
Engineered Fully Human Single-Chain Monoclonal Antibodies to PIM2 Kinase

 ,
,  ,
,  , and
, and
Abstract
:1. Introduction
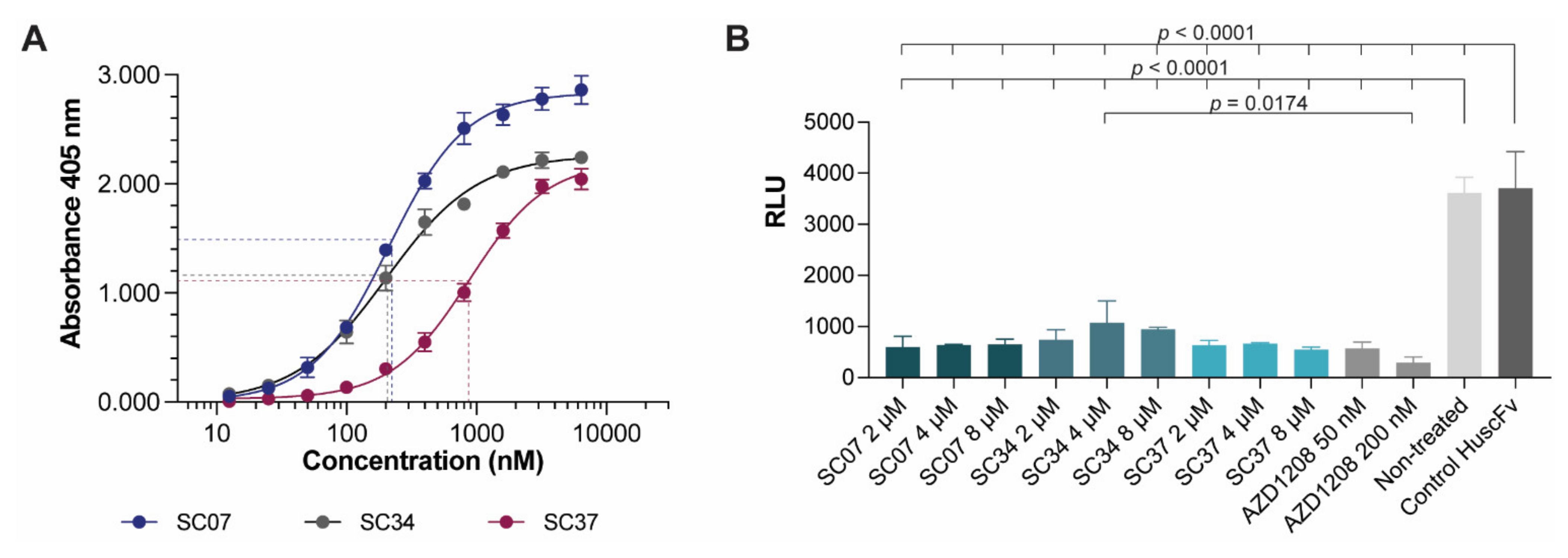
2. Results
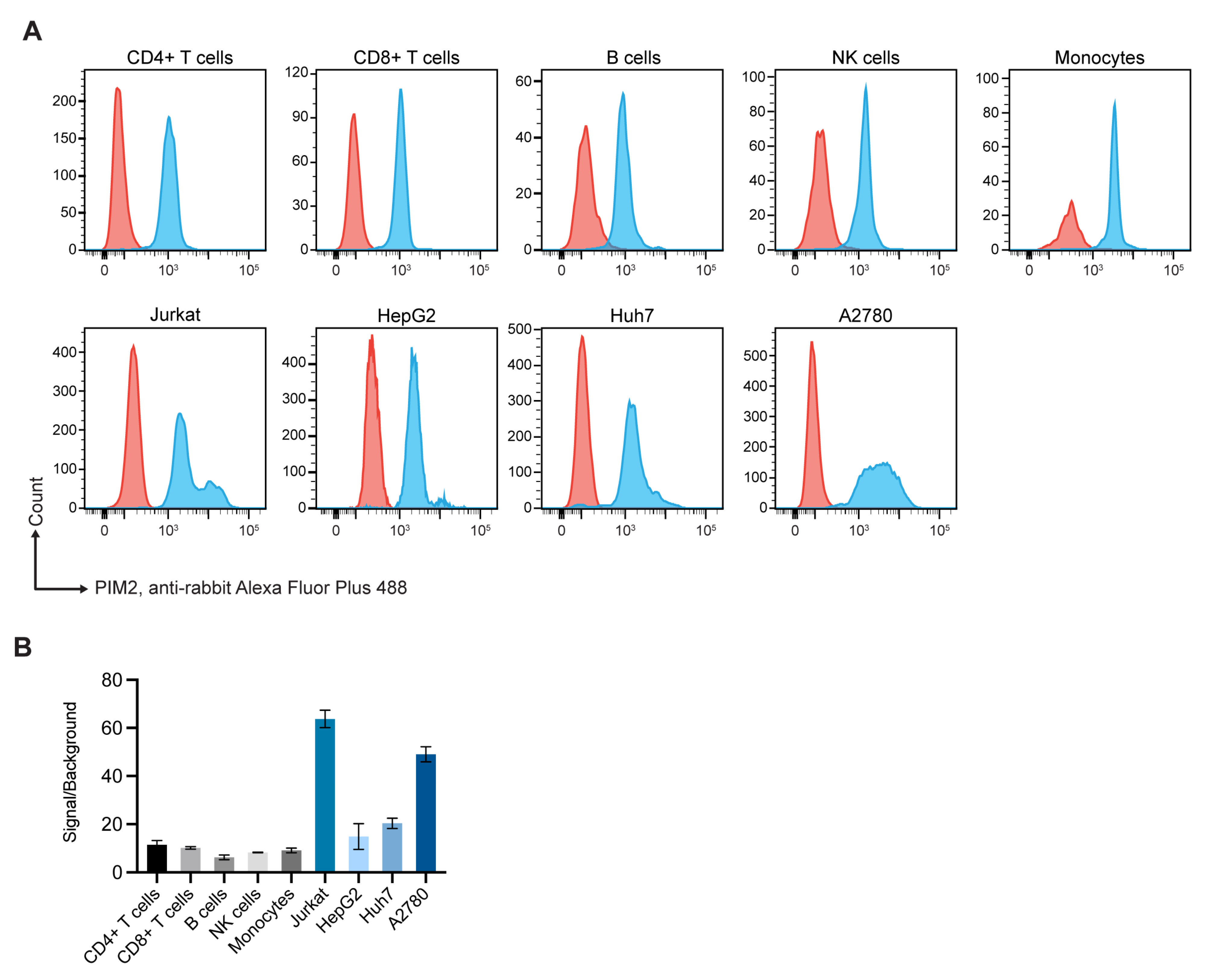
2.1. Expressions of Pim2 by Normal Blood Cell Subpopulations and Cancer Cells
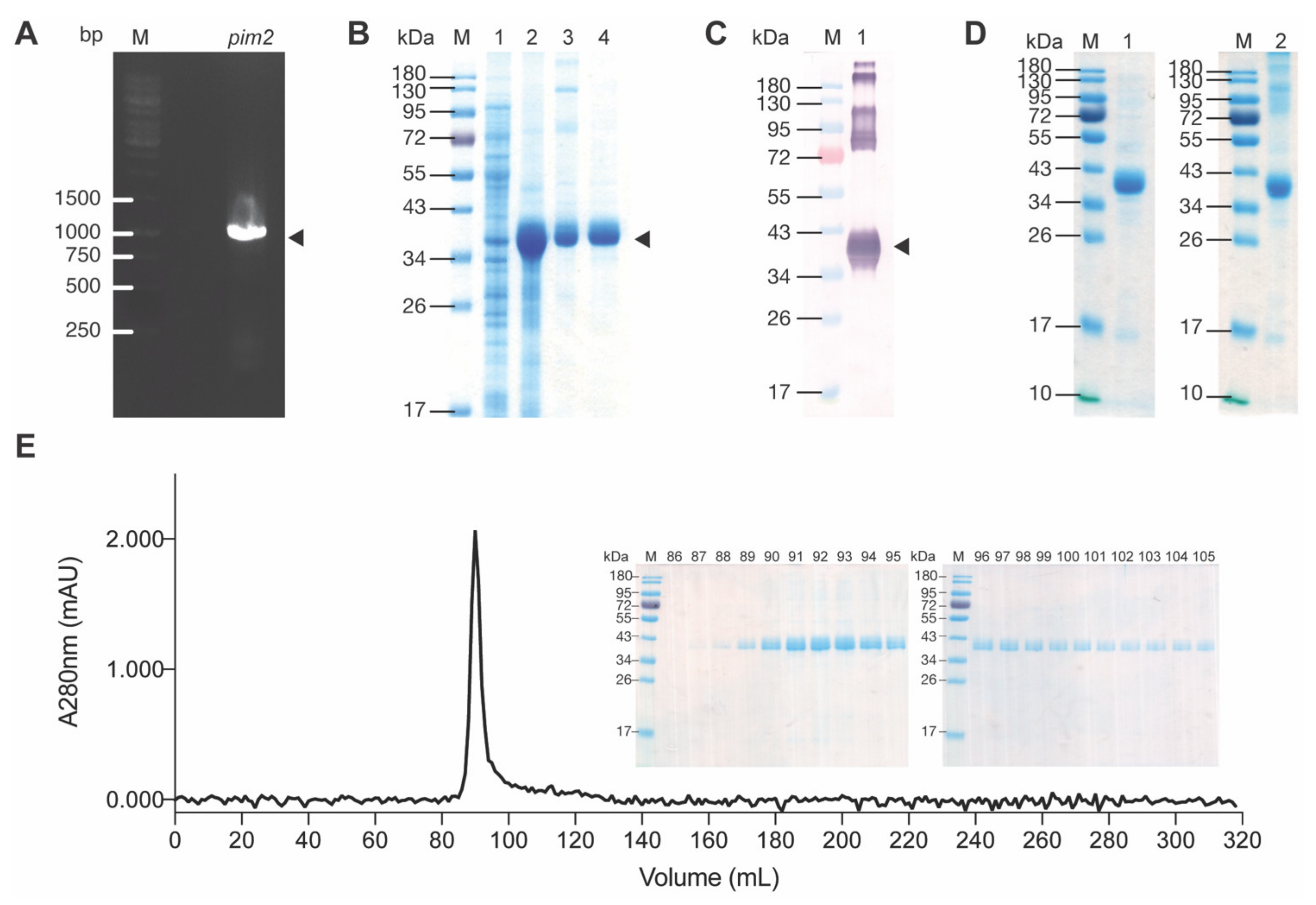
2.2. Recombinant PIM2
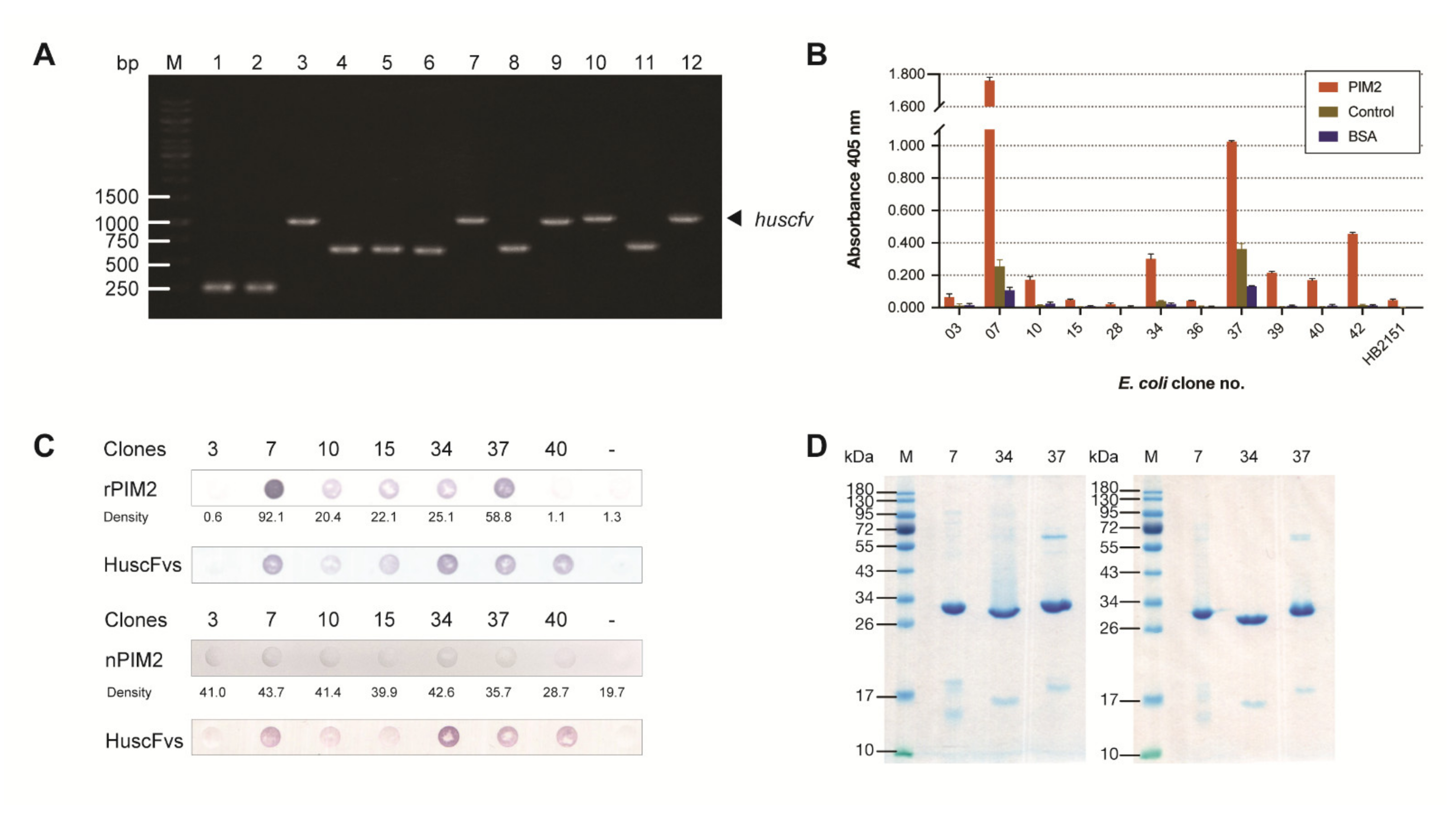
2.3. Production of HuscFvs to Recombinant PIM2 (rPIM2) and Binding of the HuscFvs to rPIM2 and Native PIM2
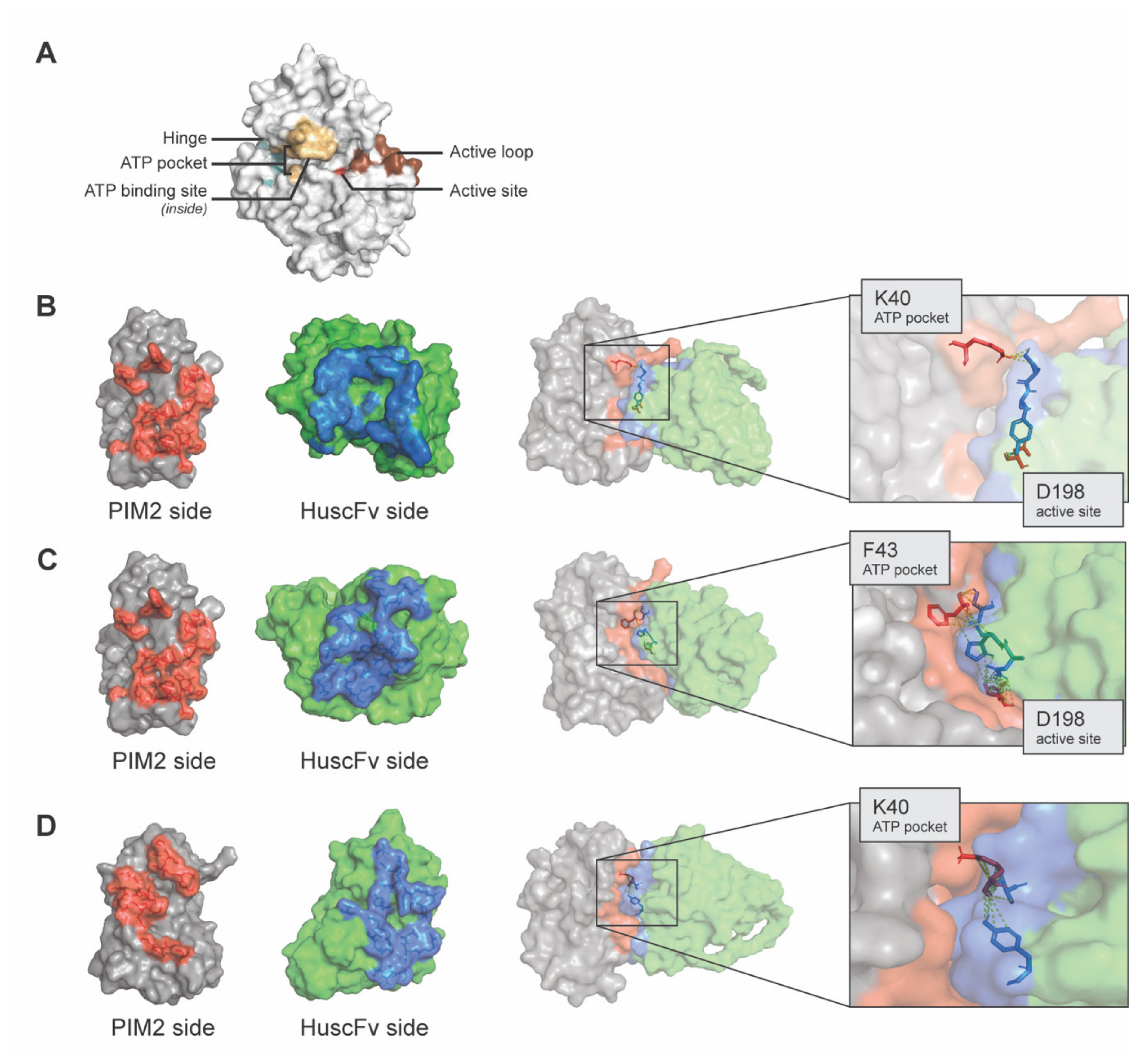
2.4. Computerized Simulation for Determining Presumptive Region(s) and Residues of PIM2 That Were Bound by the HuscFvs
2.5. Effective Concentration 50 (EC50) of HuscFvs and HuscFv-mediated Inhibition of PIM2 Kinase Activity
3. Discussion
4. Materials and Methods
4.1. Verification of PIM2 Upregulation in Cancer Cells
4.2. Preparation of Recombinant PIM2 (rPIM2)
4.3. SDS-PAGE, Native-PAGE and Western Blot Analysis
4.4. Size Exclusion Column Chromatography (SEC)
4.5. HuscFv Phage Display Library
4.6. Production of HuscFvs to rPIM2
4.7. Binding of the HuscFvs to Recombinant and Native PIM2
4.8. Large Scale Production of Soluble HuscFvs
4.9. Computerized Simulation for Determining Presumptive Residues of PIMs Bound by the HuscFvs to PIM2
4.10. Determination of Effective Concentration-50 (EC50) of the HuscFvs
4.11. Kinase and Kinase Inhibition Assays
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- van der Lugt, N.M.; Domen, J.; Verhoeven, E.; Linders, K.; van der Gulden, H.; Allen, J.; Berns, A. Proviral tagging in E mu-myc transgenic mice lacking the Pim-1 proto-oncogene leads to compensatory activation of Pim-2. EMBO J. 1995, 14, 2536–2544. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.D.; Verhoeven, E.; Domen, J.; van der Valk, M.; Berns, A. Pim-2 transgene induces lymphoid tumors, exhibiting potent synergy with c-myc. Oncogene 1997, 15, 1133–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarado, Y.; Giles, F.J.; Swords, R.T. The PIM kinases in hematological cancers. Expert Rev. Hematol. 2012, 5, 81–96. [Google Scholar] [CrossRef] [PubMed]
