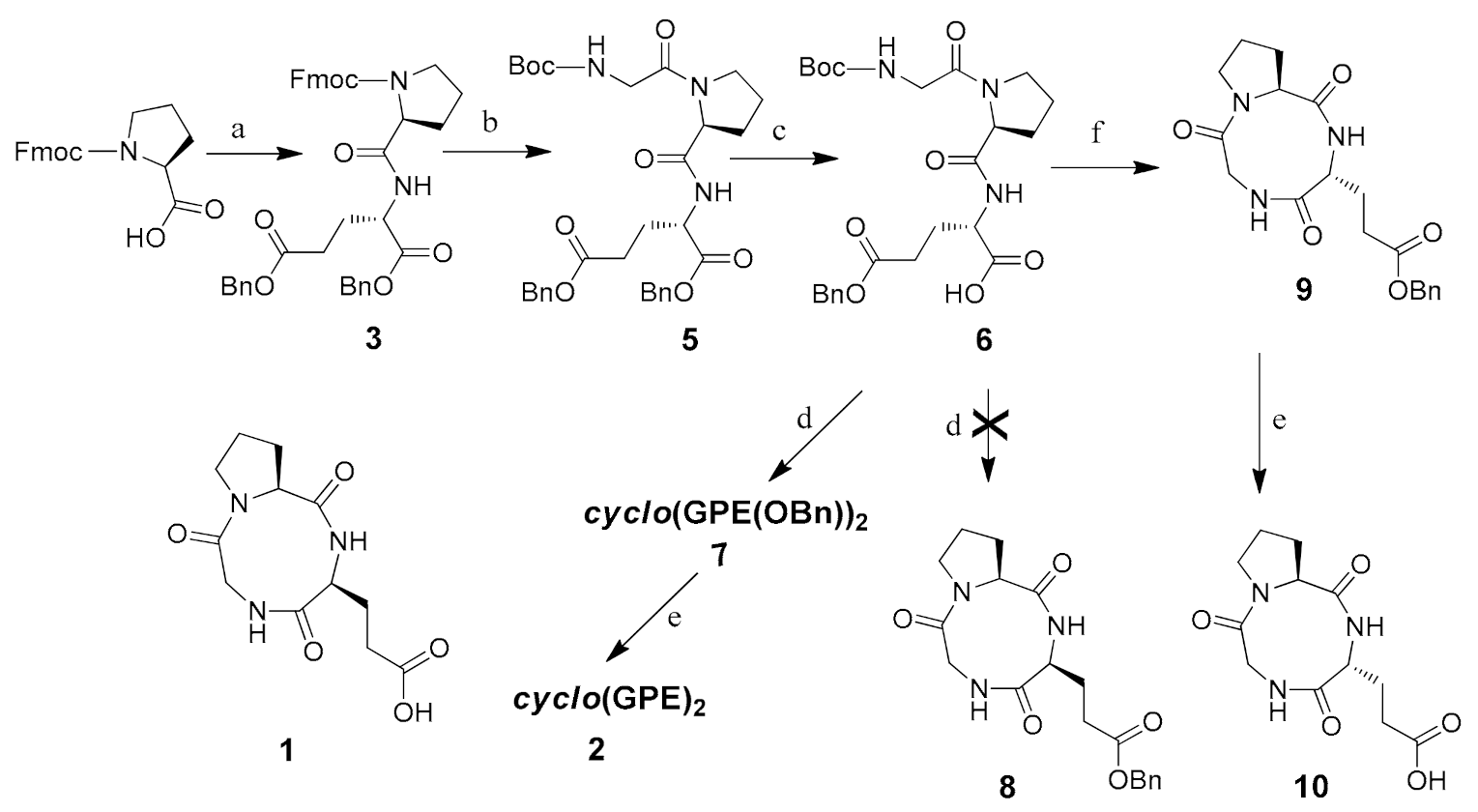
To synthesize the cyclic tripeptide, linear precursor Boc-G-P-E(OBn)2 5 (Scheme 1) was synthesized using standard solution-phase peptide coupling protocols from Fmoc-l-proline. Subsequently, regioselective enzymatic hydrolysis of the α-benzyl ester on glutamate was achieved [32]. The linear precursor was then activated with pentafluorophenol and cyclized in pyridine at high dilution (2 mM, RT). The cyclization ended up with cyclo(GPE(OBn))2 7. Formation of dimer was confirmed by high-resolution mass spectrometry (supplementary Figures S1–S3). Then, deprotection of the benzyl group furnished the synthesis of cyclo(GPE)2 2. The results indicate that GPE sequence is much easier to produce cyclic products, but others are probably more difficult to be cyclized. In contrast, Yudin et al. [33] used Boc-protected β-amino imide as a model substrate to demonstrate that medium-sized rings could be constructed through the collapse of cyclol intermediates derived from the intramolecular cyclization of β-amino imides upon heating to 50 °C for 4 h. We also monitored the cyclization step by using RP-HPLC to analyze the compounds. Pure cyclo(GPE)2OBn showed peak at retention time 3.78, and the reaction mixture showed one peak correspond to cyclo(GPE)2OBn and another was salt of TFA.
Analysis of spectroscopic data of reported natural product 1 and synthetic cyclic hexapeptide 2, especially 1H and 13C-NMR data, creates a doubt about the correctness of the natural product structure. Table 1 shows the recorded carbonyl peaks for reported cyclic peptides in 13C-NMR. All the ring carbonyl peak values of reported cyclic tripeptides are lower than 170 ppm [34,35,36]. Carbonyl peak of proline in cyclo(l-Pro-l-BnG-d-Pro) [34], cyclo(Pro-Pro-BnG) [35], cyclo(l-Pro-l-Pro-d-Pro) [35] and cyclo(l-Pro-l-Pro-l-Pro) [36] showed peaks between δ 167 and 169 ppm. Glycine carbonyl [36], just like proline, also appeared in the range around δ 166 ppm.
