
Deprotometalation-Iodolysis and Direct Iodination of 1-Arylated 7-Azaindoles: Reactivity Studies and Molecule Properties

 , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
2.1. 1-Arylation of 7-Azaindole
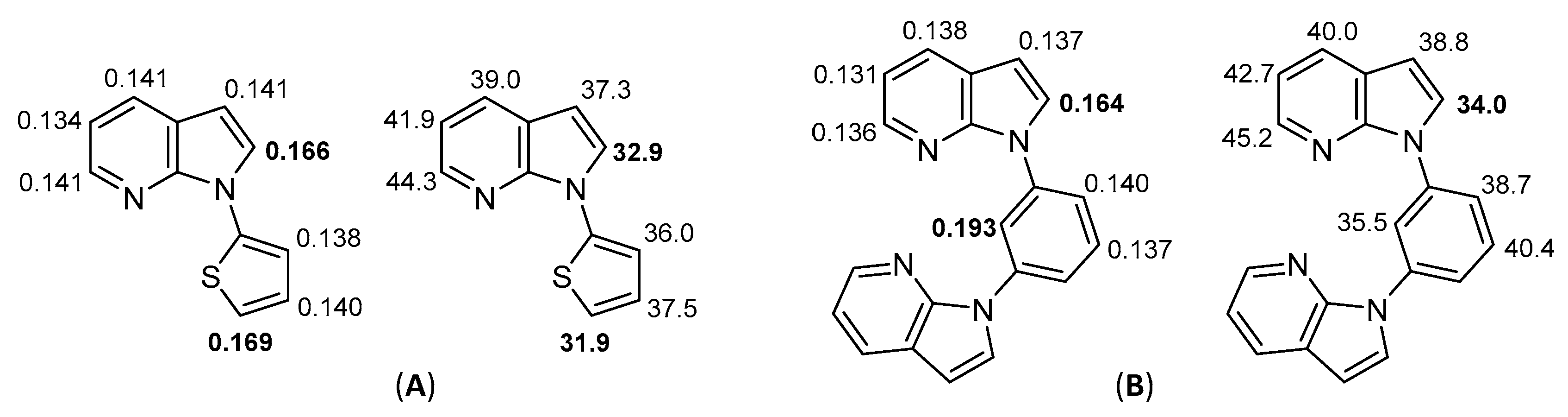
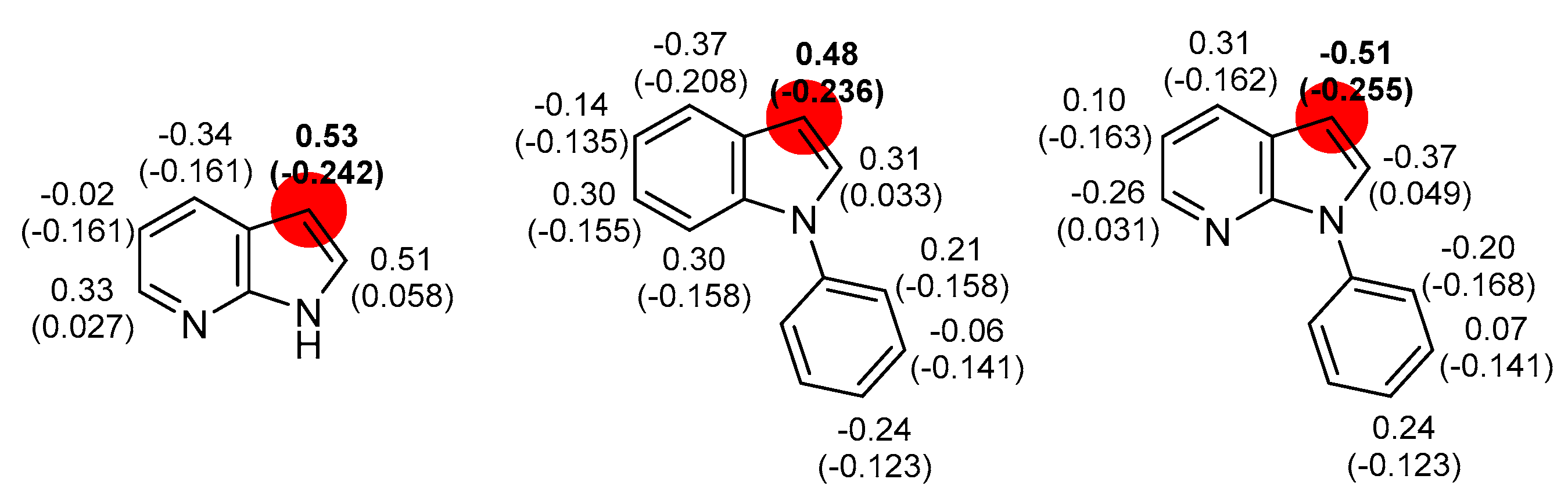
2.2. Deprotometalation-Iodolysis of 1-Arylated 7-Azaindoles
2.3. Direct Iodination of 1-Arylated 7-Azaindoles
2.4. N-Arylation of Indole by a Few of the Prepared Iodides
2.5. Biological Evaluation
3. Materials and Methods
3.1. General Information
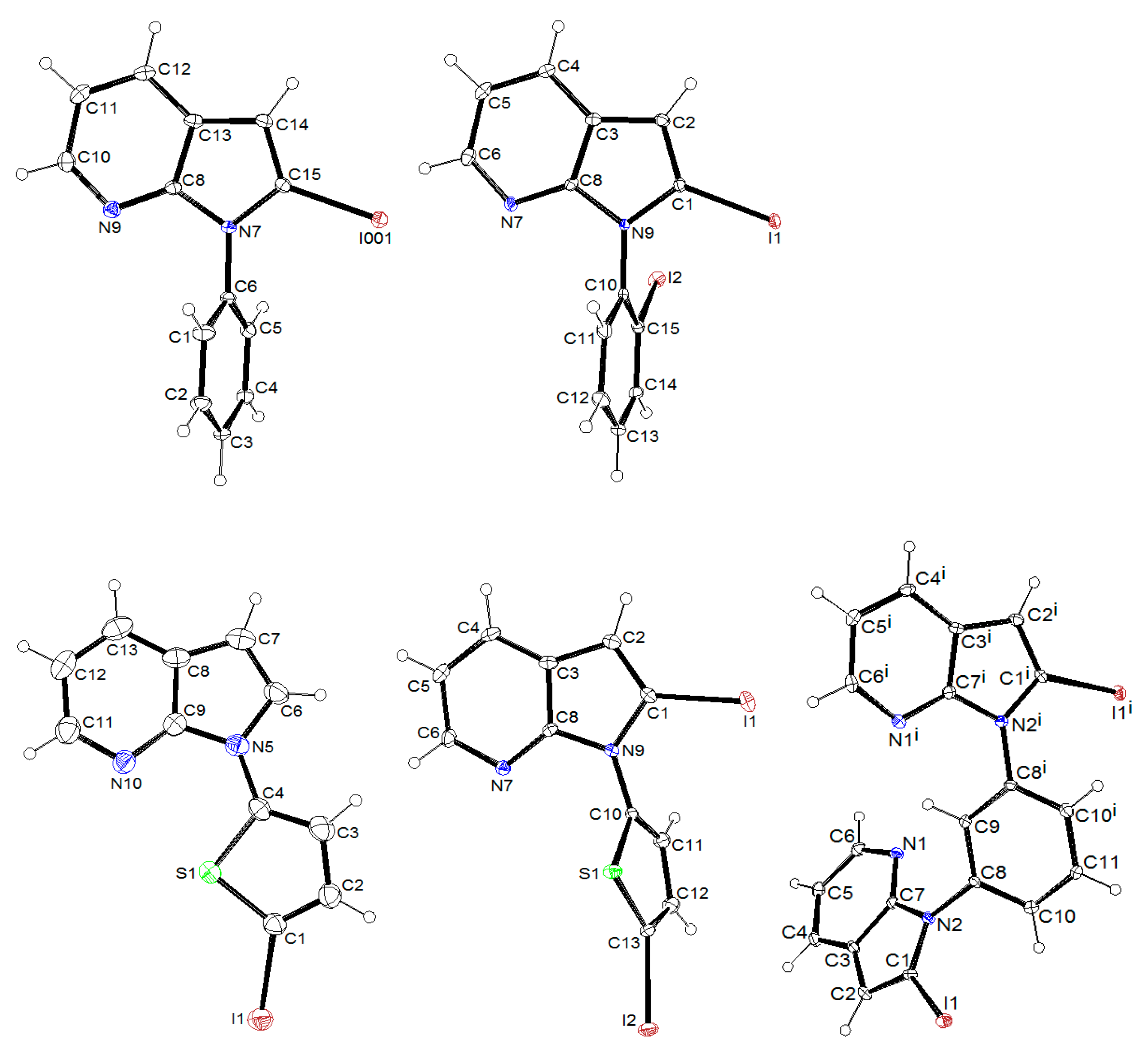
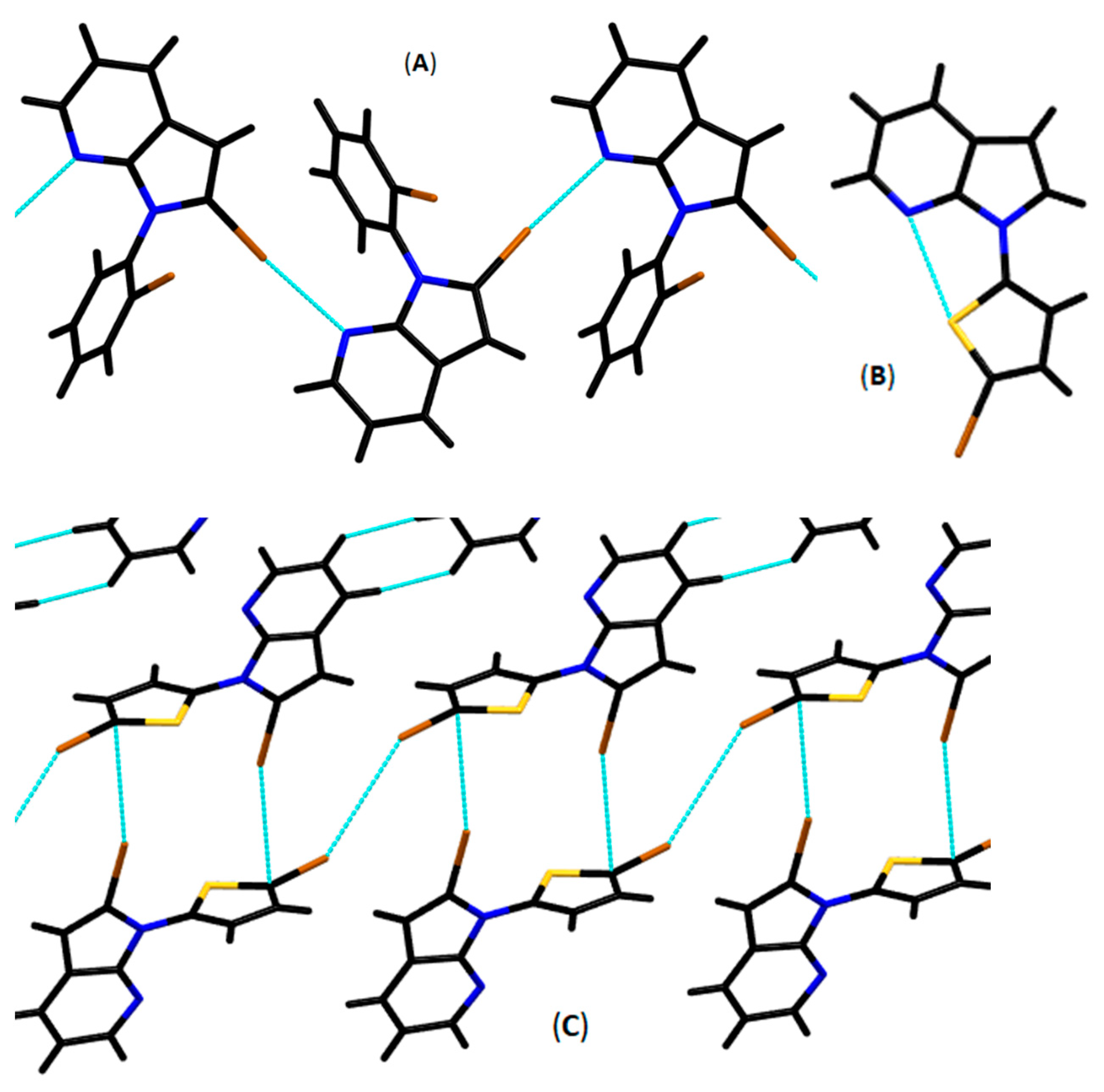
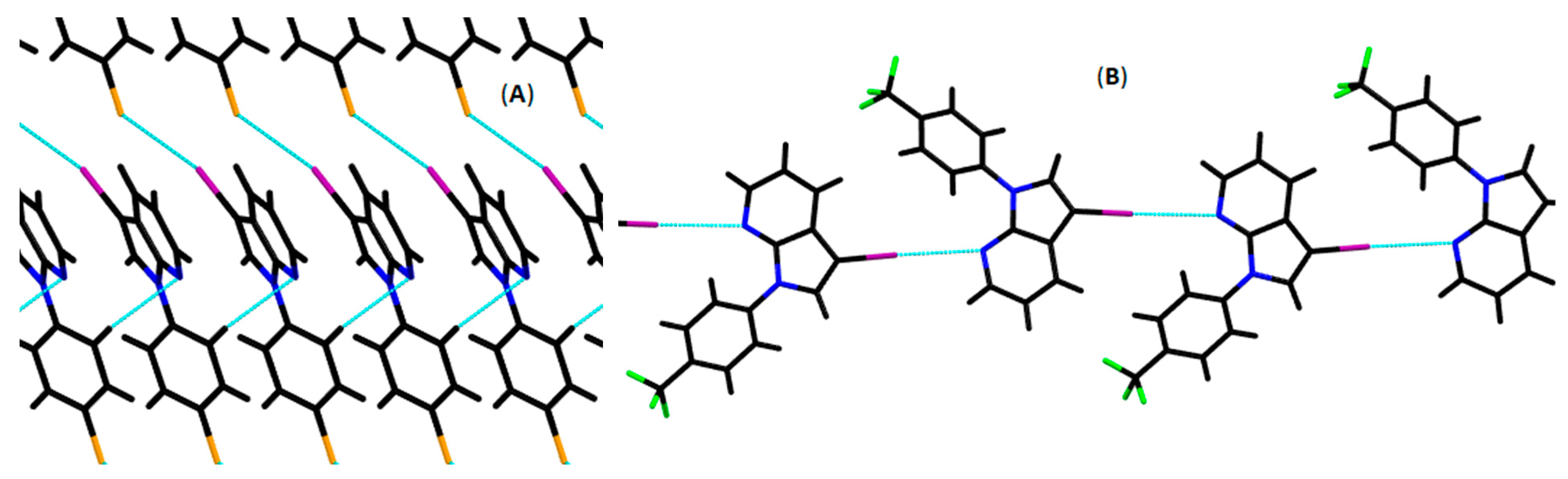
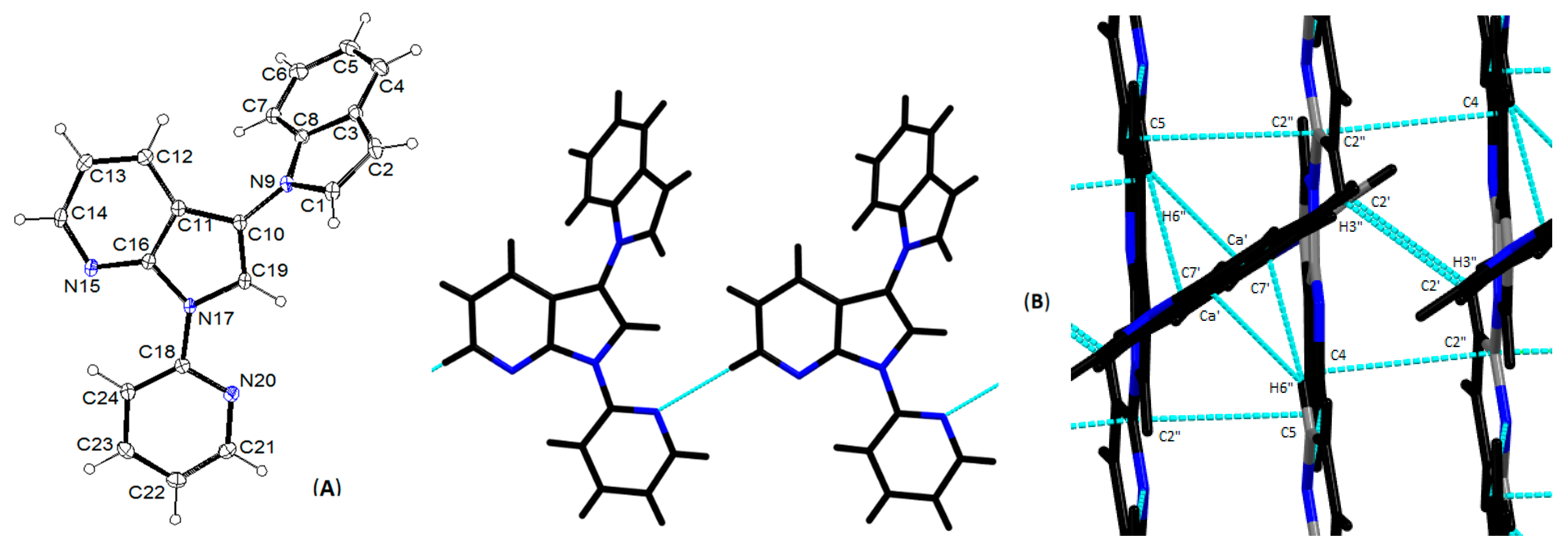
3.2. Crystallography
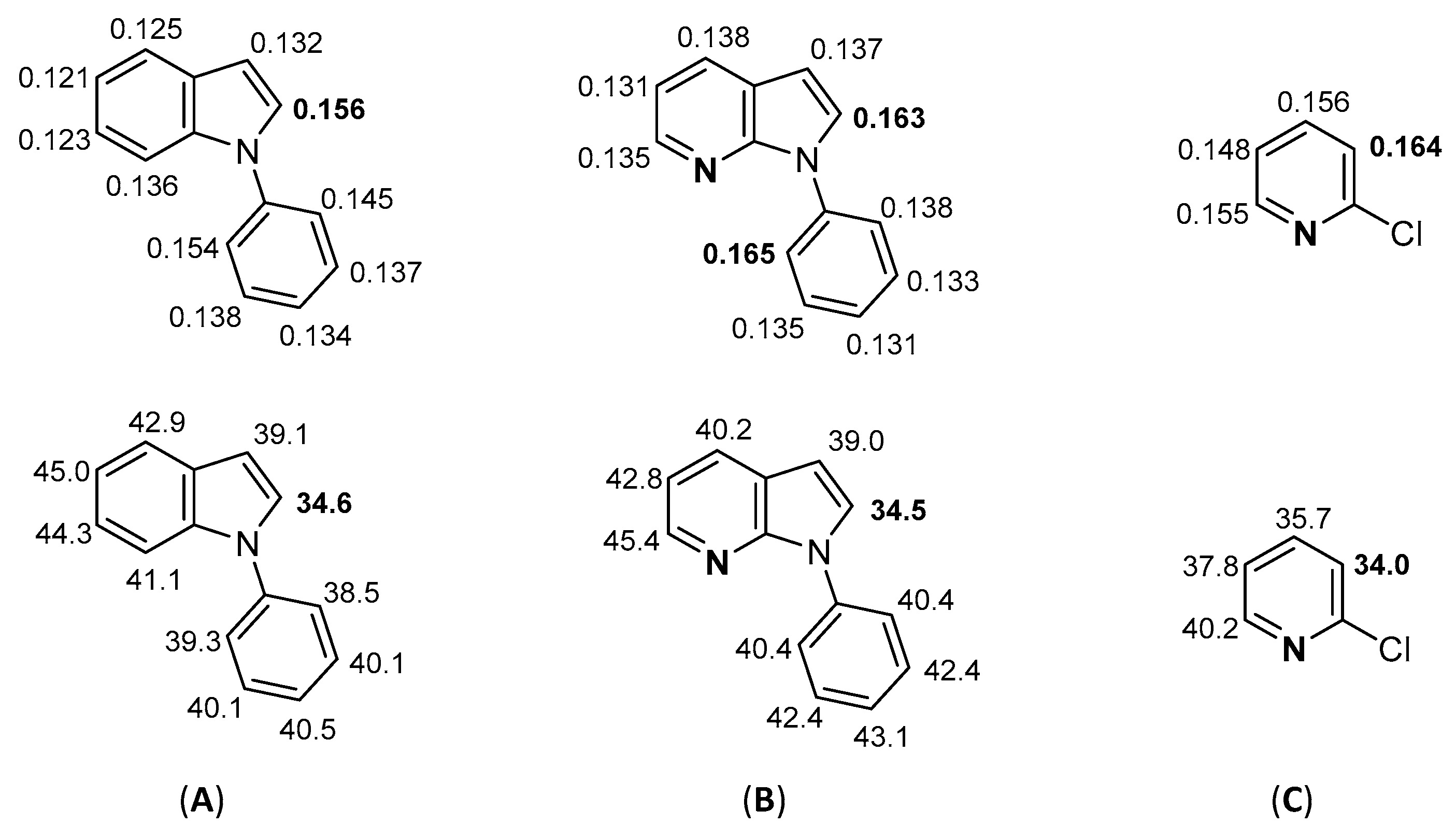
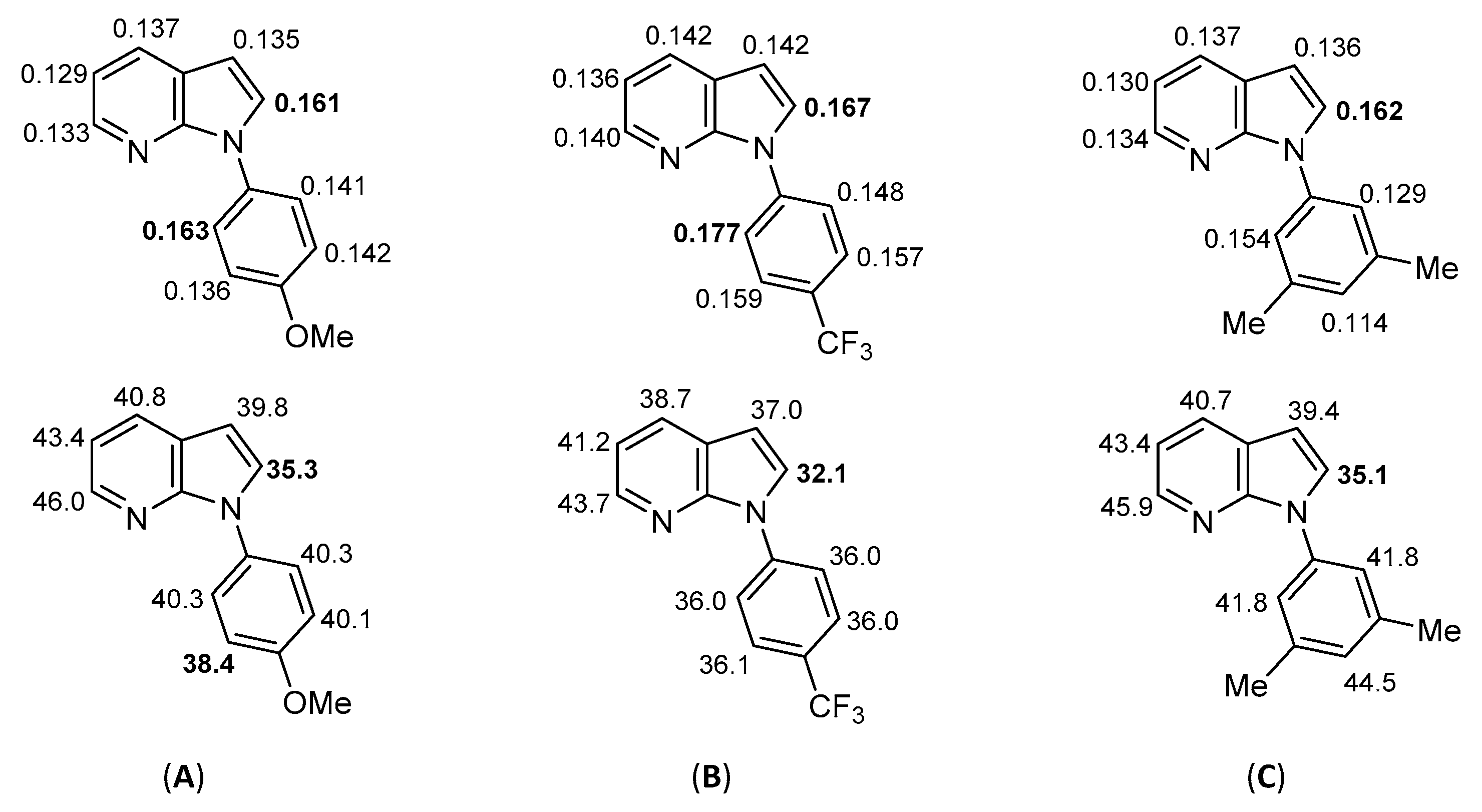
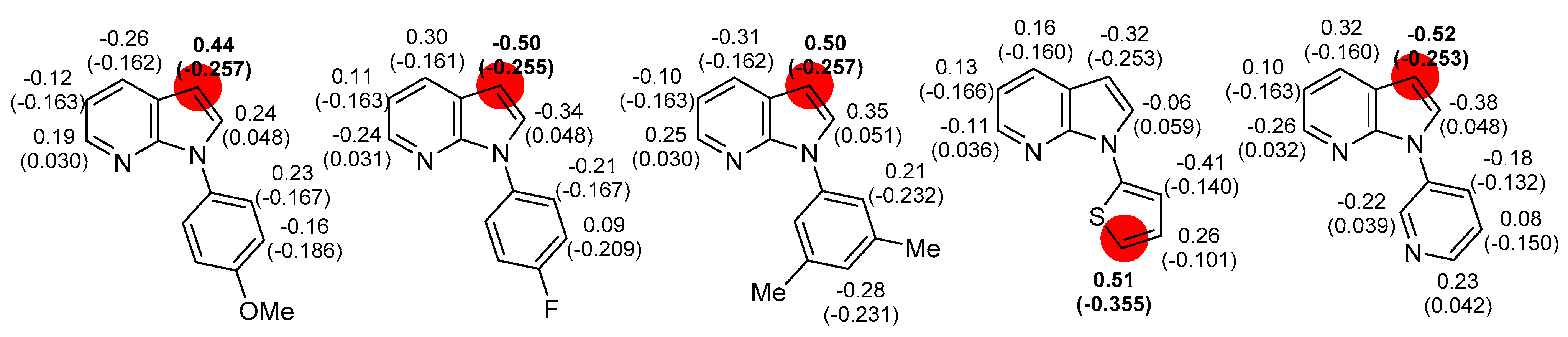
3.3. Computational Details
3.4. 1-Arylation of 7-Azaindole
3.4.1. General Procedure 1 Using Copper
3.4.2. General Procedure 2 Using Copper(I) Iodide without Ligand
3.4.3. General Procedure 3 Using Copper(I) Iodide with Ligand
3.4.4. General Procedure 4 Using Copper(I) Oxide
3.4.5. General Procedure 5 Using Copper(I) Iodide and Microwaves
3.4.6. 1-Phenyl-1H-pyrrolo[2,3-b]pyridine or 1-Phenyl-7-azaindole (1a)
3.4.7. 1-(4-Methoxyphenyl)-7-azaindole (1b)
3.4.8. 1-(4-Chlorophenyl)-7-azaindole (1c)
3.4.9. 1-(4-Fluorophenyl)-7-azaindole (1d)
3.4.10. 1-(4-(Trifluoromethyl)phenyl)-7-azaindole (1e)
3.4.11. 1-(3,5-Dimethylphenyl)-7-azaindole (1f)
3.4.12. 1-(2-Thienyl)-7-azaindole (1g)
3.4.13. 1-(3-Pyridyl)-7-azaindole (1h)
3.4.14. 1-(2-Pyridyl)-7-azaindole (1i)
3.4.15. 1-(4-Pyridyl)-7-azaindole (1j)
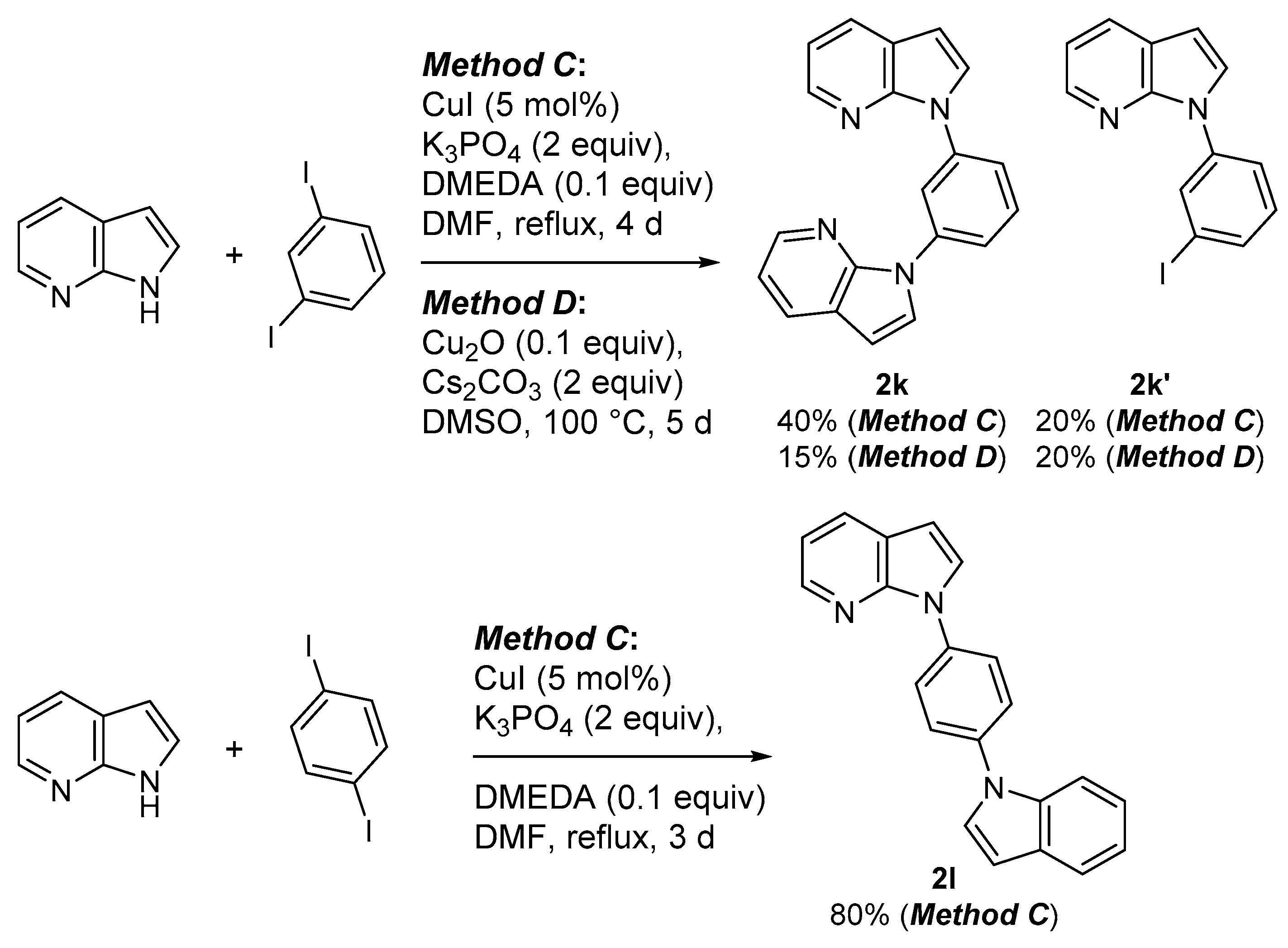
3.4.16. 1,1′-(1,3-Phenylene)bis(7-azaindole) (2k)
3.4.17. 1-(3-Iodophenyl)-7-azaindole (2k′)
3.4.18. 1,1′-(1,4-Phenylene)bis(7-azaindole) (2l)
3.5. Deprotometalation-Iodolysis of the Different 1-Arylated 7-Azaindoles
3.5.1. General Procedure 6
3.5.2. 2-Iodo-1-phenyl-7-azaindole (3a)
3.5.3. 2-Iodo-1-(2-iodophenyl)-7-azaindole (3a′)
3.5.4. 2-Iodo-1-(4-(trifluoromethyl)phenyl)-7-azaindole (3e)
3.5.5. 2-Iodo-1-(3-iodo-4-(trifluoromethyl)phenyl)-7-azaindole (3e′)
3.5.6. 1-(3,5-Dimethylphenyl)-2-iodo-7-azaindole (3f)
3.5.7. 1-(5-Iodo-2-thienyl)-7-azaindole (3g)
3.5.8. 2-Iodo-1-(5-iodo-2-thienyl)-7-azaindole (3g′)
3.5.9. 2-Iodo-1-(3-(7-aza-1-indolyl)phenyl)-7-azaindole (3k)
3.5.10. 1,1′-(1,3-Phenylene)bis(2-iodo-7-azaindole) (3k′)
3.5.11. 1,1′-(1-Iodo-2,4-phenylene)bis(2-iodo-7-azaindole (3k″)
3.6. Direct Iodination of the Different 1-Arylated 7-Azaindoles
3.6.1. General Procedure 7
3.6.2. General Procedure 8
3.6.3. 3-Iodo-1-phenyl-7-azaindole (4a)
3.6.4. 3-Iodo-1-(4-methoxyphenyl)-7-azaindole (4b)
3.6.5. 1-(4-Chlorophenyl)-3-iodo-7-azaindole (4c)
3.6.6. 1-(4-Fluorophenyl)-3-iodo-7-azaindole (4d)
3.6.7. 3-Iodo-1-(4-(trifluoromethyl)phenyl)-7-azaindole (4e)
3.6.8. 3-Iodo-1-(3,5-dimethylphenyl)-7-azaindole (4f)
3.6.9. 1-(5-Iodo-2-thienyl)-7-azaindole (3g)
3.6.10. 3-Iodo-1-(3-pyridyl)-7-azaindole (4h)
3.6.11. 3-Iodo-1-(2-pyridyl)-7-azaindole (4i)
3.7. N-Arylation of Azoles by Using 1-Arylated 3-Iodo-7-azaindoles
3.7.1. 3-(1-Indolyl)-1-(4-(trifluoromethyl)phenyl)-7-azaindole (5e)
3.7.2. General Procedure 9 Using Copper(I) Iodide with Ligand
3.7.3. 3-(1-Indolyl)-1-(2-pyridyl)-7-azaindole (5i)
3.7.4. 2-(1-Indolyl)-1-(5-(1-indolyl)-2-thienyl)-7-azaindole (6g′)
3.8. Evaluation of the Biological Properties
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Prokopov, A.A.; Yakhontov, L.N. Chemistry of the azaindoles (review). Pharm. Chem. J. 1994, 28, 471–506. [Google Scholar] [CrossRef]
- Mérour, J.-Y.; Joseph, B. Synthesis and reactivity of 7-azaindoles (1H-pyrrolo[2,3-b]pyridines). Curr. Org. Chem. 2001, 5, 471–506. [Google Scholar] [CrossRef]
