
Modulation and Pharmacology of the Mitochondrial Permeability Transition: A Journey from F-ATP Synthase to ANT


Abstract
:1. Introduction
2. Modulation of the PTP
3. Cyclophilin D: A Master PTP Regulator
4. Mitochondrial Permeability Pathways: The ANT and the F-ATP Synthase Hypotheses
4.1. The ANT Pore
4.2. The F-ATP Synthase Pore
4.3. F-ATP Synthase and ANT Mediate Distinct Permeability Pathways
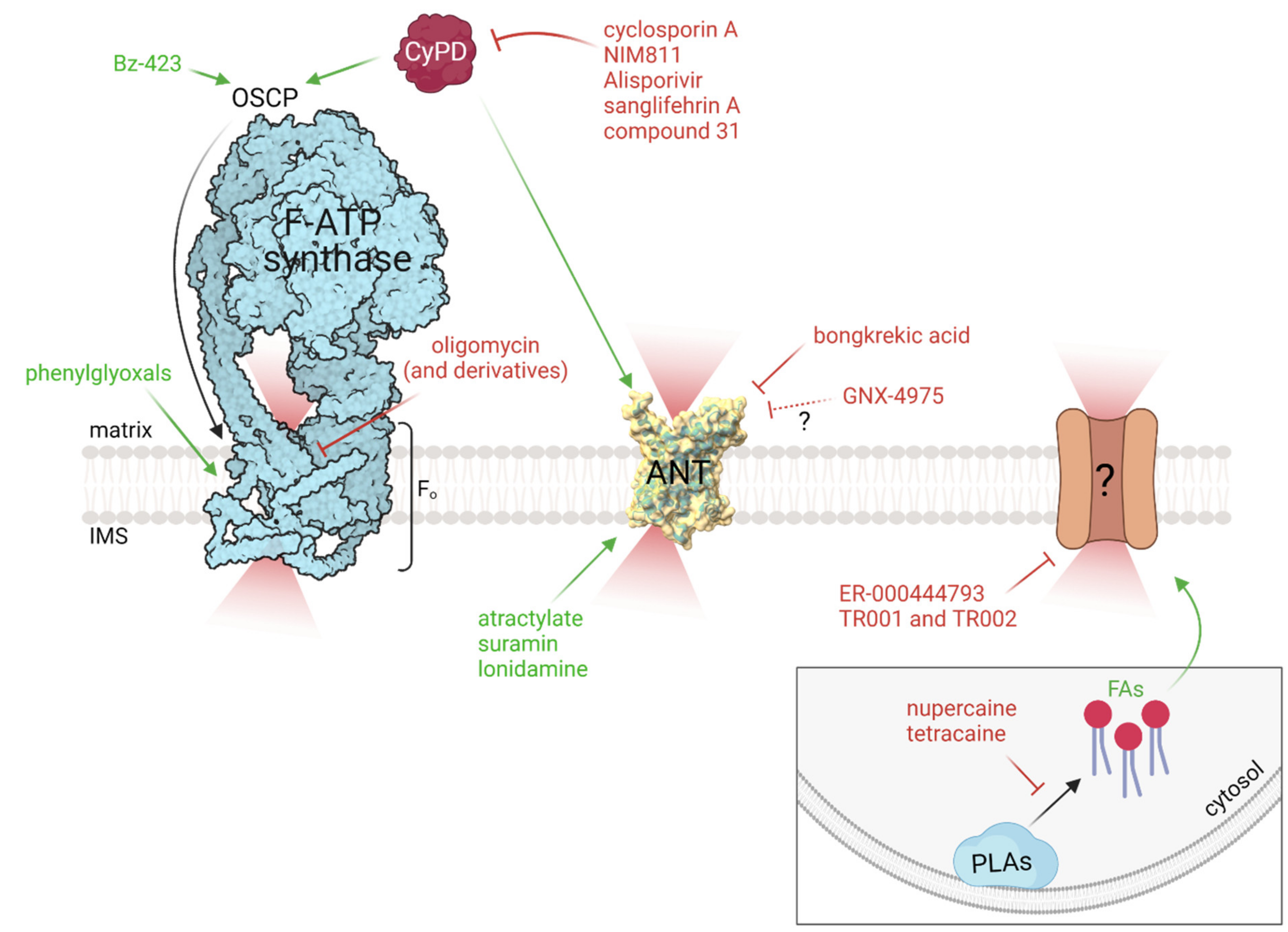
5. Pharmacology of the PTP: Hunting for the Target
5.1. F-ATP Synthase-Targeting Compounds
5.1.1. Benzodiazepine-423
5.1.2. Phenylglyoxals
5.1.3. Oligomycin and Related Compounds
5.2. ANT-Targeting Compounds
5.3. CyPD-Independent PTP Inhibitors
5.4. Fatty Acids and Phospholipase Inhibitors
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pfeiffer, D.R.; Schmid, P.C.; Beatrice, M.C.; Schmid, H.H. Intramitochondrial Phospholipase Activity and the Effects of Ca2+ plus N-Ethylmaleimide on Mitochondrial Function. J. Biol. Chem. 1979, 254, 11485–11494. [Google Scholar] [CrossRef]
- Hunter, D.R.; Haworth, R.A. The Ca2+-Induced Membrane Transition in Mitochondria. I. The Protective Mechanisms. Arch. Biochem. Biophys. 1979, 195, 453–459. [Google Scholar] [CrossRef]
- Haworth, R.A.; Hunter, D.R. The Ca2+-Induced Membrane Transition in Mitochondria. II. Nature of the Ca2+ Trigger Site. Arch. Biochem. Biophys. 1979, 195, 460–467. [Google Scholar] [CrossRef]
- Hunter, D.R.; Haworth, R.A. The Ca2+-Induced Membrane in Mitochondria Mitochondria. III. Transitional Ca2+ Release. Arch. Biochem. Biophys. 1979, 195, 468–477. [Google Scholar] [CrossRef]
- Massari, S.; Frigeri, L.; Azzone, G.F. A Quantitative Correlation between the Kinetics of Solutes and Water Translocation in Liver Mitochondria. J. Membr. Biol. 1972, 9, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Kinnally, K.W.; Campo, M.L.; Tedeschi, H. Mitochondrial Channel Activity Studied by Patch-Clamping Mitoplasts. J. Bioenerg. Biomembr. 1989, 21, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Petronilli, V.; Szabò, I.; Zoratti, M. The Inner Mitochondrial Membrane Contains Ion-Conducting Channels Similar to Those Found in Bacteria. FEBS Lett. 1989, 259, 137–143. [Google Scholar] [CrossRef] [Green Version]
- Szabó, I.; Zoratti, M. The Mitochondrial Megachannel Is the Permeability Transition Pore. J. Bioenerg. Biomembr. 1992, 24, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Beutner, G.; Rück, A.; Riede, B.; Brdiczka, D. Complexes between Porin, Hexokinase, Mitochondrial Creatine Kinase and Adenylate Translocator Display Properties of the Permeability Transition Pore. Implication for Regulation of Permeability Transition by the Kinases. Biochim. Biophys. Acta-Biomembr. 1998, 1368, 7–18. [Google Scholar] [CrossRef] [Green Version]
- Bernardi, P.; Rasola, A.; Forte, M.; Lippe, G. The Mitochondrial Permeability Transition Pore: Channel Formation by F-ATP Synthase, Integration in Signal Transduction, and Role in Pathophysiology. Physiol. Rev. 2015, 95, 1111–1155. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P.; Vassanelli, S.; Veronese, P.; Colonna, R.; Szabo, I.; Zoratti, M. Modulation of the Mitochondrial Permeability Transition Pore. Effect of Protons and Divalent Cations. J. Biol. Chem. 1992, 267, 2934–2939. [Google Scholar] [CrossRef]
- Szabo, I.; Bernardi, P.; Zoratti, M. Modulation of the Mitochondrial Megachannel by Divalent Cations and Protons. J. Biol. Chem. 1992, 267, 2940–2946. [Google Scholar] [CrossRef]