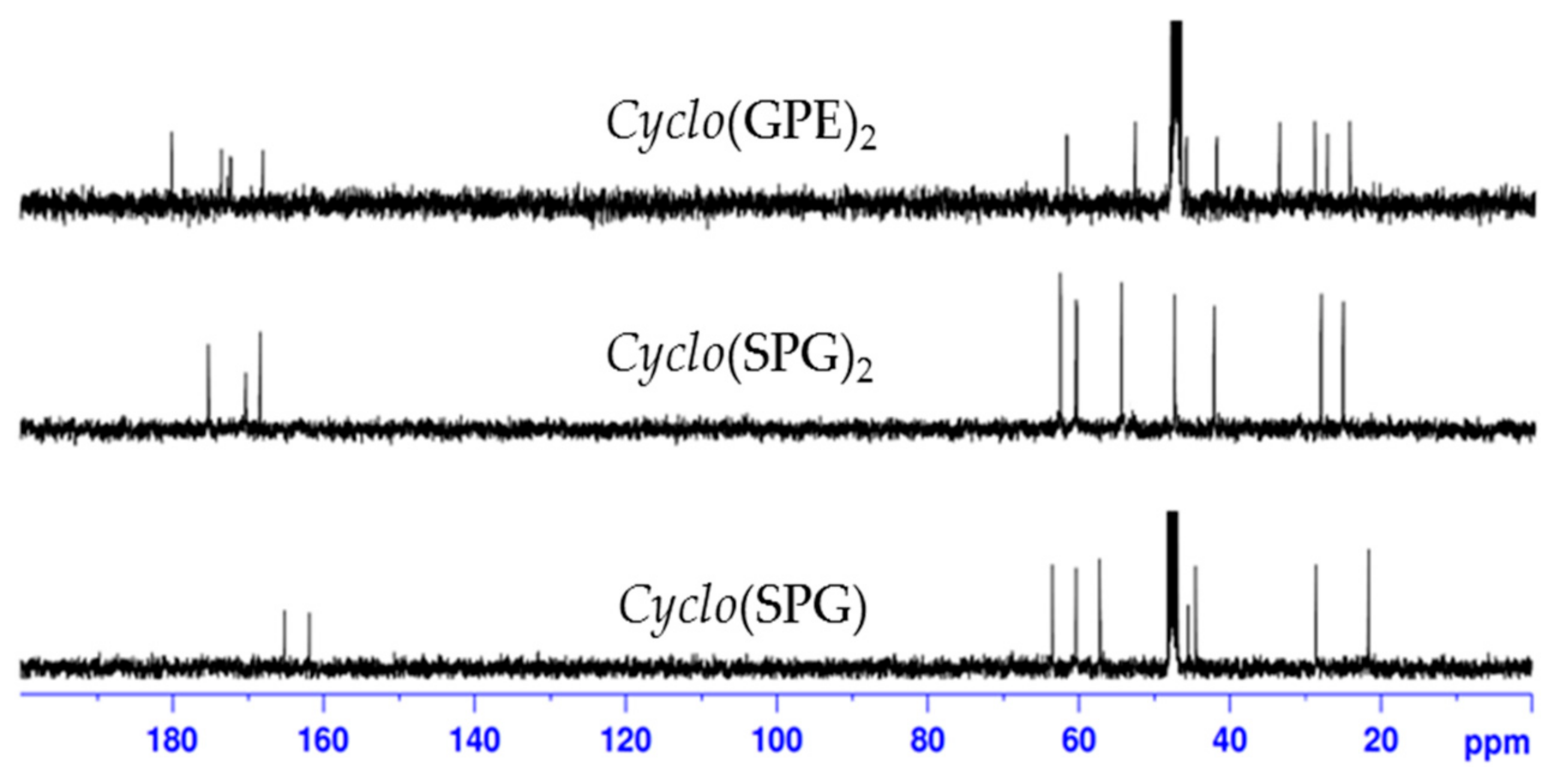
In contrast, in the case of reported natural product 1, ring carbonyl peak values for proline and glycine are much higher (δ 174.8 and 169.8 ppm) than for other reported cyclic tripeptides [34,35,36] and closer to our synthetic cyclic hexapeptide cyclo(GPE)2 2 (δ 173.8 and 169.6 ppm). Table 2 shows complete comparison of 13C chemical shift values of cyclo(GPE) 1 [8] and cyclo(GPE)2 2. To verify the spectra of cyclic monomer and dimer containing same amino acid sequence, we synthesized cyclo (serine-proline-glycine) (SPG) and cyclo(SPG)2. Compared with natural products, cyclo(SPG) containing l-serine instated l-glutamic acid in its amino acid sequence. Both the monomer and dimer showed completely different 1H and 13C-NMR spectra (unpublished work). 13C peak values of cyclo(SPG)2 are also quite similar to our synthesized hexapeptide cyclo(GPE)2 2 (Figure 1). We used a 3:1 CD3OD/D2O ratio for NMR, which was the cause of little variation of NMR peaks.
Cyclization of fully deprotected linear G-P-E(OBn)-OH in DMF at various dilution conditions with different coupling reagent combinations such as PyBOP-HOBt, HATU-HOAt and BOP-HOBt also gave dimeric compound. After failure to acquire a monomeric compound, we prepared two other linear precursors, Boc-PE(OBn)G and Boc-E(OBn)GP. These two compounds were cyclized using the same procedure and producing the same dimer 2 with lower cyclization yield. We also tried microwave assisted cyclization of linear precursor with a series of coupling reagents in DMF as well as different activated ester compounds in pyridine with variation of temperature, power and dilution failed to give cyclic tripeptide 1.
To identify the specific functional groups in our synthesized compound, an infrared spectrum was also useful to check the carbonyl position. Amide stretching of small cyclic peptides was around 1675 cm−1 and usually shifted to 1635 cm−1 for larger rings. Unfortunately, the reported cyclo(GPE) did not provide this information [8]. It is well known that cyclic tripeptide can be formed only when all peptide bonds are in crown form with cis-linkage [37,38]. Small ring size and crown structure will bring all the carbonyls closer and parallel to each other, which will increase the shielding effect of one carbonyl to another. As a result, carbonyls on the cyclic tripeptide ring shift upfield (lower than 170 ppm in 13C-NMR). Conversely, carbonyl peaks of cyclic hexapeptide usually appeared in the downfield region (higher than 170 ppm), because flexible cyclic hexapeptide contains both cis and trans links [39]. Their cis trans combination keeps ring carbonyls apart from each other, which results in a less shielding effect. Carbonyl peaks of reported natural product 1 along with our synthetic cyclic hexapeptide cyclo(GPE)2 2 showed similar peak patterns to previously synthesized [40] and isolated cyclic hexapeptides [41,42,43].
The 1H-NMR of synthesized cyclic hexapeptide also showed a similar splitting pattern to reported natural product 1 in the same duterum solvent (Table 3). Only one set of GPE protons was observed for synthetic cyclo(GPE)2. All α-hydrogens of cyclic hexapeptide showed the same splitting (Gly dd, Pro dd, Glu dd) and coupling constants as reported natural product 1. In addition to splitting, chemical shift values of α-hydrogens (Gly δ 4.33, 4.20, Pro δ 4.51, Glu δ 4.64) were also identical to the reported data. High-resolution mass spectra show a mass ion peak at 566.2349 corresponding to cyclo(GPE)2 and a peak at 283 corresponding to cyclo(GPE). In addition, our cyclic hexapeptide ([α]28D −33 (c 0.1, MeOH)) also has similar optical rotation as reported ([α]25D −27.8 (c 0.0010, MeOH)) [8]. These results demonstrated that reported natural product 1 is perhaps a cyclic hexapeptide.