- Song, J.J.; Reeves, J.T.; Gallou, F.; Tan, Z.; Yee, N.K.; Senanayake, C.H. Organometallic methods for the synthesis and functionalization of azaindoles. Chem. Soc. Rev. 2007, 36, 1120–1132. [Google Scholar] [CrossRef] [PubMed]
- Popowycz, F.; Routier, S.; Joseph, B.; Mérour, J.-Y. Synthesis and reactivity of 7-azaindole (1H-pyrrolo[2,3-b]pyridine). Tetrahedron 2007, 63, 1031–1064. [Google Scholar] [CrossRef]
- Mérour, J.-Y.; Routier, S.; Suzenet, F.; Joseph, B. Recent advances in the synthesis and properties of 4-, 5-, 6- or 7-azaindoles. Tetrahedron 2013, 69, 4767–4834. [Google Scholar] [CrossRef]
- Mérour, J.-Y.; Buron, F.; Plé, K.; Bonnet, P.; Routier, S. The azaindole framework in the design of kinase inhibitors. Molecules 2014, 19, 19935–19979. [Google Scholar] [CrossRef] [Green Version]
- Santos, A.S.; Mortinho, A.C.; Marques, M.M.B. Metal-catalyzed cross-coupling reactions on azaindole synthesis and functionalization. Molecules 2018, 23, 2673. [Google Scholar] [CrossRef] [Green Version]
- Kannaboina, P.; Mondal, K.; Laha, J.K.; Das, P. Recent advances in the global ring functionalization of 7-azaindoles. Chem. Commun. 2020, 56, 11749–11762. [Google Scholar] [CrossRef] [PubMed]
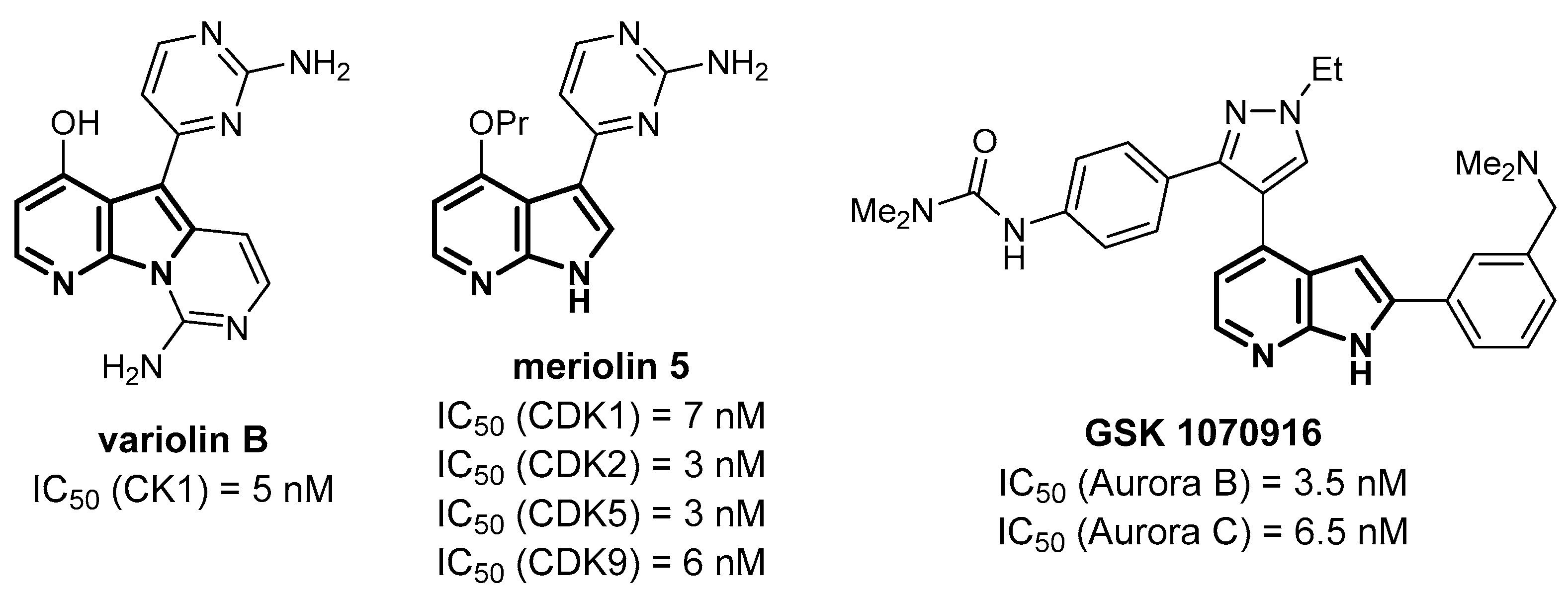
- Echalier, A.; Bettayeb, K.; Ferandin, Y.; Lozach, O.; Clelment, M.; Valette, A.; Liger, F.; Marquet, B.; Morris, J.C.; Endicott, J.A.; et al. Meriolins (3-(pyrimidin-4-yl)-7-azaindoles): Synthesis, kinase inhibitory activity, cellular effects, and structure of a CDK2/cyclin A/meriolin complex. J. Med. Chem. 2008, 51, 737–751. [Google Scholar] [CrossRef]
- Jarry, M.; Lecointre, C.; Malleval, C.; Desrues, L.; Schouft, M.-T.; Lejoncour, V.; Liger, F.; Lyvinec, G.; Joseph, B.; Loaec, N.; et al. Impact of meriolins, a new class of cyclin-dependent kinase inhibitors, on malignant glioma proliferation and neo-angiogenesis. Neuro-Oncol. 2014, 16, 1484–1498. [Google Scholar] [CrossRef] [Green Version]
- Hardwicke, M.A.; Oleykowski, C.A.; Plant, R.; Wang, J.; Liao, Q.; Moss, K.; Newlander, K.; Adams, J.L.; Dhanak, D.; Yang, J.; et al. GSK1070916, a potent Aurora B/C kinase inhibitor with broad antitumor activity in tissue culture cells and human tumor xenograft models. Mol. Cancer Ther. 2009, 8, 1808–1817. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.-B.; Wang, S. Luminescence and reactivity of 7-azaindole derivatives and complexes. Chem. Soc. Rev. 2010, 39, 3142–3156. [Google Scholar] [CrossRef]
- Li, S.-S.; Wang, C.-Q.; Lin, H.; Zhang, X.-M.; Dong, L. Rhodium(III)-catalyzed oxidative annulation of 7-azaindoles and alkynes via double C-H activation. Org. Lett. 2015, 17, 3018–3021. [Google Scholar] [CrossRef]
- Li, S.-S.; Liu, C.-F.; Xia, Y.-Q.; Li, W.-H.; Zhang, G.-T.; Zhang, X.-M.; Dong, L. A unique annulation of 7-azaindoles with alkenyl esters to produce π-conjugated 7-azaindole derivatives. Org. Biomol. Chem. 2016, 14, 5214–5218. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Li, R.; Zhan, W.; Wang, X.; Ge, Z.; Li, R. Rh(iii)-catalyzed c-h oxidative ortho-olefination of arenes using 7-azaindole as a directing group and utilization in the construction of new tetracyclic heterocycles containing a 7-azaindole skeleton. RSC Adv. 2016, 6, 48205–48211. [Google Scholar] [CrossRef]
- Jeon, M.; Park, J.; Dey, P.; Oh, Y.; Oh, H.; Han, S.; Um, S.H.; Kim, H.S.; Mishra, N.K.; Kim, I.S. Site-selective rhodium(iii)-catalyzed c-h amination of 7-azaindoles with anthranils: Synthesis and anticancer evaluation. Adv. Synth. Catal. 2017, 359, 3471–3478. [Google Scholar] [CrossRef]
- Zhang, G.-T.; Zhang, J.; Xu, Y.-J.; Dong, L. Metal-free [3+2] tandem cyclization synthesis of unique 11h-pyrido[3′,2′:4,5]pyrrolo [3,2-b] indolizine from 7-azaindoles and pyridotriazoles. Eur. J. Org. Chem. 2018, 2018, 4197–4201. [Google Scholar] [CrossRef]