- Halestrap, A.P.; Woodfield, K.Y.; Connern, C.P. Oxidative Stress, Thiol Reagents, and Membrane Potential Modulate the Mitochondrial Permeability Transition by Affecting Nucleotide Binding to the Adenine Nucleotide Translocase. J. Biol. Chem. 1997, 272, 3346–3354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolli, A.; Petronilli, V.; Bernardi, P. Modulation of the Mitochondrial Cyclosporin A-Sensitive Permeability Transition Pore by Matrix pH. Evidence That the Pore Open-Closed Probability Is Regulated by Reversible Histidine Protonation. Biochemistry 1993, 32, 4461–4465. [Google Scholar] [CrossRef]
- Kristián, T.; Bernardi, P.; Siesjö, B.K. Acidosis Promotes the Permeability Transition in Energized Mitochondria: Implications for Reperfusion Injury. J. Neurotrauma 2001, 18, 1059–1074. [Google Scholar] [CrossRef]
- Chalmers, S.; Nicholls, D.G. The Relationship between Free and Total Calcium Concentrations in the Matrix of Liver and Brain Mitochondria. J. Biol. Chem. 2003, 278, 19062–19070. [Google Scholar] [CrossRef] [Green Version]
- Strubbe-Rivera, J.O.; Schrad, J.R.; Pavlov, E.V.; Conway, J.F.; Parent, K.N.; Bazil, J.N. The Mitochondrial Permeability Transition Phenomenon Elucidated by Cryo-EM Reveals the Genuine Impact of Calcium Overload on Mitochondrial Structure and Function. Sci. Rep. 2021, 11. [Google Scholar] [CrossRef]
- Abramov, A.Y.; Fraley, C.; Diao, C.T.; Winkfein, R.; Colicos, M.A.; Duchen, M.R.; French, R.J.; Pavlov, E. Targeted Polyphosphatase Expression Alters Mitochondrial Metabolism and Inhibits Calcium-Dependent Cell Death. Proc. Natl. Acad. Sci. USA 2007, 104, 18091–18096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basso, E.; Petronilli, V.; Forte, M.A.; Bernardi, P. Phosphate Is Essential for Inhibition of the Mitochondrial Permeability Transition Pore by Cyclosporin A and by Cyclophilin D Ablation. J. Biol. Chem. 2008, 283, 26307–26311. [Google Scholar] [CrossRef] [Green Version]
- Von Stockum, S.; Basso, E.; Petronilli, V.; Sabatelli, P.; Forte, M.A.; Bernardi, P. Properties of Ca2+ Transport in Mitochondria of Drosophila Melanogaster. J. Biol. Chem. 2011, 286, 41163–41170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, D.W.; Bradshaw, P.C.; Pfeiffer, D.R. Properties of a Cyclosporin-Insensitive Permeability Transition Pore in Yeast Mitochondria. J. Biol. Chem. 1997, 272, 21104–21112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, A.; Yamamoto, T.; Yoshimura, Y.; Gouda, S.; Kawashima, S.; Yamazaki, N.; Yamashita, K.; Kataoka, M.; Nagata, T.; Terada, H.; et al. Ca2+-Induced Permeability Transition Can Be Observed Even in Yeast Mitochondria under Optimized Experimental Conditions. Biochim. Biophys. Acta -Bioenerg. 2009, 1787, 1486–1491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carraro, M.; Giorgio, V.; Sileikyte, J.; Sartori, G.; Forte, M.; Lippe, G.; Zoratti, M.; Szabò, I.; Bernardi, P. Channel Formation by Yeast F-ATP Synthase and the Role of Dimerization in the Mitochondrial Permeability Transition. J. Biol. Chem. 2014, 289, 15980–15985. [Google Scholar] [CrossRef] [Green Version]
- Petronilli, V.; Cola, C.; Massari, S.; Colonna, R.; Bernardi, P. Physiological Effectors Modify Voltage Sensing by the Cyclosporin A- Sensitive Permeability Transition Pore of Mitochondria. J. Biol. Chem. 1993, 268, 21939–21945. [Google Scholar] [CrossRef]
- Eriksson, O.; Fontaine, E.; Bernardi, P. Chemical Modification of Arginines by 2,3-Butanedione and Phenylglyoxal Causes Closure of the Mitochondrial Permeability Transition Pore. J. Biol. Chem. 1998, 273, 12669–12674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linder, M.D.; Morkunaite-Haimi, S.; Kinnunen, P.K.J.; Bernardi, P.; Eriksson, O. Ligand-Selective Modulation of the Permeability Transition Pore by Arginine Modification: Opposing Effects of p-Hydroxyphenylglyoxal and Phenylglyoxal. J. Biol. Chem. 2002, 277, 937–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kokoszka, J.E.; Waymire, K.G.; Levy, S.E.; Sligh, J.E.; Cai, J.; Jones, D.P.; MacGregor, G.R.; Wallace, D.C. The ADP/ATP Translocator Is Not Essential for the Mitochondrial Permeability Transition Pore. Nature 2004, 427, 461–465. [Google Scholar] [CrossRef] [PubMed]