- Bullock, A.N.; Russo, S.; Amos, A.; Pagano, N.; Bregman, H.; Debreczeni, J.E.; Lee, W.H.; von Delft, F.; Meggers, E.; Knapp, S. Crystal structure of the PIM2 kinase in complex with an organoruthenium inhibitor. PLoS ONE 2009, 4, e7112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, C.J.; Hammerman, P.S.; Cinalli, R.M.; Master, S.R.; Chodosh, L.A.; Thompson, C.B. The serine/threonine kinase Pim-2 is a transcriptionally regulated apoptotic inhibitor. Genes Dev. 2003, 17, 1841–1854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Abad, C.; Pisonero, H.; Blanco-Aparicio, C.; Roncador, G.; Gonzalez-Menchen, A.; Martinez-Climent, J.A.; Mata, E.; Rodriguez, M.E.; Munoz-Gonzalez, G.; Sanchez-Beato, M.; et al. PIM2 inhibition as a rational therapeutic approach in B-cell lymphoma. Blood 2011, 118, 5517–5527. [Google Scholar] [CrossRef] [Green Version]
- Yan, B.; Zemskova, M.; Holder, S.; Chin, V.; Kraft, A.; Koskinen, P.J.; Lilly, M. The PIM-2 kinase phosphorylates BAD on serine 112 and reverses BAD-induced cell death. J. Biol. Chem. 2003, 278, 45358–45367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Wang, Z.; Li, X.; Magnuson, N.S. Pim kinase-dependent inhibition of c-Myc degradation. Oncogene 2008, 27, 4809–4819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clemens, M.J.; Bushell, M.; Jeffrey, I.W.; Pain, V.M.; Morley, S.J. Translation initiation factor modifications and the regulation of protein synthesis in apoptotic cells. Cell Death Differ. 2000, 7, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Sonenberg, N.; Gingras, A.C.; Peterson, M.; Avdulov, S.; Polunovsky, V.A.; Bitterman, P.B. Translational control of cell fate: Availability of phosphorylation sites on translational repressor 4E-BP1 governs its proapoptotic potency. Mol. Cell. Biol. 2002, 22, 2853–2861. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Zavorotinskaya, T.; Dai, Y.; Niu, X.H.; Castillo, J.; Sim, J.; Yu, J.; Wang, Y.; Langowski, J.L.; Holash, J.; et al. Pim2 is required for maintaining multiple myeloma cell growth through modulating TSC2 phosphorylation. Blood 2013, 122, 1610–1620. [Google Scholar] [CrossRef] [PubMed]
- Hospital, M.A.; Jacquel, A.; Mazed, F.; Saland, E.; Larrue, C.; Mondesir, J.; Birsen, R.; Green, A.S.; Lambert, M.; Sujobert, P.; et al. RSK2 is a new Pim2 target with pro-survival functions in FLT3-ITD-positive acute myeloid leukemia. Leukemia 2018, 32, 597–605. [Google Scholar] [CrossRef] [PubMed]