- Ahn, S.; Hong, M.; Sundararajan, M.; Ess, D.H.; Baik, M.-H. Design and optimization of catalysts based on mechanistic insights derived from quantum chemical reaction modeling. Chem. Rev. 2019, 119, 6509–6560. [Google Scholar] [CrossRef]
- Chen, K.; Shi, G.; Zhang, W.; Li, H.; Wang, C. Computer-assisted design of ionic liquids for efficient synthesis of 3(2h)-furanones: A domino reaction triggered by CO2. J. Am. Chem. Soc. 2016, 138, 14198–14201. [Google Scholar] [CrossRef]
- Sorokin, V.I. Copper(I) catalyzed N-arylation of azoles, the recent developments. Mini-Rev. Org. Chem. 2008, 5, 323–330. [Google Scholar] [CrossRef]
- Monnier, F.; Taillefer, M. Catalytic C–C, C–N, and C–O Ullmann-type coupling reactions. Angew. Chem. Int. Ed. 2009, 48, 6954–6971. [Google Scholar] [CrossRef]
- Beletskaya, I.P.; Cheprakov, A.V. The complementary competitors: Palladium and copper in C–N cross-coupling reactions. Organometallics 2012, 31, 7753–7808. [Google Scholar] [CrossRef]
- Bariwal, J.; Van der Eycken, E. C–N bond forming cross-coupling reactions: An overview. Chem. Soc. Rev. 2013, 42, 9283–9303. [Google Scholar] [CrossRef]
- Monnier, F.; Taillefer, M. Copper-catalyzed C(aryl)–N bond formation. Top. Organomet. Chem. 2013, 46, 173–204. [Google Scholar] [CrossRef]
- Sambiagio, C.; Marsden, S.P.; Blacker, A.J.; McGowan, P.C. Copper catalysed Ullmann type chemistry: From mechanistic aspects to modern development. Chem. Soc. Rev. 2014, 43, 3525–3550. [Google Scholar] [CrossRef] [PubMed]
- Yong, F.-F.; Azri, M.; Darrell Lim, Y.-E.; Teo, Y.-C. Efficient copper-catalyzed cross-coupling of nitrogen nucleophiles with N,N-dibenzyl-4-iodobenzenesulfonamide and its application in the synthesis of celecoxib intermediate. Tetrahedron 2020, 76, 131680. [Google Scholar] [CrossRef]
- Zhu, R.; Xing, L.; Wang, X.; Cheng, C.; Su, D.; Hu, Y. Highly practical “ligand-free-like” copper-catalyzed N-arylation of azoles in lower nitrile solvents. Adv. Synth. Catal. 2008, 350, 1253–1257. [Google Scholar] [CrossRef]
- Hong, C.S.; Seo, J.Y.; Yum, E.K. N-arylation of azaindoles in LiCl-mediated catalytic CuI reactions. Tetrahedron Lett. 2007, 48, 4831–4833. [Google Scholar] [CrossRef]
- Antilla, J.C.; Baskin, J.M.; Barder, T.E.; Buchwald, S.L. Copper-diamine-catalyzed N-arylation of pyrroles, pyrazoles, indazoles, imidazoles, and triazoles. J. Org. Chem. 2004, 69, 5578–5587. [Google Scholar] [CrossRef]
- Teo, Y.-C.; Yong, F.-F.; Sim, S. Ligand-free Cu2O-catalyzed cross coupling of nitrogen heterocycles with iodopyridines. Tetrahedron 2013, 69, 7279–7284. [Google Scholar] [CrossRef]
- Kwon, J.K.; Cho, J.H.; Ryu, Y.-S.; Oh, S.H.; Yum, E.K. N-arylation of carbazole by microwave-assisted ligand-free catalytic CuI reaction. Tetrahedron 2011, 67, 4820–4825. [Google Scholar] [CrossRef]
- Kadari, L.; Erb, W.; Roisnel, T.; Radha Krishna, P.; Mongin, F. Iodoferrocene as a partner in N-arylation of amides. New J. Chem. 2020, 44, 15928–15941. [Google Scholar] [CrossRef]
- Kadari, L.; Erb, W.; Halauko, Y.S.; Ivashkevich, O.A.; Matulis, V.E.; Lyakhov, D.; Roisnel, T.; Radha Krishna, P.; Mongin, F. On the N-arylation of acetamide using 2-, 3- and 1′-substituted iodoferrocenes. Eur. J. Inorg. Chem. 2021, 2021, 377–391. [Google Scholar] [CrossRef]
- Desarbre, E.; Coudret, S.; Meheust, C.; Mérour, J.-Y. Synthesis of 2-substituted-1H-pyrrolo[2,3-b]pyridines: Preparation of 7-azaolivacine analog and 7-azaindolopyridopyrimidine derivatives. Tetrahedron 1997, 53, 3637–3648. [Google Scholar] [CrossRef]
- Slocum, D.W.; Jennings, C.A. Directed metalation reactions. 6. Competition of substituents for ortho direction of metalation in substituted anisoles. J. Org. Chem. 1976, 41, 3653–3664. [Google Scholar] [CrossRef]
- Naka, H.; Akagi, Y.; Yamada, K.; Imahori, T.; Kasahara, T.; Kondo, Y. Fluorous synthesis of yuehchukene by alpha-lithiation of perfluoroalkyl-tagged 1-(arylsulfonyl)indole with mesityllithium. Eur. J. Org. Chem. 2007, 2007, 4635–4637. [Google Scholar] [CrossRef]