As shown in Scheme 1, droping the α-pentafluorophenol ester of G-P-E(OBn)-OH to pyridine at 50 °C resulted in racemization to form cyclo(GPDE(OBn)) 9 (27%). Removal of the benzyl protecting group on glutamate generated a cyclic tripeptide cyclo(Gly-l-Pro-d-Glu) 10. The analysis of 13C-NMR (Figure 2) and ESIMS data (284.1241 m/z) indicated that the synthesized cyclic tripeptide 10 was not closely related to reported natural product 1 or synthesized cyclic hexapeptide 2. Their 1H-NMR data are shown in Table 4. As compared with the optical rotation, our synthesized cyclic tripeptide 10 has a distinct value ([α]24D −150 (c 0.01, MeOH)), which is different to reported natural product 1 ([α]25D −27.8 (c 0.0010, MeOH)) or synthesized compound 2 ([α]28D −33 (c 0.1, MeOH)). Our results suggest that this cyclic tripeptide is cyclo(Gly-l-Pro-d-Glu), cyclo(GPDE) 10, not cyclo(Gly-l-Pro-l-Glu) 1. These results also point out that GPE sequence is much easier to cycle than other precursors.
The antibiotic activity of natural product 1 was reported [8]. To date, we have not found any cyclic tripeptide with antibiotic activity [21,22]. We have shown that the structure–activity relationship cannot be neglected. The size of the cyclic peptide is very critical. Previous studies have disclosed the importance of ring size between 4 and 14 residues on the antimicrobial activity [44,45], such as cyclic hexapeptides [40,46,47,48].
3. Materials and Methods
All reactions involving air or moisture sensitive reagents were carried out under a dry argon or nitrogen atmosphere using freshly distilled solvents. Pyridine and dioxane were distilled from CaH2. The NMR spectra were recorded on Bruker DRX 400 (1H at 400.13 MHz, and 13C at 100.03 MHz). Chemical shifts (δ) are reported in ppm relatively to the residual solvent peak (CHCl3, δ = 7.26, 13CDCl3, δ = 77.0; CD2HOD, δ = 3.34, 13CD3OD, δ = 49.0; D2O, δ = 4.47. MALDI TOF was performed on a Bruker Autoflex MALDI-TOF mass spectrometer (Bruker Daltonics, Breman, Germany). High-resolution electrospray ionization mass spectrometry (ESI-MS) was performed on a Shimadzu-LCMS-IT-TOF mass spectrometer (Shumadzu, Kyoto, Japan). Infrared spectra were recorded on a Perkin Elmer Spectrum one FT-IR spectrometer (PerkinElmer, Shelton, CT, USA) using KBr pellets (4000–400 cm−1).
3.1. Fmoc-l-Prolyl-l-Glutamyl Dibenzyl Ester (3)
To a solution of dibenzyl L-glutamyl p-toluenesulphonate (8.88 g, 17.79 mmol) and triethylamine (3 g, 29.65 mmol) in dichloromethane (20 mL) in 50 mL round bottom flask Fmoc-l-proline (4 g, 11.86 mmol), DCC (2447 mg, 11.86 mmol) and HOBt (1601 mg, 11.86 mmol) was added at 0 °C. The reaction mixture was stirred for 2 h at 0 °C and 17 h at room temperature. The reaction was monitored by TLC and the resultant white mixture was filtered to remove 1,3-dicyclohexylurea (DCU). After evaporation of the filtrate, the resultant mass was dissolved in ethyl acetate and washed successively with 5% citric acid solution, water, 5% sodium bicarbonate solution and water. The organic extract was concentrated and purified by column chromatography using hexane/ethyl acetate (70:30) as eluent to obtain compound 3 as a white solid (6 g, 78%). [α]22D = −56 (c 0.1, CHCl3). 1H-NMR (CDCl3, 400 MHz) δ 1.88–1.99 (m, 4H), 2.25–2.40 (m, 4H), 3.46–3.56 (m, 2H), 4.24–4.34 (m, 3H), 4.41–4.46 (m, 2H), 4.61–4.69 (m, 1H), 5.04–5.17 (m, 4H), 7.30–7.39 (m, 14H), 7.59 (s, 2H), 7.75–7.77 (d, 2H, J = 7.16 Hz). 13C-NMR (CDCl3, 100 MHz) δ 27.2, 28.5, 30.1, 34.0, 47.1, 47.3, 51.8, 60.4, 66.5, 67.3, 67.7, 120.0, 125.1, 125.2, 127.1, 127.7, 128.2, 128.3, 128.5, 128.5, 128.6, 135.3, 135.8, 143.8, 144.0, 171.4, 171.7, 172.5. IR (KBr) 3326, 3065, 2952, 1736, 1704, 1532, 1451, 1418, 1353, 1262, 1169, 1119, 1089, 988, 758, 740, 698, 621 cm−1. MALDI TOF: m/z calcd. for C39H38N2O7Na [M + Na]+ 669.257; found 669.170.
3.2. l-Prolyl-l-Glutamyl Dibenzyl Ester (4)
To a solution of Fmoc-l-prolyl-l-glutamyl dibenzyl ester 3 (6 g, 9.28 mmol) in dichloromethane (16 mL) in a round bottom flask piperidine was added (4 mL). The mixture was stirred for 2 h. The reaction was monitored by TLC which showed the complete disappearance of starting material. The reaction mixture was concentrated and purified by column chromatography using DCM/MeOH (97:3) as eluent to obtain compound 4 as a pale yellow liquid (3.7 g, 94%). 1H-NMR (CDCl3, 400 MHz) δ 1.65–1.70 (m, 4H), 1.85–1.88 (m, 1H), 2.00–2.10 (m, 2H), 2.29–2.41 (m, 3H), 2.87–2.99 (m, 2H), 3.69–3.73 (m, 1H), 4.62–4.64 (m, 1H), 5.00–5.14 (m, 4H), 7.32 (s, 10H), 8.13–8.15 (d, 1H, J = 8.48 Hz). 13C-NMR (CDCl3, 100 MHz) δ 26.2, 27.6, 30.3, 30.9, 47.3, 51.1, 60.5, 66.5, 67.2, 128.2, 128.3, 128.4, 128.6, 128.6, 135.3, 135.8, 171.7, 172.3, 175.5. MALDI TOF: m/z calcd. for C24H29N2O5 [M + H]+ 425.207; found 425.267.