- Ren, K.; Duan, W.; Shi, Y.; Li, B.; Liu, Z.; Gong, J. Ectopic over-expression of oncogene Pim-2 induce malignant transformation of nontumorous human liver cell line L02. J. Korean Med. Sci. 2010, 25, 1017–1023. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, S.; Kaneita, Y.; Aoki, Y.; Seto, M.; Mori, S.; Moriyama, M. Identification of heterologous translocation partner genes fused to the BCL6 gene in diffuse large B-cell lymphomas: 5’-RACE and LA - PCR analyses of biopsy samples. Oncogene 1999, 18, 7994–7999. [Google Scholar] [CrossRef] [Green Version]
- Amson, R.; Sigaux, F.; Przedborski, S.; Flandrin, G.; Givol, D.; Telerman, A. The human protooncogene product p33pim is expressed during fetal hematopoiesis and in diverse leukemias. Proc. Natl. Acad. Sci. USA 1989, 86, 8857–8861. [Google Scholar] [CrossRef] [Green Version]
- Claudio, J.O.; Masih-Khan, E.; Tang, H.; Goncalves, J.; Voralia, M.; Li, Z.H.; Nadeem, V.; Cukerman, E.; Francisco-Pabalan, O.; Liew, C.C.; et al. A molecular compendium of genes expressed in multiple myeloma. Blood 2002, 100, 2175–2186. [Google Scholar] [CrossRef] [PubMed]
- Dhanasekaran, S.M.; Barrette, T.R.; Ghosh, D.; Shah, R.; Varambally, S.; Kurachi, K.; Pienta, K.J.; Rubin, M.A.; Chinnaiyan, A.M. Delineation of prognostic biomarkers in prostate cancer. Nature 2001, 412, 822–826. [Google Scholar] [CrossRef] [PubMed]
- Neill, G.W.; Kelsell, D.P. Spotting prostate cancer. Trends Mol. Med. 2001, 7, 432. [Google Scholar] [CrossRef]
- Fujii, C.; Nakamoto, Y.; Lu, P.; Tsuneyama, K.; Popivanova, B.K.; Kaneko, S.; Mukaida, N. Aberrant expression of serine/threonine kinase Pim-3 in hepatocellular carcinoma development and its role in the proliferation of human hepatoma cell lines. Int. J. Cancer 2005, 114, 209–218. [Google Scholar] [CrossRef] [Green Version]
- Kapelko-Slowik, K.; Owczarek, T.B.; Grzymajlo, K.; Urbaniak-Kujda, D.; Jazwiec, B.; Slowik, M.; Kuliczkowski, K.; Ugorski, M. Elevated PIM2 gene expression is associated with poor survival of patients with acute myeloid leukemia. Leuk. Lymphoma 2016, 57, 2140–2149. [Google Scholar] [CrossRef]
- Aziz, A.U.R.; Farid, S.; Qin, K.; Wang, H.; Liu, B. PIM kinases and their relevance to the PI3K/AKT/mTOR pathway in the regulation of ovarian cancer. Biomolecules 2018, 8, 7. [Google Scholar] [CrossRef] [Green Version]
- Lu, R.M.; Hwang, Y.C.; Liu, I.J.; Lee, C.C.; Tsai, H.Z.; Li, H.J.; Wu, H.C. Development of therapeutic antibodies for the treatment of diseases. J. Biomed. Sci. 2020, 27, 1. [Google Scholar] [CrossRef] [PubMed]
- Kulkeaw, K.; Sakolvaree, Y.; Srimanote, P.; Tongtawe, P.; Maneewatch, S.; Sookrung, N.; Tungtrongchitr, A.; Tapchaisri, P.; Kurazono, H.; Chaicumpa, W. Human monoclonal ScFv neutralize lethal Thai cobra, Naja kaouthia, neurotoxin. J. Proteomics 2009, 72, 270–282. [Google Scholar] [CrossRef]