- Katritzky, A.R.; Akutagawa, K. Carbon dioxide: A reagent for simultaneous protection of nucleophilic centers and the activation of alternative locations to electrophilic attack. V. Activation of the 2-alkyl group of a 2-alkylindole toward proton loss and subsequent electrophilic substitution. J. Am. Chem. Soc. 1986, 108, 6808–6809. [Google Scholar] [CrossRef]
- Curtis, N.R.; Kulagowski, J.J.; Leeson, P.D.; Ridgill, M.P.; Emms, F.; Freedman, S.B.; Patel, S.; Patel, S. Synthesis and SAR of 2- and 3-substituted 7-azaindoles as potential dopamine D4 ligands. Bioorg. Med. Chem. Lett. 1999, 9, 585–588. [Google Scholar] [CrossRef]
- Hogan, M.; Cotter, J.; Claffey, J.; Gleeson, B.; Wallis, D.; O’Shea, D.; Tacke, M. Synthesis and cytotoxicity studies of new (dimethylamino)-functionalized and 7-azaindole-substituted ‘titanocene’ anticancer agents (7-azaindole = 1H-pyrrolo[2,3-b]pyridine). Helv. Chim. Acta 2008, 91, 1787–1797. [Google Scholar] [CrossRef]
- Heinrich, T.; Seenisamy, J.; Emmanuvel, L.; Kulkarni, S.S.; Bomke, J.; Rohdich, F.; Greiner, H.; Esdar, C.; Krier, M.; Graedler, U.; et al. Fragment-based discovery of new highly substituted 1H-pyrrolo[2,3-b]- and 3H-imidazolo[4,5-b]-pyridines as focal adhesion kinase inhibitors. J. Med. Chem. 2013, 56, 1160–1170. [Google Scholar] [CrossRef]
- Juchum, M.; Günther, M.; Döring, E.; Sievers-Engler, A.; Lämmerhofer, M.; Laufer, S. Trisubstituted imidazoles with a rigidized hinge binding motif act as single digit nm inhibitors of clinically relevant egfr l858r/t790m and l858r/t790m/c797s mutants: An example of target hopping. J. Med. Chem. 2017, 60, 4636–4656. [Google Scholar] [CrossRef]
- L’Heureux, A.; Thibault, C.; Ruel, R. Synthesis of functionalized 7-azaindoles via directed ortho-metalations. Tetrahedron Lett. 2004, 45, 2317–2319. [Google Scholar] [CrossRef]
- Schneider, C.; David, E.; Toutov, A.A.; Snieckus, V. In situ anionic shielding for regioselective metalation: Directed peri and iterative metalation routes to polyfunctionalized 7-azaindoles. Angew. Chem. Int. Ed. 2012, 51, 2722–2726. [Google Scholar] [CrossRef]
- Dalziel, M.E.; Patel, J.J.; Kaye, M.K.; Cosman, J.L.; Kitching, M.O.; Snieckus, V. Regioselective functionalization of 7-azaindole by controlled annular isomerism: The directed metalation-group dance. Angew. Chem., Int. Ed. 2019, 58, 7313–7317. [Google Scholar] [CrossRef]
- Zhao, S.-B.; Wang, R.-Y.; Wang, S. Intramolecular C–H activation directed self-assembly of an organoplatinum(II) molecular square. J. Am. Chem. Soc. 2007, 129, 3092–3093. [Google Scholar] [CrossRef] [PubMed]
- Barberis, C.; Moorcroft, N.; Arendt, C.; Levit, M.; Moreno-Mazza, S.; Batchelor, J.; Mechin, I.; Majid, T. Discovery of N-substituted 7-azaindoles as PIM1 kinase inhibitors—Part I. Bioorg. Med. Chem. Lett. 2017, 27, 4730–4734. [Google Scholar] [CrossRef] [PubMed]
- L’Helgoual’ch, J.M.; Seggio, A.; Chevallier, F.; Yonehara, M.; Jeanneau, E.; Uchiyama, M.; Mongin, F. Deprotonative metalation of five-membered aromatic heterocycles using mixed lithium-zinc species. J. Org. Chem. 2008, 73, 177–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snégaroff, K.; L’Helgoual’ch, J.-M.; Bentabed-Ababsa, G.; Nguyen, T.T.; Chevallier, F.; Yonehara, M.; Uchiyama, M.; Derdour, A.; Mongin, F. Deprotonative metalation of functionalized aromatics using mixed lithium-cadmium, lithium-indium, and lithium-zinc species. Chem. Eur. J. 2009, 15, 10280–10290. [Google Scholar] [CrossRef] [PubMed]
- Akimoto, G.; Otsuka, M.; Takita, R.; Uchiyama, M.; Hedidi, M.; Bentabed-Ababsa, G.; Lassagne, F.; Erb, W.; Mongin, F. Deprotonative metalation of methoxy-substituted arenes using lithium 2,2,6,6-tetramethylpiperidide: Experimental and computational study. J. Org. Chem. 2018, 83, 13498–13506. [Google Scholar] [CrossRef] [PubMed]