3.3. Boc-Glycyl-l-Prolyl-l-Glutamyl Dibenzyl Ester (5)
Dichloromethane (20 mL) and triethylamine (953 mg, 9.43 mmol) were taken in a round bottom flask and cooled at 0 °C. To this, l-prolyl-l-glutamyl dibenzyl ester 4 (2 g, 4.71 mmol) was added and stirred for 5 min. Boc-glycine (825 mg, 4.71 mmol), DCC (972 mg, 4.71 mmol) and HOBt (636 mg, 4.71 mmol) were added. The reaction mixture was stirred 2 h at 0 °C and 17 h at rt. The reaction was monitored by TLC which showed the complete disappearance of both starting materials. The resultant white mixture was filtered to remove 1,3-dicyclohexyleurea (DCU). Filtrate was dried, and residue was dissolved in ethyl acetate. The organic layer was washed with 5% aq. citric acid solution followed by water and then with 5% aq. NaHCO3 solution followed by water. The organic layer was dried over Na2SO4, and the mixture was purified by column chromatography using hexane/ethyl acetate (65:35) as eluent to obtain compound 5 as colorless liquid. (2.25 g, 82%). [α]22D = −66 (c 0.1, CHCl3). 1H-NMR (CDCl3, 400 MHz) δ 1.4 (s, 9H), 1.95–2.06 (m, 4H), 1.97–2.04 (m, 2H), 2.17–2.24 (m, 2H), 2.36–2.41 (m, 2H), 3.31–3.37 (m, 1H), 3.44–3.49 (m, 1H), 3.82 (dd, 1H, J = 3.92 Hz, J = 17.2 Hz), 3.96 (dd, 1H, J = 4.5 Hz, J = 17.4 Hz), 4.49–4.53 (m, 1H), 4.54–4.58 (m, 1H), 5.06–5.16 (m, 4H), 5.25–5.26 (d, 1H, J = 1.76 Hz), 5.47 (s, 1H), 7.26–7.38 (m, 10H). 13C-NMR (CDCl3, 100 MHz) δ 24.8, 26.9, 27.9, 28.4, 30.2, 43.1, 46.3, 51.9, 60.1, 66.5, 67.2, 79.7, 135.3, 135.8, 155.8, 168.5, 171.2, 171.4, 172.7. IR (KBr) 3316, 3034, 2977, 1733, 1653, 1528, 1455, 1391, 1367, 1251, 1168, 1057, 1029, 971, 866, 751, 699, 581 cm−1. HRMS (ESI): m/z calcd. for C31H40N3O8 [M + H]+ 582.2810; found 582.2814.
3.4. Boc-Glycyl-l-Prolyl-l-Glutamic Acid γ-Benzyl Ester (6)
To a solution of 0.1 M, pH 7 phosphate buffer (32 mL) containing B. subtilis protease (Sigma type–VIII) (10 mg) in a 100 mL round bottom flask a solution of Boc-glycyl-l-prolyl-l-glutamyl dibenzyl ester 5 (1 g, 1.72 mmol) in acetone was added drop by drop (8 mL). The reaction mixture was stirred at 35 °C overnight. The reaction was monitored by TLC which showed the complete disappearance of the starting material. Then, the pH of the solution was basified to 8 and unchanged ester was extracted with ethyl acetate. The aqueous layer was acidified to pH 2, centrifuged to remove enzyme and extracted with ethyl acetate. The resultant organic layer was dried over Na2SO4, concentrated and purified by column chromatography using DCM/MeOH (97:3) as eluent to obtain compound 6 as a white solid. (727 mg, 86%). [α]22D = −64 (c 0.1, CHCl3). 1H-NMR (CDCl3, 400 MHz) δ 1.41 (s, 9H), 1.88–1.93 (m, 2H), 2.16–2.25 (m, 2H), 2.37–2.50 (m, 2H), 3.39–3.41 (m, 1H), 3.51–3.55 (m, 1H), 3.84 (dd, 1H, J = 3.84 Hz, J = 17.28 Hz), 4.00 (dd, 1H, J = 5.64 Hz, J = 17.48 Hz), 4.48–4.53 (m, 2H), 5.07 (s, 2H), 5.65 (s, 1H), 7.28–7.33 (m, 5H), 7.46–7.48 (d, 1H, J = 7.56 Hz), 8.20 (br s, 1H). 13C-NMR (CDCl3, 100 MHz) δ 24.8, 26.8, 28.3, 30.3, 43.0, 46.5, 51.9, 53.5, 60.4, 66.5, 79.9, 128.2, 128.3, 128.6, 135.6, 135.8, 156.1, 169.2, 171.6, 173.2, 173.6. IR (KBr) 3327, 2978, 1731, 1652, 1531, 1454, 1392, 1367, 1252, 1167, 1057, 865, 749, 699 cm−1. HRMS (ESI): m/z calcd. for C24H33N3O8 [M]+ 491.2268; found 491.2268.
3.5. Cyclo(Glycyl-l-Prolyl-l-Glutamyl(OBn))2 (7)
Boc-glycyl-l-prolyl-l-glutamyl γ-benzyl ester α-acid 6 (491.53 mg, 1 mmol) was added to a 10 (mL) round bottom flask containing DCM (2 mL) and cooled at 0 °C. Pentafluorophenol (202 mg, 1.1 mmol) and DCC (206 mg, 1 mmol) were added. The reaction mixture was stirred for 1 h at 0 °C and 22 h at rt. The solvent was evaporated, and the residue was dissolved in ethyl acetate and filtered to remove 1,3 dicyclohexyleurea (DCU). The filtrate was evaporated to obtain Boc-glycyl-l-prolyl-l-glutamyl γ-benzyl ester α-pentafluorophenol ester. Boc-glycyl-l-prolyl-l-glutamyl γ-benzyl ester α-pentafluorophenol ester (658 mg, 1 mmol) was taken in a (25 mL) round bottom flask and cooled to 0 °C. TFA/DCM (1:1) (4 mL) was added and stirred 1 h at 0 °C. The solvents were evaporated and triturated with ether followed by decanting to remove free pentafluorophenol. The residue was dried in vacuum, which was used directly in the ensuing cyclization procedure. Glycyl-l-prolyl-l-glutamyl γ-benzyl ester α-pentafluorophenol ester (492 mg, 1 mmol) was dissolved in dioxane (20 mL). This solution was added dropwise with efficient stirring to a 1 L round bottom flask containing 500 mL pyridine. Addition was completed after 6 h. After stirring over 36 h at rt, the solvent was distilled off. Residue was dissolved in DCM. The organic layer was washed with 1 N HCl solution, followed by water and then with 5% sodium bicarbonate solution, followed by water. The organic layer was dried over sodium sulphate, and the mixture was purified by column chromatography using DCM/MeOH (95:5) as eluent to obtain compound 7 as a white solid. (110 mg, 30%). [α]22D = −46 (c 0.5, CHCl3). 1H-NMR (CDCl3, 400 MHz) δ 1.93–1.96 (m, 8H), 2.22–2.29 (m, 4H), 2.38–2.45 (m, 4H), 3.44–3.46 (m, 2H), 3.75–3.81 (m, 4H), 4.20–4.21 (d, 1H, J = 4.72 Hz), 4.24–4.25 (d, 1H, J = 4.96 Hz), 4.33 (dd, 2H, J = 4.04 Hz), 4.44–4.49 (m, 2H), 5.07 (s, 4H), 7.13–7.15 (d, 2H, J = 8.24 Hz), 7.29–7.34 (m, 10H), 7.73 (s, 2H). 13C-NMR (CDCl3, 100 MHz) δ 24.9, 26.9, 29.7, 30.6, 42.9, 46.9, 52.1, 61.4, 66.5, 128.2, 128.3, 128.4, 128.6, 135.8, 168.2, 171.5, 171.8, 173.2. IR (KBr) 3329, 3065, 2951, 1732, 1647, 1516, 1454, 1386, 1325, 1260, 1165, 1004, 738, 699, 597 cm−1. ESI: m/z calcd. for C38H47N6O10 [M + H]+ 747.33; found 747.27.