- Wang, Y.; Xiu, J.; Ren, C.; Yu, Z. Protein kinase PIM2: A simple PIM family kinase with complex functions in cancer metabolism and therapeutics. J. Cancer 2021, 12, 2570–2581. [Google Scholar] [CrossRef] [PubMed]
- Luszczak, S.; Kumar, C.; Sathyadevan, V.K.; Simpson, B.S.; Gately, K.A.; Whitaker, H.C.; Heavey, S. PIM kinase inhibition: Co-targeted therapeutic approaches in prostate cancer. Signal Transduct. Target. Ther. 2020, 5, 7. [Google Scholar] [CrossRef] [PubMed]
- Sawaguchi, Y.; Yamazaki, R.; Nishiyama, Y.; Mae, M.; Abe, A.; Nishiyama, H.; Nishisaka, F.; Ibuki, T.; Sasai, T.; Matsuzaki, T. Novel pan-Pim kinase inhibitors with imidazopyridazine and thiazolidinedione structure exert potent antitumor activities. Front. Pharmacol. 2021, 12, 672536. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.W.; Beharry, Z.M.; Hill, E.G.; Song, J.H.; Wang, W.; Xia, Z.; Zhang, Z.; Aplan, P.D.; Aster, J.C.; Smith, C.D.; et al. A small molecule inhibitor of Pim protein kinases blocks the growth of precursor T-cell lymphoblastic leukemia/lymphoma. Blood 2010, 115, 824–833. [Google Scholar] [CrossRef] [Green Version]
- Fujii, S.; Nakamura, S.; Oda, A.; Miki, H.; Tenshin, H.; Teramachi, J.; Hiasa, M.; Bat-Erdene, A.; Maeda, Y.; Oura, M.; et al. Unique anti-myeloma activity by thiazolidine-2,4-dione compounds with Pim inhibiting activity. Br. J. Haematol. 2018, 180, 246–258. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.S.; Redkar, S.; Taverna, P.; Cortes, J.E.; Gandhi, V. Mechanisms of cytotoxicity to Pim kinase inhibitor, SGI-1776, in acute myeloid leukemia. Blood 2011, 118, 693–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foulks, J.M.; Carpenter, K.J.; Luo, B.; Xu, Y.; Senina, A.; Nix, R.; Chan, A.; Clifford, A.; Wilkes, M.; Vollmer, D.; et al. A small-molecule inhibitor of PIM kinases as a potential treatment for urothelial carcinomas. Neoplasia 2014, 16, 403–412. [Google Scholar] [CrossRef] [Green Version]
- Nair, J.R.; Caserta, J.; Belko, K.; Howell, T.; Fetterly, G.; Baldino, C.; Lee, K.P. Novel inhibition of PIM2 kinase has significant anti-tumor efficacy in multiple myeloma. Leukemia 2017, 31, 1715–1726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santio, N.M.; Vahakoski, R.L.; Rainio, E.M.; Sandholm, J.A.; Virtanen, S.S.; Prudhomme, M.; Anizon, F.; Moreau, P.; Koskinen, P.J. Pim-selective inhibitor DHPCC-9 reveals Pim kinases as potent stimulators of cancer cell migration and invasion. Mol. Cancer 2010, 9, 279. [Google Scholar] [CrossRef] [Green Version]