- García-Álvarez, P.; Mulvey, R.E.; Parkinson, J.A. “LiZn(TMP)3”, a zincate or a turbo-lithium amide reagent? Dosy NMR spectroscopic evidence. Angew. Chem. Int. Ed. 2011, 50, 9668–9671. [Google Scholar] [CrossRef]
- Mokhtari Brikci-Nigassa, N.; Bentabed-Ababsa, G.; Erb, W.; Mongin, F. In situ ‘trans-metal trapping’: An efficient way to extend the scope of aromatic deprotometalation. Synthesis 2018, 50, 3615–3633. [Google Scholar] [CrossRef]
- Ameur Messaoud, M.Y.; Bentabed-Ababsa, G.; Hedidi, M.; Derdour, A.; Chevallier, F.; Halauko, Y.S.; Ivashkevich, O.A.; Matulis, V.E.; Picot, L.; Thiéry, V.; et al. Deproto-metalation of N-arylated pyrroles and indoles using a mixed lithium-zinc base and regioselectivity-computed CH acidity relationship. Beilstein J. Org. Chem. 2015, 11, 1475–1485. [Google Scholar] [CrossRef] [Green Version]
- Snégaroff, K.; Nguyen, T.T.; Marquise, N.; Halauko, Y.S.; Harford, P.J.; Roisnel, T.; Matulis, V.E.; Ivashkevich, O.A.; Chevallier, F.; Wheatley, A.E.H.; et al. Deprotonative metalation of chloro- and bromopyridines using amido-based bimetallic species and regioselectivity-computed CH acidity relationships. Chem. Eur. J. 2011, 17, 13284–13297. [Google Scholar] [CrossRef] [PubMed]
- Fraser, R.R.; Mansour, T.S.; Savard, S. Acidity measurements in THF. V. Heteroaromatic compounds containing 5-membered rings. Can. J. Chem. 1985, 63, 3505–3509. [Google Scholar] [CrossRef]
- Nagaradja, E.; Chevallier, F.; Roisnel, T.; Dorcet, V.; Halauko, Y.S.; Ivashkevich, O.A.; Matulis, V.E.; Mongin, F. Deproto-metallation using a mixed lithium-zinc base and computed CH acidity of 1-aryl 1H-benzotriazoles and 1-aryl 1H-indazoles. Org. Biomol. Chem. 2014, 12, 1475–1487. [Google Scholar] [CrossRef] [PubMed]
- Gros, P.; Choppin, S.; Fort, Y. Lithiation of 2-chloro- and 2-methoxypyridine with lithium dialkylamides: Initial ortho-direction or subsequent lithium ortho-stabilization? J. Org. Chem. 2003, 68, 2243–2247. [Google Scholar] [CrossRef] [PubMed]
- Chevallier, F.; Halauko, Y.S.; Pecceu, C.; Nassar, I.F.; Dam, T.U.; Roisnel, T.; Matulis, V.E.; Ivashkevich, O.A.; Mongin, F. N-aryl pyrazoles: DFT calculations of CH acidity and deprotonative metallation using a combination of lithium and zinc amides. Org. Biomol. Chem. 2011, 9, 4671–4684. [Google Scholar] [CrossRef] [Green Version]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The halogen bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [Green Version]
- Tilly, D.; Chevallier, F.; Mongin, F. Synthesis of heterobiaryls featuring 1,4 N…S inter-ring interactions. Synthesis 2016, 48, 184–199. [Google Scholar] [CrossRef]
- Merkul, E.; Schäfer, E.; Müller, T.J.J. Rapid synthesis of bis(hetero)aryls by one-pot Masuda borylation-suzuki coupling sequence and its application to concise total syntheses of meridianins A and G. Org. Biomol. Chem. 2011, 9, 3139–3141. [Google Scholar] [CrossRef]
- Koller, M.; Carcache, D.A.; Orain, D.; Ertl, P.; Behnke, D.; Desrayaud, S.; Laue, G.; Vranesic, I. Discovery of 1H-pyrrolo[2,3-c]pyridine-7-carboxamides as novel, allosteric mGluR5 antagonists. Bioorg. Med. Chem. Lett. 2012, 22, 6454–6459. [Google Scholar] [CrossRef]
- Hong, S.; Kim, J.; Seo, J.H.; Jung, K.H.; Hong, S.-S.; Hong, S. Design, synthesis, and evaluation of 3,5-disubstituted 7-azaindoles as Trk inhibitors with anticancer and antiangiogenic activities. J. Med. Chem. 2012, 55, 5337–5349. [Google Scholar] [CrossRef]
- Kannaboina, P.; Anilkumar, K.; Aravinda, S.; Vishwakarma, R.A.; Das, P. Direct C-2 arylation of 7-azaindoles: Chemoselective access to multiarylated derivatives. Org. Lett. 2013, 15, 5718–5721. [Google Scholar] [CrossRef] [PubMed]