- Doczi, J.; Torocsik, B.; Echaniz-Laguna, A.; De Camaret, B.M.; Starkov, A.; Starkova, N.; Gál, A.; Molńr, M.J.; Kawamata, H.; Manfredi, G.; et al. Alterations in Voltage-Sensing of the Mitochondrial Permeability Transition Pore in ANT1-Deficient Cells. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wojtczak, L.; Lehninger, A.L. Formation and Disappearance of an Endogenous Uncoupling Factor during Swelling and Contraction of Mitochondria. BBA-Biochim. Biophys. Acta 1961, 51, 442–456. [Google Scholar] [CrossRef]
- Petronilli, V.; Costantini, P.; Scorrano, L.; Colonna, R.; Passamonti, S.; Bernardi, P. The Voltage Sensor of the Mitochondrial Permeability Transition Pore Is Tuned by the Oxidation-Reduction State of Vicinal Thiols. Increase of the Gating Potential by Oxidants and Its Reversal by Reducing Agents. J. Biol. Chem. 1994, 269, 16638–16642. [Google Scholar] [CrossRef]
- Costantini, P.; Chernyak, B.V.; Petronilli, V.; Bernardi, P. Modulation of the Mitochondrial Permeability Transition Pore by Pyridine Nucleotides and Dithiol Oxidation at Two Separate Sites. J. Biol. Chem. 1996, 271, 6746–6751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costantini, P.; Colonna, R.; Bernardi, P. Induction of the Mitochondrial Permeability Transition by N-Ethylmaleimide Depends on Secondary Oxidation of Critical Thiol Groups. Potentiation by Copper-Ortho-Phenanthroline without Dimerization of the Adenine Nucleotide Translocase. Biochim. Biophys. Acta-Bioenerg. 1998, 1365, 385–392. [Google Scholar] [CrossRef]
- De Marchi, U.; Basso, E.; Szabò, I.; Zoratti, M. Electrophysiological Characterization of the Cyclophilin D-Deleted Mitochondrial Permeability Transition Pore. Mol. Membr. Biol. 2006, 23, 521–530. [Google Scholar] [CrossRef] [PubMed]
- Baines, C.P.; Kaiser, R.A.; Purchell, N.H.; Blair, N.S.; Osinska, H.; Hambleton, M.A.; Brunskill, E.W.; Sayen, M.R.; Gottlieb, R.A.; Dorn II, G.W.; et al. Loss of Cyclophilin D Reveals a Critical Role for Mitochondrial Permeability Transition in Cell Death. Nature 2005, 434, 658–662. [Google Scholar] [CrossRef]
- Nakayama, H.; Chen, X.; Baines, C.P.; Klevitsky, R.; Zhang, X.; Zhang, H.; Jaleel, N.; Chua, B.H.L.; Hewett, T.E.; Robbins, J.; et al. Ca2+- and Mitochondrial-Dependent Cardiomyocyte Necrosis as a Primary Mediator of Heart Failure. J. Clin. Invest. 2007, 117, 2431–2444. [Google Scholar] [CrossRef] [PubMed]
- Millay, D.P.; Sargent, M.A.; Osinska, H.; Baines, C.P.; Barton, E.R.; Vuagniaux, G.; Sweeney, H.L.; Robbins, J.; Molkentin, J.D. Genetic and Pharmacologic Inhibition of Mitochondrial-Dependent Necrosis Attenuates Muscular Dystrophy. Nat. Med. 2008, 14, 442–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palma, E.; Tiepolo, T.; Angelin, A.; Sabatelli, P.; Maraldi, N.M.; Basso, E.; Forte, M.A.; Bernardip, P.; Bonaldo, P. Genetic Ablation of Cyclophilin D Rescues Mitochondrial Defects and Prevents Muscle Apoptosis in Collagen VI Myopathic Mice. Hum. Mol. Genet. 2009, 18, 2024–2031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crompton, M.; Virji, S.; Ward, J.M. Cyclophilin-D Binds Strongly to Complexes of the Voltage-Dependent Anion Channel and the Adenine Nucleotide Translocase to Form the Permeability Transition Pore. Eur. J. Biochem. 1998, 258, 729–735. [Google Scholar] [CrossRef] [PubMed]
- Woodfield, K.; Rück, A.; Brdiczka, D.; Halestrap, A.P. Direct Demonstration of a Specific Interaction between Cyclophilin-D and the Adenine Nucleotide Translocase Confirms Their Role in the Mitochondrial Permeability Transition. Biochem. J. 1998, 336, 287–290. [Google Scholar] [CrossRef]