- Keeton, E.K.; McEachern, K.; Dillman, K.S.; Palakurthi, S.; Cao, Y.; Grondine, M.R.; Kaur, S.; Wang, S.; Chen, Y.; Wu, A.; et al. AZD1208, a potent and selective pan-Pim kinase inhibitor, demonstrates efficacy in preclinical models of acute myeloid leukemia. Blood 2014, 123, 905–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haddach, M.; Michaux, J.; Schwaebe, M.K.; Pierre, F.; O’Brien, S.E.; Borsan, C.; Tran, J.; Raffaele, N.; Ravula, S.; Drygin, D.; et al. Discovery of CX-6258. A potent, selective, and orally efficacious pan-Pim kinases inhibitor. ACS Med. Chem. Lett. 2012, 3, 135–139. [Google Scholar] [CrossRef]
- Raab, M.S.; Thomas, S.K.; Ocio, E.M.; Guenther, A.; Goh, Y.-T.; Talpaz, M.; Hohmann, N.; Zhao, S.; Xiang, F.; Simon, C.; et al. The first-in-human study of the pan-PIM kinase inhibitor PIM447 in patients with relapsed and/or refractory multiple myeloma. Leukemia 2019, 33, 2924–2933. [Google Scholar] [CrossRef] [PubMed]
- Cervantes-Gomez, F.; Chen, L.S.; Orlowski, R.Z.; Gandhi, V. Biological effects of the Pim kinase inhibitor, SGI-1776, in multiple myeloma. Clin. Lymphoma Myeloma Leuk. 2013, 13 Suppl 2, S317–S329. [Google Scholar] [CrossRef] [Green Version]
- Lin, A.; Giuliano, C.J.; Palladino, A.; John, K.M.; Abramowicz, C.; Yuan, M.L.; Sausville, E.L.; Lukow, D.A.; Liu, L.; Chait, A.R.; et al. Off-target toxicity is a common mechanism of action of cancer drugs undergoing clinical trials. Sci. Trans. Med. 2019, 11, eaaw8412. [Google Scholar] [CrossRef] [PubMed]
- Bruggemann, M.; Osborn, M.J.; Ma, B.; Hayre, J.; Avis, S.; Lundstrom, B.; Buelow, R. Human antibody production in transgenic animals. Arch. Immunol. Ther. Exp. (Warsz.) 2015, 63, 101–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinitz, M. Production of human monoclonal antibodies by the epstein-barr virus method. Methods Mol. Biol. 2014, 1060, 111–122. [Google Scholar] [CrossRef]