- Cardoza, S.; Das, P.; Tandon, V. Pd-catalyzed sequential arylation of 7-azaindoles: Aggregate-induced emission of tetra-aryl 7-azaindoles. J. Org. Chem. 2019, 84, 14015–14029. [Google Scholar] [CrossRef]
- Dar, M.O.; Bharatam, P.V.; Das, P.; Kukreti, S.; Tandon, V. Cu(II)-catalyzed sulfonylation of 7-azaindoles using DABSO as SO2-source and its mechanistic study. Tetrahedron 2020, 76, 131337. [Google Scholar] [CrossRef]
- Qian, G.; Hong, X.; Liu, B.; Mao, H.; Xu, B. Rhodium-catalyzed regioselective C-H chlorination of 7-azaindoles using 1,2-dichloroethane. Org. Lett. 2014, 16, 5294–5297. [Google Scholar] [CrossRef] [PubMed]
- Begunov, R.S.; Sokolov, A.A.; Belova, V.O.; Solov’ev, M.E. Quantum chemical study of regioselectivity of reactions of substituted pyrido[1,2-a]benzimidazoles with electrophiles. Russ. Chem. Bull. 2016, 65, 644–647. [Google Scholar] [CrossRef]
- Lassagne, F.; Sims, J.M.; Erb, W.; Mongin, O.; Richy, N.; El Osmani, N.; Fajloun, Z.; Picot, L.; Thiéry, V.; Robert, T.; et al. Thiazolo[5,4-f]quinoxalines, oxazolo[5,4-f]quinoxalines and pyrazino[b,e]isatins: Synthesis from 6-aminoquinoxalines and properties. Eur. J. Org. Chem. 2021, 2021, 2756–2763. [Google Scholar] [CrossRef]
- Fukui, K.; Yonezawa, T.; Nagata, C.; Shingu, H. Molecular-orbital theory of orientation in aromatic, heteroaromatic, and other conjugated molecules. J. Chem. Phys. 1954, 22, 1433–1442. [Google Scholar] [CrossRef] [Green Version]
- Carissan, Y.; Hagebaum-Reignier, D.; Goudard, N.; Humbel, S. Hückel-Lewis projection method: A “weights watcher” for mesomeric structures. J. Phys. Chem. A 2008, 112, 13256–13262. [Google Scholar] [CrossRef]
- Carissan, Y.; Hagebaum-Reignier, D.; Goudard, N.; Humbel, S. HuLiS Program: Lewis-Embedded in Hückel Theory. Available online: http://www.hulis.free.fr (accessed on 25 March 2021).
- Carissan, Y.; Hagebaum-Reignier, D.; Goudard, N.; Humbel, S. Weight Watchers électronique: Calculez votre poids de formes résonantes. L’Act. Chim. 2016, 406, 36–40. [Google Scholar]
- Goudard, N.; Carissan, Y.; Hagebaum-Reignier, D.; Humbel, S. HuLiS 3.3.4. Available online: http://ism2.univ-amu.fr/hulis (accessed on 23 March 2021).
- Hong, J.S.; Shim, H.S.; Kim, T.-J.; Kang, Y. (N-7-Azaindolyl)oligothiophenes: Synthesis, characterization, and photophysical properties. Tetrahedron 2007, 63, 8761–8769. [Google Scholar] [CrossRef]
- Hedidi, M.; Bentabed-Ababsa, G.; Derdour, A.; Roisnel, T.; Dorcet, V.; Chevallier, F.; Picot, L.; Thiéry, V.; Mongin, F. Synthesis of C,N′-linked bis-heterocycles using a deprotometalation-iodination-N-arylation sequence and evaluation of their antiproliferative activity in melanoma cells. Bioorg. Med. Chem. 2014, 22, 3498–3507. [Google Scholar] [CrossRef] [Green Version]
- Hedidi, M.; Erb, W.; Bentabed-Ababsa, G.; Chevallier, F.; Picot, L.; Thiery, V.; Bach, S.; Ruchaud, S.; Roisnel, T.; Dorcet, V.; et al. Synthesis of N-pyridyl azoles using a deprotometalation-iodolysis-N-arylation sequence and evaluation of their antiproliferative activity in melanoma cells. Tetrahedron 2016, 72, 6467–6476. [Google Scholar] [CrossRef] [Green Version]
- Amara, R.; Bentabed-Ababsa, G.; Hedidi, M.; Khoury, J.; Awad, H.; Nassar, E.; Roisnel, T.; Dorcet, V.; Chevallier, F.; Fajloun, Z.; et al. Synthesis of N-aryl and N-heteroaryl γ-, δ-, and ε-lactams using deprotometalation-iodination and N-arylation, and properties thereof. Synthesis 2017, 49, 4500–4516. [Google Scholar] [CrossRef] [Green Version]
- Hedidi, M.; Maillard, J.; Erb, W.; Lassagne, F.; Halauko, Y.S.; Ivashkevich, O.A.; Matulis, V.E.; Roisnel, T.; Dorcet, V.; Hamzé, M.; et al. Fused systems based on 2-aminopyrimidines: Synthesis combining deprotolithiation-in situ zincation with N-arylation reactions and biological properties. Eur. J. Org. Chem. 2017, 2017, 5903–5915. [Google Scholar] [CrossRef]