- Giorgio, V.; Bisetto, E.; Soriano, M.E.; Dabbeni-Sala, F.; Basso, E.; Petronilli, V.; Forte, M.A.; Bernardi, P.; Lippe, G. Cyclophilin D Modulates Mitochondrial FOF1-ATP Synthase by Interacting with the Lateral Stalk of the Complex. J. Biol. Chem. 2009, 284, 33982–33988. [Google Scholar] [CrossRef] [Green Version]
- Amanakis, G.; Murphy, E.; Cyclophilin, D. An Integrator of Mitochondrial Function. Frontiers in Physiology. 2020, 11, 595. [Google Scholar] [CrossRef] [PubMed]
- Rasola, A.; Sciacovelli, M.; Chiara, F.; Pantic, B.; Brusilow, W.S.; Bernardi, P. Activation of Mitochondrial ERK Protects Cancer Cells from Death through Inhibition of the Permeability Transition. Proc. Natl. Acad. Sci. USA 2010, 107, 726–731. [Google Scholar] [CrossRef] [Green Version]
- Teodoro, J.S.; Varela, A.T.; Duarte, F.V.; Gomes, A.P.; Palmeira, C.M.; Rolo, A.P. Indirubin and NAD+ Prevent Mitochondrial Ischaemia/Reperfusion Damage in Fatty Livers. Eur. J. Clin. Invest. 2018, 48. [Google Scholar] [CrossRef]
- Miura, T.; Nishihara, M.; Miki, T. Drug Development Targeting the Glycogen Synthase Kinase-3β (GSK-3β)-Mediated Signal Transduction Pathway: Role of GSK-3β in Myocardial Protection against Ischemia/Reperfusion Injury. J. Pharmacol. Sci. 2009, 109, 162–167. [Google Scholar] [CrossRef] [Green Version]
- Hurst, S.; Gonnot, F.; Dia, M.; Crola Da Silva, C.; Gomez, L.; Sheu, S.S. Phosphorylation of Cyclophilin D at Serine 191 Regulates Mitochondrial Permeability Transition Pore Opening and Cell Death after Ischemia-Reperfusion. Cell Death Dis. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Parks, R.J.; Menazza, S.; Holmström, K.M.; Amanakis, G.; Fergusson, M.; Ma, H.; Aponte, A.M.; Bernardi, P.; Finkel, T.; Murphy, E. Cyclophilin D-Mediated Regulation of the Permeability Transition Pore Is Altered in Mice Lacking the Mitochondrial Calcium Uniporter. Cardiovasc. Res. 2019, 115, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Bochaton, T.; Crola-Da-Silva, C.; Pillot, B.; Villedieu, C.; Ferreras, L.; Alam, M.R.; Thibault, H.; Strina, M.; Gharib, A.; Ovize, M.; et al. Inhibition of Myocardial Reperfusion Injury by Ischemic Postconditioning Requires Sirtuin 3-Mediated Deacetylation of Cyclophilin D. J. Mol. Cell. Cardiol. 2015, 84, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Si, Y.; Bao, H.; Xu, Y.; Pan, X.X.; Zeng, L.; Jing, L. Regulation of Sirtuin 3-Mediated Deacetylation of Cyclophilin D Attenuated Cognitive Dysfunction Induced by Sepsis-Associated Encephalopathy in Mice. Cell. Mol. Neurobiol. 2017, 37, 1457–1464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hafner, A.V.; Dai, J.; Gomes, A.P.; Xiao, C.Y.; Palmeira, K.C.M.; Rosenzweig, A.; Sinclair, D.A. Regulation of the MPTP by SIRT3-Mediated Deacetylation of CypD at Lysine 166 Suppresses Age-Related Cardiac Hypertrophy. Aging 2010, 2, 914–923. [Google Scholar] [CrossRef] [Green Version]
- Kang, B.H.; Plescia, J.; Dohi, T.; Rosa, J.; Doxsey, S.J.; Altieri, D.C. Regulation of Tumor Cell Mitochondrial Homeostasis by an Organelle-Specific Hsp90 Chaperone Network. Cell 2007, 131, 257–270. [Google Scholar] [CrossRef] [PubMed]
- Sciacovelli, M.; Guzzo, G.; Morello, V.; Frezza, C.; Zheng, L.; Nannini, N.; Calabrese, F.; Laudiero, G.; Esposito, F.; Landriscina, M.; et al. The Mitochondrial Chaperone TRAP1 Promotes Neoplastic Growth by Inhibiting Succinate Dehydrogenase. Cell Metab. 2013, 17, 988–999. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wang, Y.; Lin, J.; Chen, Q.Z.; Zhu, N.; Jiang, D.Q.; Li, M.X. Overexpression of Mitochondrial Hsp75 Protectsneural Stem Cells against Microglia-Derived Solublefactor-Induced Neurotoxicity by Regulating Mitochondrial Permeability Transition Pore Opening in Vitro. Int. J. Mol. Med. 2015, 36, 1487–1496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Liu, L.; Li, X.; Zhang, X.; Zhao, J.; Luo, Y.; Guo, X.; Zhao, T. TRAP1 Attenuates H9C2 Myocardial Cell Injury Induced by Extracellular Acidification via the Inhibition of MPTP Opening. Int. J. Mol. Med. 2020, 46, 663–674. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhang, L.; Zhao, J.; Guo, X.; Luo, Y.; Hu, W.; Zhao, T. Tumor Necrosis Factor Receptor-Associated Protein 1 Protects against Mitochondrial Injury by Preventing High Glucose-Induced MPTP Opening in Diabetes. Oxid. Med. Cell. Longev. 2020, 2020. [Google Scholar] [CrossRef]