- Mazor, Y.; Van Blarcom, T.; Mabry, R.; Iverson, B.L.; Georgiou, G. Isolation of engineered, full-length antibodies from libraries expressed in Escherichia coli. Nat. Biotechnol. 2007, 25, 563–565. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Jacobsen, F.W.; Cai, L.; Chen, Q.; Shen, W.D. Development of a novel mammalian cell surface antibody display platform. MAbs 2010, 2, 508–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaheen, H.H.; Prinz, B.; Chen, M.-T.; Pavoor, T.; Lin, S.; Houston-Cummings, N.R.; Moore, R.; Stadheim, T.A.; Zha, D. A dual-mode surface display system for the maturation and production of monoclonal antibodies in glyco-engineered Pichia pastoris. PLoS ONE 2013, 8, e70190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiner, L.M.; Dhodapkar, M.V.; Ferrone, S. Monoclonal antibodies for cancer immunotherapy. Lancet 2009, 373, 1033–1040. [Google Scholar] [CrossRef] [Green Version]
- Young Kim, H.; Young Yum, S.; Jang, G.; Ahn, D.R. Discovery of a non-cationic cell penetrating peptide derived from membrane-interacting human proteins and its potential as a protein delivery carrier. Sci. Rep. 2015, 5, 11719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, S.L.; Fan, T.C.; Fu, H.W.; Chen, C.J.; Hwang, C.S.; Hung, T.J.; Lin, L.Y.; Chang, M.D. A novel cell-penetrating peptide derived from human eosinophil cationic protein. PLoS ONE 2013, 8, e57318. [Google Scholar] [CrossRef] [Green Version]
- Phanthong, S.; Densumite, J.; Seesuay, W.; Thanongsaksrikul, J.; Teimoori, S.; Sookrung, N.; Poovorawan, Y.; Onvimala, N.; Guntapong, R.; Pattanapanyasat, K.; et al. Human antibodies to VP4 inhibit replication of enteroviruses across subgenotypes and serotypes, and enhance host innate immunity. Front. Microbiol. 2020, 11, 562768. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, M.J.; Billingsley, M.M.; Haley, R.M.; Wechsler, M.E.; Peppas, N.A.; Langer, R. Engineering precision nanoparticles for drug delivery. Nat. Rev. Drug Discov. 2021, 20, 101–124. [Google Scholar] [CrossRef]
- Adolf-Bryfogle, J.; Xu, Q.; North, B.; Lehmann, A.; Dunbrack, R.L., Jr. PyIgClassify: A database of antibody CDR structural classifications. Nucleic Acids Res. 2015, 43, D432–D438. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Zhang, Y. Improving the physical realism and structural accuracy of protein models by a two-step atomic-level energy minimization. Biophys. J. 2011, 101, 2525–2534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Liang, Y.; Zhang, Y. Atomic-level protein structure refinement using fragment-guided molecular dynamics conformation sampling. Structure 2011, 19, 1784–1795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozakov, D.; Hall, D.R.; Xia, B.; Porter, K.A.