- Amara, R.; Awad, H.; Chaker, D.; Bentabed-Ababsa, G.; Lassagne, F.; Erb, W.; Chevallier, F.; Roisnel, T.; Dorcet, V.; Fajloun, Z.; et al. Conversion of isatins to tryptanthrins, heterocycles endowed with a myriad of bioactivities. Eur. J. Org. Chem. 2019, 2019, 5302–5312. [Google Scholar] [CrossRef]
- Gottlieb, H.E.; Kotlyar, V.; Nudelman, A. NMR chemical shifts of common laboratory solvents as trace impurities. J. Org. Chem. 1997, 62, 7512–7515. [Google Scholar] [CrossRef]
- Kjonaas, R.A.; Hoffer, R.K. Regiospecific 1,4-addition with Grignard-derived mixed triorganozincate reagents. J. Org. Chem. 1988, 53, 4133–4135. [Google Scholar] [CrossRef]
- Altomare, A.; Burla, M.C.; Camalli, M.; Cascarano, G.L.; Giacovazzo, C.; Guagliardi, A.; Moliterni, A.G.G.; Polidori, G.; Spagna, R. SIR97: A new tool for crystal structure determination and refinement. J. Appl. Crystallogr. 1999, 32, 115–119. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrugia, L.J. WinGX and ORTEP for windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Crystallogr. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with shelxl. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J. ORTEP-3 for windows—A version of ORTEP-III with a graphical user interface (GUI). J. Appl. Crystallogr. 1997, 30, 565. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision A.02; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Cances, E.; Mennucci, B.; Tomasi, J. A new integral equation formalism for the polarizable continuum model: Theoretical background and applications to isotropic and anisotropic dielectrics. J. Chem. Phys. 1997, 107, 3032–3041. [Google Scholar] [CrossRef]
- Hodgkinson, R.C.; Schulz, J.; Willis, M.C. Palladium-catalyzed tandem alkenyl- and aryl-C-N bond formation: A cascade N-annulation route to 1-functionalized 7-azaindoles. Tetrahedron 2009, 65, 8940–8949. [Google Scholar] [CrossRef]
- Phillips, D.P.; Zhu, X.-F.; Lau, T.L.; He, X.; Yang, K.; Liu, H. Copper-catalyzed C-N coupling of amides and nitrogen-containing heterocycles in the presence of cesium fluoride. Tetrahedron Lett. 2009, 50, 7293–7296. [Google Scholar] [CrossRef]
- Pai, G.; Chattopadhyay, A.P. N-arylation of nitrogen containing heterocycles with aryl halides using copper nanoparticle catalytic system. Tetrahedron Lett. 2016, 57, 3140–3145. [Google Scholar] [CrossRef]
- Klapars, A.; Antilla, J.C.; Huang, X.; Buchwald, S.L. A general and efficient copper catalyst for the amidation of aryl halides and the N-arylation of nitrogen heterocycles. J. Am. Chem. Soc. 2001, 123, 7727–7729. [Google Scholar] [CrossRef]
- Teo, Y.-C.; Yong, F.-F.; Lim, G.S. A manganese/copper bimetallic catalyst for C–N coupling reactions under mild conditions in water. Tetrahedron Lett. 2011, 52, 7171–7174. [Google Scholar] [CrossRef]
- Wu, Q.; Lavigne, J.A.; Tao, Y.; D’Iorio, M.; Wang, S. Blue-luminescent/electroluminescent Zn(II) compounds of 7-azaindole and N-(2-pyridyl)-7-azaindole: Zn(7-azaindole)2(CH3COO)2, Zn(NPA)(CH3COO)2, and Zn(NPA)((S)-(+)-CH3CH2CH(CH3)COO)2 (NPA = N-(2-pyridyl)-7-azaindole). Inorg. Chem. 2000, 39, 5248–5254. [Google Scholar] [CrossRef] [PubMed]
- Tani, K.; Sakurai, H.; Fujii, H.; Hirao, T. Synthesis of Re(I) complexes bearing tridentate 2,6-bis(7′-azaindolyl)phenyl ligand with green emission properties. J. Organomet. Chem. 2004, 689, 1665–1674. [Google Scholar] [CrossRef]
- Lee, E.; Thirupathaiah, B.; Han, J.; Jung, D.; Kwon, G.; Kim, C.; Seo, S. Novel organic semiconductors based on phenyl and phenylthienyl derivatives for organic thin-film transistors. J. Nanosci. Nanotechnol. 2016, 16, 910–919. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Ar-X | Method (Reaction Time) | Product (E), Yield (%) 1 |
|---|---|---|---|
| 1 2 3 4 5 |  | A (29 h) B (24 h) C (30 h) D (24 h) E (15 min) | 1a, 20 1a, 15 1a, 75 1a, 75 1a, 18 |