- Laquatra, C.; Sanchez-Martin, C.; Dinarello, A.; Cannino, G.; Minervini, G.; Moroni, E.; Schiavone, M.; Tosatto, S.; Argenton, F.; Colombo, G.; et al. HIF1α-Dependent Induction of the Mitochondrial Chaperone TRAP1 Regulates Bioenergetic Adaptations to Hypoxia. Cell Death Dis. 2021, 12, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Lu, Y.; Yu, D.; Zhang, D.; Hu, W. TRAP1 Provides Protection Against Myocardial Ischemia-Reperfusion Injury by Ameliorating Mitochondrial Dysfunction. Cell. Physiol. Biochem. 2015, 36, 2072–2082. [Google Scholar] [CrossRef]
- Xu, L.; Voloboueva, L.A.; Ouyang, Y.B.; Emery, J.F.; Giffard, R.G. Overexpression of Mitochondrial Hsp70/Hsp75 in Rat Brain Protects Mitochondria, Reduces Oxidative Stress, and Protects from Focal Ischemia. J. Cereb. Blood Flow Metab. 2009, 29, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.; Dai, L.; Liu, Y.; Lee, J.; Ghahhari, N.M.; Segala, G.; Beebe, K.; Jenkins, L.M.; Lyons, G.C.; Bernasconi, L.; et al. The Mitochondrial HSP90 Paralog TRAP1 Forms an OXPHOS-Regulated Tetramer and Is Involved in Mitochondrial Metabolic Homeostasis. BMC Biol. 2020, 18, 1–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Martin, C.; Moroni, E.; Ferraro, M.; Laquatra, C.; Cannino, G.; Masgras, I.; Negro, A.; Quadrelli, P.; Rasola, A.; Colombo, G. Rational Design of Allosteric and Selective Inhibitors of the Molecular Chaperone TRAP1. Cell Rep. 2020, 31, 107531. [Google Scholar] [CrossRef] [PubMed]
- Fournier, N.; Ducet, G.; Crevat, A. Action of Cyclosporine on Mitochondrial Calcium Fluxes. J. Bioenerg. Biomembr. 1987, 19, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Crompton, M.; Ellinger, H.; Costi, A. Inhibition by Cyclosporin A of a Ca2+-Dependent Pore in Heart Mitochondria Activated by Inorganic Phosphate and Oxidative Stress. Biochem. J. 1988, 255, 357–360. [Google Scholar]
- Broekemeier, K.M.; Pfeiffer, D.R. Cyclosporin A-Sensitive and Insensitive Mechanisms Produce the Permeability Transition in Mitochondria. Biochem. Biophys. Res. Commun. 1989, 163, 561–566. [Google Scholar] [CrossRef]
- Li, B.; Chauvin, C.; De Paulis, D.; De Oliveira, F.; Gharib, A.; Vial, G.; Lablanche, S.; Leverve, X.; Bernardi, P.; Ovize, M.; et al. Inhibition of Complex I Regulates the Mitochondrial Permeability Transition through a Phosphate-Sensitive Inhibitory Site Masked by Cyclophilin D. Biochim. Biophys. Acta-Bioenerg. 2012, 1817, 1628–1634. [Google Scholar] [CrossRef]
- Liu, J.; Albers, M.W.; Wandless, T.J.; Luán, S.; Alberg, D.G.; Belshaw, P.J.; Schreiber, S.L.; MacKintosh, C.; Cohen, P.; Klee, C.B. Inhibition of T Cell Signaling by Immunophilin-Ligand Complexes Correlates with Loss of Calcineurin Phosphatase Activity. Biochemistry 1992, 31, 3896–3901. [Google Scholar] [CrossRef]
- Clipstone, N.A.; Crabtree, G.R. Identification of Calcineurin as a Key Signalling Enzyme in T Lymphocyte Activation. Nature 1992, 356, 695–697. [Google Scholar] [CrossRef] [PubMed]
- Walsh, C.T.; Zydowsky, L.D.; McKeon, F.D. Cyclosporin A, the Cyclophilin Class of Peptidylprolyl Isomerases, and Blockade of T Cell Signal Transduction. J. Biol. Chem. 1992, 267, 13115–13118. [Google Scholar] [CrossRef]