3.6. Cyclo(Glycyl-l-Prolyl-l-Glutamyl)2 (2)
Cyclo(glycyl-l-prolyl-l-glutamyl(OBn))27 (200 mg, 0.27 mmol) was dissolved in 4 mL of MeOH in a 10 mL round bottom flask and 20% palladium hydroxide on carbon was added under nitrogen. The vessel was puged three times with nitrogen and three times with hydrogen, and the reaction mixture was then stirred for 2 h under hydrogen at atmospheric pressure. The catalyst was removed by filtration, and the filtrate was evaporated to obtain compound 2 as a white solid. (144 mg, 95%). [α]28D = −33 (c 0.1, MeOH). 1H-NMR (CD3OD, D2O, 400 MHz) δ 2.13–2.21 (m, 6H), 2.27–2.38 (m, 6H), 2.41–2.53 (m, 4H), 3.71–3.77 (m, 2H), 3.94–3.99 (m, 2H), 4.2 (d, 2H, J = 17.32 Hz), 4.33 (d, 2H, J = 17.56 Hz), 4.51 (dd, 2H, J = 8.72 Hz, J = 6.12 Hz), 4.64 (dd, 2H, J = 10.28 Hz, J = 4.08 Hz). 13C-NMR (CD3OD, D2O, 400 MHz) δ 26.1, 29.0, 30.7, 35.4, 43.7, 47.7, 54.5, 63.5, 169.6, 173.8, 175.0, 181.6. IR (KBr) 3405, 2944, 1635, 1564, 1404, 1328, 1189, 1111, 1050, 919, 599 cm−1. HRMS (ESI): m/z calcd. for C24H34N6O10 [M]+ 566.2336; found 566.2349.
3.7. Cyclo(Glycyl-l-Prolyl-d-Glutamyl) (10)
Boc-glycyl-l-prolyl-l-glutamyl γ-benzyl ester α-acid 6 (100 mg, 0.2 mmol) was added to a 10 (mL) round bottom flask containing DCM (2 mL), pentafluorophenol (41.5 mg, 0.22 mmol) and DCC (41.3 mg, 0.3 mmol). Reaction mixture was stirred for 1.5 h at rt. The solvent was evaporated, and the residue was dissolved in ethyl acetate and filtered to remove 1,3 dicyclohexyleurea (DCU). The filtrate was evaporated to obtain Boc-glycyl-l-prolyl-l-glutamyl γ-benzyl ester α-pentafluorophenol ester. Boc-glycyl-l-prolyl-l-glutamyl γ-benzyl ester α-pentafluorophenol ester (131.5 mg, 0.2 mmol) was taken in TFA/DCM (1:1) (2 mL) and stirred for 1 h at 0 °C. The solvents were evaporated to remove TFA. The residue was dried in vacuum, which was used directly in the ensuing cyclization procedure. The TFA salt of glycyl-l-prolyl-l-glutamyl γ-benzyl ester α-pentafluorophenol ester (111.5 mg, 0.2 mmol) was dissolved in 1,4-dioxane. This solution was added dropwise with efficient stirring to a 1 L round bottom flask containing 500 mL pyridine at 50 °C. After dropping was completed, the temperature was lowered to rt and stirring over 60 h at rt, the solvent was distilled off. The residue mass was dissolved in DCM, hexane, ethyl ester and washed with ethyl acetate to obtain cyclo(glycyl-l-prolyl-d-glutamyl(OBn)) (20 mg, 27%).
Cyclo(glycyl-l-prolyl-d-glutamyl(OBn)) (20 mg, 0.053 mmol) was dissolved in 2 mL MeOH and DMF in a 10 mL round bottom flask, and palladium hydroxide on carbon was added under nitrogen. The reaction mixture was then stirred for 2 h. The catalyst was removed by filtration, and the filtrate was evaporated to obtain a white solid compound (10.5 mg, 70%). [α]24D = −150 (c 0.01, MeOH). 1H-NMR (D2O, 400 MHz) δ 2.06–2.18 (m, 3H), 2.19–2.24 (m, 2H), 2.32–2.42 (m, 2H), 2.64–2.70 (m, 1H), 3.35–3.59 (m, 1H), 3.67–3.72 (m, 1H), 3.79 (t, 1H, J = 6.4 Hz), 4.10 (dd, 1H, J = 17.1 Hz, 3.3 Hz), 4.17 (t, dd, 1H, J = 4.6 Hz), 4.24 (dd, 1H, J = 17.4 Hz, 7.6 Hz). 13C-NMR (D2O, 100 MHz) δ 22.1, 26.4, 27.8, 32.0, 45.0, 45.6, 57.2, 57.7, 161.4, 164.9, 174.4. IR (KBr) 3375, 2927, 2328, 2858, 1652, 1430, 1314, 1151, 1097, 985, 874, 751, 631 cm−1. HRMS(ESI): m/z calcd. for C12H18N3O5 [M + H]+ 284.1242; found 284.1242.
4. Conclusions
In summary, an α-cyclic hexapeptide cyclo(GPE)2 2 was synthesized via cyclization of fully deprotected linear precursor with a series of coupling reagents in DMF. Additionally, we were able to obtain cyclo(GPDE) with much synthetic effort. Even though the characterization data of the reported natural product and our synthetic cyclo(GPE)2 indicate that the reported compound may be a dimer of cyclo(GPE), this strategy still provided a way to synthesize cyclic peptide of varying ring size. In light of the cyclic peptide synthesis using a solution phase method, it can also provide opportunities to explore the synthesis of cyclic products with various functionalities. The unusual antibiotic activity of natural product 1 raised our suspicion as a cyclic tripeptide. The characterization data of the reported natural product and our synthetic cyclo(GPE)2 also indicate that the reported compound may be a dimer of cyclo(GPE).
Supplementary Materials
The following are available online, Figure S1: 1H NMR spectrum of cyclo(Gly-l-Pro-l-Glu(OBn))2 7 in CDCl3, Figure S2: 13C NMR spectrum of cyclo(Gly-l-Pro-l-Glu(OBn))2 7 in CDCl3, Figure S3: HRMS(ESI) spectrum of cyclo(Gly-l-Pro-l-Glu(OBn))2 7, Figure S4: 1H NMR spectrum of cyclo(Gly-l-Pro-l-Glu)2 2 in CD3OD/D2O, Figure S5: 13C NMR spectrum of cyclo(Gly-l-Pro-l-Glu)2 2 in CD3OD/D2O, Figure S6: IR spectrum of cyclo(Gly-l-Pro-l-Glu)2 2, Figure S7: HRMS(ESI) spectrum of cyclo(Gly-l-Pro-l-Glu)2 2, Figure S8: 1H NMR spectrum of cyclo(Gly-l-Pro-d-Glu(Bn)) 9 in CDCl3, Figure S9: 13C NMR spectrum of cyclo(Gly-l-Pro-d-Glu(Bn)) 9 in CDCl3, Figure S10: IR spectrum of cyclo(Gly-l-Pro-d-Glu(Bn)) 9, Figure S11: HRMS(ESI) spectrum of cyclo(Gly-l-Pro-d-Glu(Bn)) 9, Figure S12: 1H NMR spectrum of cyclo(Gly-l-Pro-d-Glu) 10 in D2O, Figure S13: 13C NMR spectrum of cyclo(Gly-l-Pro-d-Glu) 10 in D2O, Figure S14: IR spectrum of cyclo(Gly-l-Pro-d-Glu) 10, Figure S15: HRMS(ESI) spectrum of cyclo(Gly-l-Pro-d-Glu) 10.