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The ClusPro web server for protein-protein docking. Nat. Protoc. 2017, 12, 255–278. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.C.; Rodrigues, J.P.; Kastritis, P.L.; Bonvin, A.M.; Vangone, A. PRODIGY: A web server for predicting the binding affinity of protein-protein complexes. Bioinformatics 2016, 32, 3676–3678. [Google Scholar] [CrossRef]
- Xiong, P.; Zhang, C.; Zheng, W.; Zhang, Y. BindProfX: Assessing mutation-induced binding affinity change by protein interface profiles with pseudo-counts. J. Mol. Biol. 2017, 429, 426–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| E. coli Clone No. | Ig Domain | Closest Human V Region | Identity (%) | Amino Acid Homology with Human FRs (%) | ||
|---|---|---|---|---|---|---|
| FR1 | FR2 | FR3 | ||||
| 3 | VH | M99649 IGHV3-7*01 | 96.53 | 92.00 | 100.00 | 92.11 |
| VL | Z00023 IGKV4-1801 | 97.64 | 100.00 | 100.00 | 100.00 | |
| 7 | VH | M99660 IGHV3-23*01 | 100.00 | 100.00 | 100.00 | 100.00 |
| VL | X01668 IGKV3-11*01 | 97.85 | 100.00 | 100.00 | 94.44 | |
| 10 | VH | J04096 IGHV6-1*01 | 100.00 | 100.00 | 100.00 | 100.00 |
| VL | Z00013 IGKV1-0*01 | 93.19 | 84.62 | 94.12 | 97.22 | |
| 15 | VH | M99641 IGHV-18*01 | 98.61 | 96.00 | 100.00 | 100.00 |
| VL | X59315 IGKV1-39*01 | 98.92 | 92.31 | 100.00 | 100.00 | |
| 28 | VH | X62112 IGHV4-4*07 | 99.30 | 100.00 | 100.00 | 128.57 |
| VL | X59315 IGKV1-39*01 | 96.77 | 92.31 | 100.00 | 97.22 | |
| 34 | VH | X92255 IGHV4-34*03 | 97.89 | 100.00 | 100.00 | 94.59 |
| VL | X12686 IGKV3-20*01 | 91.49 | 96.15 | 88.24 | 91.67 | |
| 37 | VH | AC245166 IGHV3-23*04 | 100.00 | 100.00 | 100.00 | 100.00 |
| VL | M23090 IGKV3-15*01 | 95.70 | 96.15 | 94.12 | 94.44 | |
| 40 | VH | M99663 IGHV3-30*03 | 97.92 | 80.00 | 100.00 | 100.00 |
| VL | X12686 IGKV3-20*01 | 99.29 | 100.00 | 100.00 | 100.00 | |
| 42 | VH | X92343 IGHV1-46*01 | 99.65 | 96.00 | 100.00 | 100.00 |
| VL | M23090 IGKV3-15*01 | 96.06 | 96.15 | 100.00 | 88.89 | |
| PIM2 | HuscFv7 | Interactive Bond | ||
|---|---|---|---|---|
| Residue | Region | Residue | Domain | |
| Y214 | T28 | VH-CDR1 | Hydrogen | |
| H215 | T28 | VH-CDR1 | Hydrogen | |
| A216 | T28 | VH-CDR1 | Hydrogen | |
| A187 | S54 | VHCDR2 | Hydrogen | |
| R65 | Y59 | VH-CDR2 | Hydrogen | |
| I63 | Y105 | VH-CDR3 | Pi-alkyl | |
| P64 | Y105 | VH-CDR3 | Pi-alkyl | |
| K40 | ATP pocket | S106 | VH-CDR3 | Hydrogen |