- Cereghetti, G.M.; Stangherlin, A.; Martins De Brito, O.; Chang, C.R.; Blackstone, C.; Bernardi, P.; Scorrano, L. Dephosphorylation by Calcineurin Regulates Translocation of Drp1 to Mitochondria. Proc. Natl. Acad. Sci. USA 2008, 105, 15803–15808. [Google Scholar] [CrossRef] [Green Version]
- Ma, S.; Boerner, J.E.; TiongYip, C.L.; Weidmann, B.; Ryder, N.S.; Cooreman, M.P.; Lin, K. NIM811, a Cyclophilin Inhibitor, Exhibits Potent in Vitro Activity against Hepatitis C Virus Alone or in Combination with Alpha Interferon. Antimicrob. Agents Chemother. 2006, 50, 2976–2982. [Google Scholar] [CrossRef] [Green Version]
- Paeshuyse, J.; Kaul, A.; De Clercq, E.; Rosenwirth, B.; Dumont, J.M.; Scalfaro, P.; Bartenschlager, R.; Neyts, J. The Non-Immunosuppressive Cyclosporin DEBIO-025 Is a Potent Inhibitor of Hepatitis C Virus Replication in Vitro. Hepatology 2006, 43, 761–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ptak, R.G.; Gallay, P.A.; Jochmans, D.; Halestrap, A.P.; Ruegg, U.T.; Pallansch, L.A.; Bobardt, M.D.; De Béthune, M.P.; Neyts, J.; De Clercq, E.; et al. Inhibition of Human Immunodeficiency Virus Type 1 Replication in Human Cells by Debio-025, a Novel Cyclophilin Binding Agent. Antimicrob. Agents Chemother. 2008, 52, 1302–1317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Baradie, K.B.Y.; Khan, M.B.; Mendhe, B.; Waller, J.; O’Brien, F.; Hamrick, M.W. The Cyclophilin Inhibitor NIM-811 Increases Muscle Cell Survival with Hypoxia in Vitro and Improves Gait Performance Following Ischemia–Reperfusion in Vivo. Sci. Rep. 2021, 11, 1–13. [Google Scholar] [CrossRef]
- Zulian, A.; Rizzo, E.; Schiavone, M.; Palma, E.; Tagliavini, F.; Blaauw, B.; Merlini, L.; Maraldi, N.M. ari.; Sabatelli, P.; Braghetta, P.; et al. NIM811, a Cyclophilin Inhibitor without Immunosuppressive Activity, Is Beneficial in Collagen VI Congenital Muscular Dystrophy Models. Hum. Mol. Genet. 2014, 23, 5353–5363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mbye, L.H.; Singh, I.N.; Sullivan, P.G.; Springer, J.E.; Hall, E.D. Attenuation of Acute Mitochondrial Dysfunction after Traumatic Brain Injury in Mice by NIM811, a Non-Immunosuppressive Cyclosporin A Analog. Exp. Neurol. 2008, 209, 243–253. [Google Scholar] [CrossRef]
- Tóth, E.; Maléth, J.; Závogyán, N.; Fanczal, J.; Grassalkovich, A.; Erdős, R.; Pallagi, P.; Horváth, G.; Tretter, L.; Bálint, E.R.; et al. Novel Mitochondrial Transition Pore Inhibitor N-Methyl-4-Isoleucine Cyclosporin Is a New Therapeutic Option in Acute Pancreatitis. J. Physiol. 2019, 597, 5879–5898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zenke, G.; Strittmatter, U.; Fuchs, S.; Quesniaux, V.F.J.; Brinkmann, V.; Schuler, W.; Zurini, M.; Enz, A.; Billich, A.; Sanglier, J.-J.; et al. Sanglifehrin A, a Novel Cyclophilin-Binding Compound Showing Immunosuppressive Activity with a New Mechanism of Action. J. Immunol. 2001, 166, 7165–7171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, S.J.; McStay, G.P.; Halestrap, A.P. Sanglifehrin A Acts as a Potent Inhibitor of the Mitochondrial Permeability Transition and Reperfusion Injury of the Heart by Binding to Cyclophilin-D at a Different Site from Cyclosporin A. J. Biol. Chem. 2002, 277, 34793–34799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed-Belkacem, A.; Colliandre, L.; Ahnou, N.; Nevers, Q.; Gelin, M.; Bessin, Y.; Brillet, R.; Cala, O.; Douguet, D.; Bourguet, W.; et al. Fragment-Based Discovery of a New Family of Non-Peptidic Small-Molecule Cyclophilin Inhibitors with Potent Antiviral Activities. Nat. Commun. 2016, 7, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Panel, M.; Ruiz, I.; Brillet, R.; Lafdil, F.; Teixeira-Clerc, F.; Nguyen, C.T.; Calderaro, J.; Gelin, M.; Allemand, F.; Guichou, J.F.; et al. Small-Molecule Inhibitors of Cyclophilins Block Opening of the Mitochondrial Permeability Transition Pore and Protect Mice From Hepatic Ischemia/Reperfusion Injury. Gastroenterology 2019, 157, 1368–1382. [Google Scholar] [CrossRef]