Author Contributions
The listed authors contributed to this work as described in the following: conceived and designed the experiments, D.-F.T.; performed the experiments, S.C. and C.-W.W.; analyzed the data, S.C., C.-W.W. and C.-Y.L.; wrote the paper, S.C., C.-Y.L. and D.-F.T. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Ministry of Science and Technology, grant number 92-2751-B-259-001-Y, 104-2113-M-259-002, 107-2221-E-182-022, 109-2221-E-182-008-MY3 and Chang Gung Memorial Hospital, Linkou, Taiwan, grant number CMRPD1K0101.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are availiable on request from corresponding authors. The data are not publicly available due to confidentiality due diligence.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compound are not available from the authors.
References
- Friedrich, A.B.; Fischer, I.; Proksch, P.; Hacker, J.; Hentschel, U. Temporal variation of the microbial community associated with the mediterranean sponge Aplysina aerophoba. FEMS Microbiol. Ecol. 2001, 38, 105–113. [Google Scholar] [CrossRef]
- Proksch, P.; Edrada, R.A.; Ebel, R. Drugs from the seas–current status and microbiological implications. Appl. Microbiol. Biotechnol. 2002, 59, 125–134. [Google Scholar] [PubMed]
- Li, L.; Zhan, H.; Duan, P.; Liao, J.; Quan, J.; Hu, Y.; Chen, Z.; Zhu, J.; Liu, M.; Wu, Y.-D.; et al. Self-assembling nanotubes consisting of rigid cyclic γ–peptides. Adv. Funct. Mater. 2012, 22, 3051–3056. [Google Scholar] [CrossRef]
- Zainuddin, E.N.; Jansen, R.; Nimtz, M.; Wray, V.; Preisitsch, M.; Lalk, M.; Mundt, S.; Lyngbyazothrins, A.-D. antimicrobial cyclic undecapeptides from the cultured cyanobacterium Lyngbya sp. J. Nat. Prod. 2009, 72, 1373–1378. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.E.; Yu, K.; Behrisch, H.W.; Soest, R.V.; Andersen, R.J. Rolloamides A and B, cytotoxic cyclic heptapeptides isolated from the caribbean marine Sponge Eurypon laughlini. J. Nat. Prod. 2009, 72, 1253–1257. [Google Scholar] [CrossRef]
- Ford, P.W.; Gustafson, K.R.; McKee, T.C.; Shigematsu, N.; Maurizi, L.K.; Pannell, L.K.; Williams, D.E.; Silva, E.D.; Lassota, P.; Allen, T.M.; et al. HIV-inhibitory and cytotoxic depsipeptides from the Sponges Theonella mirabilis and Theonella swinhoei Collected in Papua New Guinea. J. Am. Chem. Soc. 1999, 121, 5899–5909. [Google Scholar] [CrossRef]
- Capon, R.J.; Ford, J.; Lacey, E.; Gill, J.H.; Heiland, K.; Friedel, T. Phoriospongin A and B: Two new nematocidal depsipeptides from the australian marine sponges Phoriospongia sp. and Callyspongiabilamell- ata. J. Nat. Prod. 2002, 65, 358–363. [Google Scholar] [CrossRef]
- Mitova, M.; Popov, S.; De Rosa, S. Cyclic peptides from a Ruegeria strain of bacteria associated with the sponge Suberites domuncula. J. Nat. Prod. 2004, 67, 1178–1181. [Google Scholar] [CrossRef]
- Aracil, J.M.; Badre, A.; Fadli, M.; Jeanty, G.; Banaigs, B.; Francisco, C.; Lafargue, F.; Heitz, A.; Aumelas, A. Nouveaux cyclotétrapeptides isolés de l’ascidie cystodytes delle chiajei. Tetrahedron Lett. 1991, 32, 2609–2612. [Google Scholar] [CrossRef]
- Kawagishi, H.; Somoto, A.; Kuranari, J.; Kimura, A.; Chiba, S. A novel cyclotetrapeptide produced by Lactobacillus helveticus as a tyrosinase inhibitor. Tetrahedron Lett. 1993, 34, 3439–3440. [Google Scholar] [CrossRef]
- Yang, L.; Tan, R.X.; Wang, Q.; Huang, W.Y.; Yin, Y.X. Antifungal cyclopeptides from Halobacillus litoralis YS3106 of marine origin. Tetrahedron Lett. 2002, 43, 6545–6548. [Google Scholar] [CrossRef]
- Shin, J.; Seo, Y.; Lee, H.S.; Rho, J.R.; Mo, S.J. A new cyclic peptide from a marine-derived bacterium of the genus Nocardiopsis. J. Nat. Prod. 2003, 66, 883–884. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Zou, B.; Pei, D.; Ma, D. Total synthesis of cyclic tetrapeptide FR235222, a potent immunosuppressant that inhibits mammalian histone deacetylases. Org. Lett. 2005, 7, 2775–2777. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, S.S.; Ross, N.C.; McLaughlin, J.P.; Aldrich, J.V. Synthesis of Cyclic Tetrapeptide CJ 15,208: A Novel Kappa Opioid Receptor Antagonist. Adv. Exp. Med. Biol. 2009, 611, 269. [Google Scholar] [PubMed]
- Davies, J.S. The cyclization of peptides and depsipeptides. J. Peptide Sci. 2003, 9, 471–501. [Google Scholar] [CrossRef] [PubMed]
- Hamada, Y.; Shioiri, T. Recent progress of the synthetic studies of biologically active marine cyclic peptides and depsipeptides. Chem. Rev. 2005, 105, 4441–4482. [Google Scholar] [CrossRef]
- Schmidt, U.; Langner, J. Cyclotetrapeptides and cyclopentapeptides: Occurrence and synthesis. J. Peptide Res. 1997, 49, 67–73. [Google Scholar] [CrossRef]
- Tajima, H.; Wakimoto, T.; Takada, K.; Ise, Y.; Abe, I. Revised structure of cyclolithistide A, a cyclic depsipeptide from the marine sponge Discodermia japonica. J. Nat. Prod. 2014, 77, 154–158. [Google Scholar] [CrossRef]
- Usami, Y. Recent synthetic studies leading to structural revisions of marine natural products. Mar. Drugs 2009, 7, 314–330. [Google Scholar] [CrossRef]
- Chakraborty, S.; Tyagi, P.; Tai, D.F.; Lee, G.H.; Peng, S.M. A lead (II) 3D coordination polymer based on a marine cyclic peptide motif. Molecules 2013, 18, 4972–4985. [Google Scholar] [CrossRef]
- Chakraborty, S.; Tai, D.F.; Lin, Y.C.; Chiou, T.W. Antitumor and antimicrobial activity of some cyclic tetrapeptides and tripeptides derived from marine bacteria. Mar. Drugs 2015, 13, 3029–3045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakraborty, S.; Lin, S.H.; Shiuan, D.; Tai, D.F. Syntheses of some α-cyclic tripeptides as potential inhibitors for HMG-CoA reductase. Amino Acids 2015, 47, 1495–1505. [Google Scholar] [CrossRef] [PubMed]