| 6 7 8 9 10 |  | A (6 d) B (24 h) C (6 d) D (24 h) E (25 min) | 1b, 0 1b, 50 1b, 54 1b, 60 1b, 35 |
| 11 |  | D (24 h) | 1c, 65 |
| 12 |  | D (24 h) | 1d, 80 |
| 13 14 |  | C (24 h) D (24 h) | 1e, 95 1e, 65 |
| 15 |  | C (24 h) | 1f, 97 |
| 16 17 |  | B (24 h) C (48 h) | 1g, 37 1g, 92 |
| 18 19 |  | C (48 h) D (24 h) | 1h, 80 1h, 80 |
| 20 21 |  | C (48 h) D (24 h) | 1i, 75 1i, 35 |
| 22 |  | D (24 h) | 1j, 35 |

| Entry | Substrate | Product(s) (E), Yield(s) (%) 1 | ||
|---|---|---|---|---|
| 1 |  | 1a |  | 3a, 75; 3a′, 5 |
| 2 |  | 1e |  | 3e, 10; 3e′, 20 2 |
| 3 |  | 1f |  | 3f, 40 3 |
| 4 |  | 1g |  | 3g, 30; 3g′, 40 |
| 5 |  | 2k |  | 3k, 20; 3k′, 20; 2 3k″, 14 2 |

| Entry | Substrate | n Equiv | Conditions | Product (E), Yield (%) 1 | ||
|---|---|---|---|---|---|---|
| 1 |  | 1a | 1.1 | rt, 12 h |  | 4a, 65 |
| 2 |  | 1b | 1.1 | rt, 12 h |  | 4b, 40 |
| 3 |  | 1c | 2 | 40 °C, 14 h |  | 4c, 40 |
| 4 |  | 1d | 1.1 | rt, 12 h |  | 4d, 52 |
| 5 6 |  | 1e | 1.1 2 | rt, 12 h 40 °C, 14 h |  | 4e, 33 4e, 45 |
| 7 |  | 1f | 1.1 | rt, 12 h |  | 4f, 62 |
| 8 |  | 1g | 1.1 | rt, 12 h |  | 3g, 35 |
| 9 10 |  | 1h | 1.1 2 | 40 °C, 12 h 40 °C, 10 h |  | 4h, 36 4h, 45 |
| 11 12 |  | 1i | 1.1 2 | rt, 12 h 40 °C, 14 h |  | 4i, 17 4i, 51 |

| Entry | Ar-I (n Equiv) | Method (Time) | Product (E), Yield (%) 1 | |||
|---|---|---|---|---|---|---|
| 1 |  | 4e | (1) | D (24 h) |  | 5e, 30 2 |
| 2 |  | 4i | (1) | C (24 h) |  | 5i, 50 2 |
| 3 |  | 3g′ | (0.5) | C (24 h) |  | 6g′, 20 3 |
| Compound | Amount dissolved in DMSO | Escherichia coli | Pseudomonas aeruginosa | Staphylococcus aureus | Enterococcus faecium | Listeria monocytogenes | Candida dubliniensis | Candida albicans |
|---|---|---|---|---|---|---|---|---|
| 1a | 500 μg 2 | 6 | 5 | 8 | 5 | ± | 23 | - |
| 1b | 250 μg 3 | 0 | 0 | 0 | 11 | 0 | 18 | - |
| 1c | 150 μg 4 | 0 | 0 | 0 | 0 | 0 | - | 0 |
| 1e | 500 μg 2 | ± | 0 | 0 | 0 | 0 | 0 | - |
| 1f | 200 μg 3 | 0 | 0 | 0 | 11 | 0 | 13 | - |
| 1h | 500 μg 2 | 10 | 8 | 11 | 0 | 10 | 27 | - |
| 1i | 250 μg 3 | 0 | 0 | ± | 13 | 0 | 16 | - |
| 1j | 250 μg 3 | 0 | 0 | 0 | 15 | 22 | 10 | - |
| 2k | 150 μg 4 | 0 | 0 | 0 | 0 | 0 | - | 0 |
| 2l | 500 μg 2 | 0 | 0 | 0 | 0 | 0 | 0 | - |
| 3a | 500 μg 2 | 0 | 0 | 0 | 0 | 0 | 13 | - |
| 3g | 500 μg 2 | ± | 0 | 0 | 0 | 0 | 0 | - |
| 3g′ | 500 μg 2 | ± | ± | 0 | 0 | 0 | 0 | - |
| 5i | 500 μg 2 | 0 | 0 | 0 | 0 | 0 | 0 | - |
| Reference compound | 28 5 | 28 5 | 18 6 | 24 6 | 30 7 | 10 8 | - | |
| Compound | RSA (%) 1 at t = 0 min | RSA (%) 1 at t = 30 min |
|---|---|---|
| 1b | 52 | 54 |
| 1c | 98 | 58 |
| 1f | 42 | 48 |
| 1i | 44 | 46 |
| 1j | 55 | 58 |
| 5i | 51 | 53 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ameur Messaoud, M.Y.; Bentabed-Ababsa, G.; Fajloun, Z.; Hamze, M.; Halauko, Y.S.; Ivashkevich, O.A.; Matulis, V.E.; Roisnel, T.; Dorcet, V.; Mongin, F. Deprotometalation-Iodolysis and Direct Iodination of 1-Arylated 7-Azaindoles: Reactivity Studies and Molecule Properties. Molecules 2021, 26, 6314. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26206314
Ameur Messaoud MY, Bentabed-Ababsa G, Fajloun Z, Hamze M, Halauko YS, Ivashkevich OA, Matulis VE, Roisnel T, Dorcet V, Mongin F. Deprotometalation-Iodolysis and Direct Iodination of 1-Arylated 7-Azaindoles: Reactivity Studies and Molecule Properties. Molecules. 2021; 26(20):6314. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26206314
Chicago/Turabian StyleAmeur Messaoud, Mohamed Yacine, Ghenia Bentabed-Ababsa, Ziad Fajloun, Monzer Hamze, Yury S. Halauko, Oleg A. Ivashkevich, Vadim E. Matulis, Thierry Roisnel, Vincent Dorcet, and Florence Mongin. 2021. "Deprotometalation-Iodolysis and Direct Iodination of 1-Arylated 7-Azaindoles: Reactivity Studies and Molecule Properties" Molecules 26, no. 20: 6314. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26206314