- Schiavone, M.; Zulian, A.; Menazza, S.; Petronilli, V.; Argenton, F.; Merlini, L.; Sabatelli, P.; Bernardi, P. Alisporivir Rescues Defective Mitochondrial Respiration in Duchenne Muscular Dystrophy. Pharmacol. Res. 2017, 125, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Wissing, E.R.; Millay, D.P.; Vuagniaux, G.; Molkentin, J.D. Debio-025 Is More Effective than Prednisone in Reducing Muscular Pathology in Mdx Mice. Neuromuscul. Disord. 2010, 20, 753–760. [Google Scholar] [CrossRef] [Green Version]
- Blatt, N.B.; Bednarski, J.J.; Warner, R.E.; Leonetti, F.; Johnson, K.M.; Boitano, A.; Yung, R.; Richardson, B.C.; Johnson, K.J.; Ellman, J.A.; et al. Benzodiazepine-Induced Superoxide SignalsB Cell Apoptosis: Mechanistic Insight and Potential Therapeutic Utility. J. Clin. Invest. 2002, 110, 1123–1132. [Google Scholar] [CrossRef] [PubMed]
- Urbani, A.; Giorgio, V.; Carrer, A.; Franchin, C.; Arrigoni, G.; Jiko, C.; Abe, K.; Maeda, S.; Shinzawa-Itoh, K.; Bogers, J.F.M.; et al. Purified F-ATP Synthase Forms a Ca2+-Dependent High-Conductance Channel Matching the Mitochondrial Permeability Transition Pore. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johans, M.; Milanesi, E.; Franck, M.; Johans, C.; Liobikas, J.; Panagiotaki, M.; Grecil, L.; Principato, G.; Kinnunen, P.K.J.; Bernardi, P.; et al. Modification of Permeability Transition Pore Arginine(s) by Phenylglyoxal Derivatives in Isolated Mitochondria and Mammalian Cells: Structure-Function Relationship of Arginine Ligands. J. Biol. Chem. 2005, 280, 12130–12136. [Google Scholar] [CrossRef] [Green Version]
- Eriksson, O.; Fontaine, E.; Petronilli, V.; Bernardi, P. Inhibition of the Mitochondrial Cyclosporin A-Sensitive Permeability Transition Pore by the Arginine Reagent Phenylglyoxal. FEBS Lett. 1997, 409, 361–364. [Google Scholar] [CrossRef] [Green Version]
- Speer, O.; Morkunaite-Haimi, S.; Liobikas, J.; Franck, M.; Hensbo, L.; Linder, M.D.; Kinnunen, P.K.J.; Wallimann, T.; Eriksson, O. Rapid Suppression of Mitochondrial Permeability Transition by Methylglyoxal: Role of Reversible Arginine Modification. J. Biol. Chem. 2003, 278, 34757–34763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, L.; Carraro, M.; Sartori, G.; Minervini, G.; Eriksson, O.; Petronilli, V.; Bernardi, P. Arginine 107 of Yeast ATP Synthase Subunit g Mediates Sensitivity of the Mitochondrial Permeability Transition to Phenylglyoxal. J. Biol. Chem. 2018, 293, 14632–14645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petronilli, V.; Nicolli, A.; Costantini, P.; Colonna, R.; Bernardi, P. Regulation of the Permeability Transition Pore, a Voltage-Dependent Mitochondrial Channel Inhibited by Cyclosporin A. Biochim. et Biophys. Acta (BBA)-Bioenerg. 1994, 1187, 255–259. [Google Scholar] [CrossRef]
- Mnatsakanyan, N.; Llaguno, M.C.; Yang, Y.; Yan, Y.; Weber, J.; Sigworth, F.J.; Jonas, E.A. A Mitochondrial Megachannel Resides in Monomeric F1FO ATP Synthase. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Morciano, G.; Preti, D.; Pedriali, G.; Aquila, G.; Missiroli, S.; Fantinati, A.; Caroccia, N.; Pacifico, S.; Bonora, M.; Talarico, A.; et al. Discovery of Novel 1,3,8-Triazaspiro[4.5]Decane Derivatives That Target the c Subunit of F1/FO-Adenosine Triphosphate (ATP) Synthase for the Treatment of Reperfusion Damage in Myocardial Infarction. J. Med. Chem. 2018, 61, 7131–7143. [Google Scholar] [CrossRef] [PubMed]
- Kunji, E.R.S.; Aleksandrova, A.; King, M.S.; Majd, H.; Ashton, V.L.; Cerson, E.; Springett, R.; Kibalchenko, M.; Tavoulari, S.; Crichton, P.G.; et al. The Transport Mechanism of the Mitochondrial ADP/ATP Carrier. Biochim. Biophys. Acta-Mol. Cell Res. 2016, 1863, 2379–2393. [Google Scholar] [CrossRef] [Green Version]