- New, R.; Bogus, M.; Bansal, G.S.; Dryjska, M.; Zajkowska, K.; Burnet, M. Efficacy of bioactive cyclic peptides in Rheumatoid Arthritis: Translation from in vitro to in vivo models. Molecules 2017, 22, 1613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sara, V.R.; Carlsson-Skwirut, C.; Bergman, T.; Jornvall, H.; Roberts, P.J.; Crawford, M.; Hakansson, L.N.; Civalero, I.; Nordberg, A. Identification of Gly-Pro-Glu (GPE), the aminoterminal tripeptide of insulin-like growth factor 1 which is truncated in brain, as a novel neuroactive peptide. Biochem. Biophys. Res. Commun. 1989, 165, 766–771. [Google Scholar] [CrossRef]
- Gorecki, D.C.; Beresewicz, M.; Zabłocka, B. Neuroprotective effects of short peptides derived from the Insulin-like growth factor 1. Neurochemistry International. Neurochem. Int. 2007, 51, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.; Gluckman, P.D. IGF-1 derived small neuropeptides and analogues: A novel strategy for the development of pharmaceuticals for neurological conditions. Br. J. Pharmacol. 2009, 157, 881–898. [Google Scholar] [CrossRef] [Green Version]
- Cacciatore, I.; Cornacchia, C.; Baldassarre, L.; Fornasari, E.; Mollica, A.; Stefanucci, A.; Pinnen, F. GPE and GPE analogues as promising neuroprotective agents. Mini Rev. Med. Chem. 2012, 1, 13–23. [Google Scholar] [CrossRef]
- Sizonenko, S.V.; Sirimanne, E.S.; Williams, C.E.; Gluckman, P.D. Neuroprotective effects of the N-terminal tripeptide of IGF-1, glycine-proline-glutamate, in the immature rat brain after hypoxic-ischemic injury. Brain Res. 2001, 22, 42–50. [Google Scholar]
- White, C.J.; Yudin, A.K. Contemporary strategies for peptide macrocyclization. Nat. Chem. 2011, 3, 509–524. [Google Scholar] [CrossRef]
- Kartha, G.; Ambady, G.; Shankar, P.V. Structure and conformation of a cyclic tripeptide. Nature 1974, 247, 204–205. [Google Scholar] [CrossRef]
- Meng, X.Y.; Zhang, H.X.; Mezei, M.; Cui, M. Molecular docking: A powerful approach for structure-based drug discovery. Curr. Comput. Aided Drug Des. 2011, 7, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Miyazawa, T.; Ogura, M.; Nakajo, S.; Yamada, T. Synthesis of monoesters of N-protected α-aminodicarboxylic acids via the microbial protease-catalyzed regioselective hydrolysis of their diesters. Biotechnol. Tech. 1998, 12, 431–434. [Google Scholar] [CrossRef]
- Mendoza-Sanchez, R.; Corless, V.B.; Nguyen, Q.N.; Bergeron-Brlek, M.; Frost, J.; Adachi, S.; Tantillo, D.J.; Yudin, A.K. Cyclols revisited: Facile synthesis of medium-sized cyclic peptides. Chem. Eur. J. 2017, 23, 13319–13322. [Google Scholar] [PubMed]
- Kessler, H.; Bermel, W.; Krack, G.; Bats, J.W.; Fuess, H.; Fuess, H.; Hull, W.E. Peptidkonformationen, 27. cyclo-[l-Pro-(Bzl)Gly-d-Pro]. Synthese und Konformation im Kristall und in Lösung. Chem. Ber. 1983, 116, 3164–3181. [Google Scholar]
- Kessler, H.; Schuck, R.; Siegmeier, R.; Bats, J.W.; Fuess, H.; Forster, H. Peptidkonformationen, 22. Die Konformation von cyclo-[Pro2-(NB)Gly] im Kristall und in Lösung durch Röntgenstrukturanalyse und ein- und zweidimensionale NMR-Spektroskopie. Liebigs Ann. Chem. 1983, 2, 231–247. [Google Scholar] [CrossRef]
- Kessler, H.; Kondor, P.; Krack, G.; Kramer, P. Conformational equilibrium in the backbone of cyclic tripeptides. J. Am. Chem. Soc. 1978, 100, 2548–2550. [Google Scholar]
- Venkatachalam, C.M. Stereochemical studies of cyclic peptides. II. molecular structure of cyclotriprolyl. Biochim. Biophys. Acta 1968, 168, 397–401. [Google Scholar] [CrossRef]
- Tosso, R.D.; Zamora, M.A.; Suvire, F.D.; Enriz, R.D. Ab Initio and DFT study of the conformational energy hypersurface of cyclic Gly-Gly-Gly. J. Phys. Chem. A 2009, 113, 10818–10825. [Google Scholar] [CrossRef]
- Kartha, G.; Varughese, K.I.; Aimoto, S. Conformation of cyclo(-l-Pro-Gly-)3 and its Ca2+ and Mg2+ complexes. Proc. Natl. Acad. Sci. USA 1982, 79, 4519–4522. [Google Scholar]
- Dahiya, R.; Gautam, H. Total synthesis and antimicrobial activity of a natural cyclohexapeptide of marine origin. Mar. Drugs 2010, 8, 2384–2394. [Google Scholar]
- Vera, B.; Vicente, J.; Rodriguez, A.D. Isolation and structural elucidation of euryjanicins B−D, proline-containing cycloheptapeptides from the Caribbean marine sponge Prosuberites laughlini. J. Nat. Prod. 2009, 72, 1555–1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitova, M.I.; Stuart, B.G.; Cao, G.H.; Blunt, J.W.; Cole, A.L.J.; Munro, M.H.; Chrysosporide, G.A. Cyclic pentapeptide from a New Zealand sample of the fungus Sepedonium chrysospermum. J. Nat. Prod. 2006, 69, 1481–1484. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, P.W.; Chang, F.R.; Wu, C.C.; Wu, K.Y.; Li, C.M.; Chen, S.L.; Wu, Y.C. New cytotoxic cyclic peptides and dianthramide from Dianthus superbus. J. Nat. Prod. 2004, 67, 1522–1527. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.L.; Hodges, R.S. Structure-activity relationships of de novo designed cyclic antimicrobial peptides based on gramicidin S. Biopolymers 2003, 71, 28–48. [Google Scholar] [CrossRef] [PubMed]
- Falanga, A.; Nigro, E.; De Biasi, M.G.; Daniele, A.; Morelli, G.; Galdiero, S.; Scudiero, O. Cyclic peptides as novel therapeutic microbicides: Engineering of human defensin mimetics. Molecules 2017, 22, 1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dartois, V.; Sanchez-Quesada, J.; Cabezas, E.; Chi, E.; Dubbelde, C.; Dunn, C.; Granja, J.; Gritzen, C.; Weinberger, D.; Reza Ghadiri, M.; et al. Systemic antibacterial activity of novel synthetic cyclic peptides. Antimicrob. Agents Chemother. 2005, 49, 3302–3310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheinpflug, K.; Krylova, O.; Nikolenko, H.; Thurm, C.; Dathe, M. Evidence for a novel mechanism of antimicrobial action of a cyclic R-, W-rich hexapeptide. PLoS ONE 2015, 10, e0125056. [Google Scholar] [CrossRef]
- Wiese, J.; Abdelmohsen, U.R.; Motiei, A.; Humeida, U.H.; Imhoff, J.F. Bacicyclin, a new antibacterial cyclic hexapeptide from Bacillus sp. strain BC028 isolated from Mytilus edulis. Bioorg. Med. Chem. Lett. 2018, 28, 558–561. [Google Scholar] [CrossRef]
Scheme 1.