| D198 | Active loop | Y107 | VH-CDR3 | Hydrogen |
| E239 | K111 | VH-CDR3 | Salt bridge | |
| R201 | D112 | VH-CDR3 | Hydrogen | |
| G199 | Y113 | VH-CDR3 | Hydrogen | |
| H212 | D120 | VH-CDR3 | Hydrogen | |
| R65 | W241 | VL-CDR3 | Hydrogen, Pi-alkyl | |
| HuscFv34 | ||||
| Residue | Domain | |||
| H63 | F29 | VH-FR1 | Pi-alkyl | |
| P64 | F29 | VH-FR1 | Pi-alkyl | |
| F43 | ATP pocket | S30 | VH-FR1 | Hydrogen |
| R201 | E50 | VH-CDR2 | Salt bridge, Attractive charge | |
| D198 | Active loop | N52 | VH-CDR2 | Hydrogen |
| F43 | ATP pocket | H53 | VH-CDR2 | Hydrogen |
| S185 | H53 | VH-CDR2 | Hydrogen | |
| R201 | S56 | VH-CDR2 | Hydrogen | |
| P64 | N76 | VH-CDR4 | Hydrogen | |
| H212 | R103 | VH-CDR3 | Pi-cation | |
| R211 | S163 | VL-CDR1 | Hydrogen | |
| H212 | S163 | VL-CDR1 | Hydrogen | |
| R201 | Y229 | VL-CDR3 | Hydrogen | |
| H212 | Y229 | VL-CDR3 | Hydrogen | |
| HuscFv37 | ||||
| Residue | Domain | |||
| L37 | N101 | VH-CDR3 | Hydrogen | |
| D124 | Y102 | VH-CDR3 | Hydrogen | |
| A122, Q123 | Y102 | H-CDR3 | Amide-pi | |
| E131 | F104 | H-CDR3 | Pi-anion | |
| K40 | ATP pocket | Y111 | VH-CDR3 | Hydrogen |
| E131 | R170 | VL-CDR1 | Hydrogen | |
| K132 | R170 | VL-CDR1 | Hydrogen | |
| G234 | N171 | VL-CDR1 | Hydrogen | |
| T130 | N172 | VL-CDR1 | Hydrogen | |
| K40 | ATP pocket | Y189 | VH-FR2 | Hydrogen |
| K40 | ATP pocket | T196 | VL-CDR2 | Hydrogen |
| D235 | R206 | VL-CDR4 | Hydrogen | |
| E239 | R206 | VL-CDR4 | Salt bridge | |
| S207 | S207 | VL-CDR4 | Hydrogen | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaewchim, K.; Glab-ampai, K.; Mahasongkram, K.; Chulanetra, M.; Seesuay, W.; Chaicumpa, W.; Sookrung, N. Engineered Fully Human Single-Chain Monoclonal Antibodies to PIM2 Kinase. Molecules 2021, 26, 6436. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26216436
Kaewchim K, Glab-ampai K, Mahasongkram K, Chulanetra M, Seesuay W, Chaicumpa W, Sookrung N. Engineered Fully Human Single-Chain Monoclonal Antibodies to PIM2 Kinase. Molecules. 2021; 26(21):6436. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26216436
Chicago/Turabian StyleKaewchim, Kanasap, Kittirat Glab-ampai, Kodchakorn Mahasongkram, Monrat Chulanetra, Watee Seesuay, Wanpen Chaicumpa, and Nitat Sookrung. 2021. "Engineered Fully Human Single-Chain Monoclonal Antibodies to PIM2 Kinase" Molecules 26, no. 21: 6436. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26216436