Synthesis of cyclo(GPE)2 and cyclo(GPDE). (a) Dibenzyl l-glutamate p-toluenesulphonate, DCC, HOBt, TEA, DCM, 0 °C~rt (b) (i) 20% piperidine in DCM, rt (ii) Boc-Gly-OH, DCC, HOBt, TEA, DCM, 0 °C~rt (c) B. licheniformis protease (Sigma type–VIII), pH 7 buffer, acetone, 37 °C (d) (i) DCC, pentafluorophenol, DCM (ii) TFA/DCM (1:1), 0 °C (iii) pyridine, high dilution, rt (e) H2, Pd(OH)2/C, MeOH (f) (i) DCC, pentafluorophenol, DCM (ii) TFA/DCM (1:1), rt (iii) pyridine, high dilution, rt.
Scheme 1.
Synthesis of cyclo(GPE)2 and cyclo(GPDE). (a) Dibenzyl l-glutamate p-toluenesulphonate, DCC, HOBt, TEA, DCM, 0 °C~rt (b) (i) 20% piperidine in DCM, rt (ii) Boc-Gly-OH, DCC, HOBt, TEA, DCM, 0 °C~rt (c) B. licheniformis protease (Sigma type–VIII), pH 7 buffer, acetone, 37 °C (d) (i) DCC, pentafluorophenol, DCM (ii) TFA/DCM (1:1), 0 °C (iii) pyridine, high dilution, rt (e) H2, Pd(OH)2/C, MeOH (f) (i) DCC, pentafluorophenol, DCM (ii) TFA/DCM (1:1), rt (iii) pyridine, high dilution, rt.

Figure 1.
13C-NMR spectra of cyclo(SPG), cyclo(SPG)2 and cyclo(GPE)2 in CD3OD/D2O.

Figure 2.
13C-NMR spectra of cyclo(GPDE(OBn)) and cyclo(GPDE) in D2O.


{kind=link}
{kind=link}
{kind=link}
Table 1.
13C chemical shift values for carbonyls of reported cyclic tripeptides.
| Cyclic Tripeptides | Solvents | 13C Chemical Shift Values of Carbonyls (ppm) | |
|---|---|---|---|
| Pro- | Gly- | ||
| Cyclo(l-Pro-BnG-d-Pro) [34] | C6D6 | 171.0 166.0 | 165.0 |
| Cyclo(l-Pro-l-Pro-BnG) [35] | CDCl3 | 169.6 168.8 | 165.2 |
| Cyclo(l-Pro-l-Pro-d-Pro) [35] | CDCl3 | 169.0 168.0 166.0 | |
| Cyclo(l-Pro-l-Pro-l-Pro) [36] | CDCl3 | 167.6 | |
| Cyclo (BnG-BnG-BnG) [36] | CDCl3 | 166.0 | |
| Amino Unit | Reported Data | Synthesized Product |
|---|---|---|
| Cyclo(GPE) 1 [8] | Cyclo(GPE)2 2 | |
| Glycine (carbonyl) | 169.8 | 169.6 |
| α- | 42.2 | 43.7 |
| Proline (carbonyl) | 174.8 | 173.8 |
| α- | 60.0 | 63.5 |
| β- | 29.5 | 35.4 |
| γ- | 24.9 | 26.1 |
| δ- | 47.6 | 47.7 |
| Glutamate (carbonyl) | 176.2 | 175.0 |
| Side Chain (carbonyl) | 183.1 | 181.6 |
| α- | 57.5 | 54.5 |
| β- | 25.8 | 29.0 |
| γ- | 29.5 | 30.7 |
| Amino Unit | Reported Data | Synthesized Product | |
|---|---|---|---|
| Cyclo(GPE) 1 [8] | Cyclo(GPE)2 2 | ||
| Glycine | α- | 4.20 d (17.3) | 4.33 d (17.3) |
| 4.13 d (17.3) | 4.20 d (17.3) | ||
| Proline | α- | 4.49 dd (8.6; 3.5) | 4.51 dd (8.7; 6.1) |
| β- | 2.34 m, 2.08 m | 2.41 m, 2.13 m | |
| γ- | 2.08 m | 2.13 m | |
| δ- | 3.67 m | 3.96 m | |
| Glutamate | α- | 4.42 dd (9.1; 4.9) | 4.64 dd (10.3; 4.1) |
| β- | 2.58 m, 2.14 m | 2.48 m, 2.13 m | |
| γ- | 2.47 m | 2.48 m | |
Coupling constants (hertz) are in parentheses.
| Amino Unit | Reported Data | Synthesized Product |
|---|---|---|
| Cyclo(GPE) 1 | Cyclo(GPDE) 10 | |
| Glycine | ||
| α- | 4.20 d (17.3) | 4.24 dd (17.4, 7.6) |
| 4.13 d (17.3) | 4.10 dd (17.1, 3.3) | |
| Proline | ||
| α- | 4.49 dd (8.6; 3.5) | 4.17 t (4.6) |
| β- | 2.34 m, 2.08 m | 2.67 m, 2.12 m |
| γ- | 2.08 m | 2.12 m |
| δ- | 3.67 m | 3.47 m, 3.70 m |
| Glutamate | ||
| α- | 4.42 dd (9.1; 4.9) | 3.79 t (6.4) |
| β- | 2.58 m, 2.14 m | 2.22 m |
| γ- | 2.47 m | 2.37 m |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Lin, C.-Y.; Chakraborty, S.; Wong, C.-W.; Tai, D.-F. Controversy of Peptide Cyclization from Tripeptide. Molecules 2021, 26, 389. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26020389
AMA Style
Lin C-Y, Chakraborty S, Wong C-W, Tai D-F. Controversy of Peptide Cyclization from Tripeptide. Molecules. 2021; 26(2):389. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26020389
Chicago/Turabian StyleLin, Chung-Yin, Subrata Chakraborty, Chia-Wei Wong, and Dar-Fu Tai. 2021. "Controversy of Peptide Cyclization from Tripeptide" Molecules 26, no. 2: 389. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26020389