- Brustovetsky, N.; Klingenberg, M. Mitochondrial ADP/ATP Carrier Can Be Reversibly Converted into a Large Channel by Ca2+. Biochemistry 1996, 35, 8483–8488. [Google Scholar] [CrossRef] [PubMed]
- Brustovetsky, N.; Tropschug, M.; Heimpel, S.; Heidkämper, D.; Klingenberg, M. A Large Ca2+-Dependent Channel Formed by Recombinant ADP/ATP Carrier from Neurospora Crassa Resembles the Mitochondrial Permeability Transition Pore. Biochemistry 2002, 41, 11804–11811. [Google Scholar] [CrossRef] [PubMed]
- Bernardes, C.F.; Fagian, M.M.; Meyer-Fernandes, J.R.; Castilho, R.F.; Vercesi, A.E. Suramin Inhibits Respiration and Induces Membrane Permeability Transition in Isolated Rat Liver Mitochondria Celene. Toxicology. 2001, 169, 17–23. [Google Scholar] [CrossRef]
- Kaulich, M.; Streicher, F.; Mayer, R.; Müller, I.; Müller, C.E. Flavonoids-Novel Lead Compounds for the Development of P2Y2 Receptor Antagonists. Drug Dev. Res. 2003, 59, 72–81. [Google Scholar] [CrossRef]
- Belzacq, A.S.; El Hamel, C.; La Vieira, H.; Cohen, I.; Haouzi, D.; Métivier, D.; Marchetti, P.; Brenner, C.; Kroemer, G. Adenine Nucleotide Translocator Mediates the Mitochondrial Membrane Permeabilization Induced by Lonidamine, Arsenite and CD437. Oncogene 2001, 20, 7579–7587. [Google Scholar] [CrossRef] [Green Version]
- Martin, L.J.; Fancelli, D.; Wong, M.; Niedzwiecki, M.; Ballarini, M.; Plyte, S.; Chang, Q. GNX-4728, a Novel Small Molecule Drug Inhibitor of Mitochondrial Permeability Transition, Is Therapeutic in a Mouse Model of Amyotrophic Lateral Sclerosis. Front. Cell. Neurosci. 2014, 8, 433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stocco, A.; Smolina, N.; Sabatelli, P.; Šileikytė, J.; Artusi, E.; Mouly, V.; Cohen, M.; Forte, M.; Schiavone, M.; Bernardi, P. Treatment with a Triazole Inhibitor of the Mitochondrial Permeability Transition Pore Fully Corrects the Pathology of Sapje Zebrafish Lacking Dystrophin. Pharmacol. Res. 2021, 165, 105421. [Google Scholar] [CrossRef] [PubMed]
- Antonucci, S.; Di Sante, M.; Sileikyte, J.; Deveraux, J.; Bauer, T.; Bround, M.J.; Menabò, R.; Paillard, M.; Alanova, P.; Carraro, M.; et al. A Novel Class of Cardioprotective Small-Molecule PTP Inhibitors. Pharmacol. Res. 2020, 151, 104548. [Google Scholar] [CrossRef] [PubMed]
- Briston, T.; Lewis, S.; Koglin, M.; Mistry, K.; Shen, Y.; Hartopp, N.; Katsumata, R.; Fukumoto, H.; Duchen, M.R.; Szabadkai, G.; et al. Identification of ER-000444793, a Cyclophilin D-Independent Inhibitor of Mitochondrial Permeability Transition, Using a High-Throughput Screen in Cryopreserved Mitochondria. Sci. Rep. 2016, 6, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carraro, M.; Checchetto, V.; Szabó, I.; Bernardi, P. F-ATP Synthase and the Permeability Transition Pore: Fewer Doubts, More Certainties. FEBS Letters. 2019, 593, 1542–1553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bround, M.J.; Bers, D.M.; Molkentin, J.D. A 20/20 View of ANT Function in Mitochondrial Biology and Necrotic Cell Death. J. Mol. Cell. Cardiol. 2020, 144, A3–A13. [Google Scholar] [CrossRef]
- Brustovetsky, N. The Role of Adenine Nucleotide Translocase in the Mitochondrial Permeability Transition. Cells 2020, 9, 2686. [Google Scholar] [CrossRef] [PubMed]
- Ruprecht, J.J.; King, M.S.; Zögg, T.; Aleksandrova, A.A.; Pardon, E.; Crichton, P.G.; Steyaert, J.; Kunji, E.R.S. The Molecular Mechanism of Transport by the Mitochondrial ADP/ATP Carrier. Cell 2019, 176, 435–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruprecht, J.J.; Kunji, E.R. Structural Changes in the Transport Cycle of the Mitochondrial ADP/ATP Carrier. Curr. Opin. Struct. Biology. 2019, 57, 135–144. [Google Scholar] [CrossRef] [PubMed]
- McStay, G.P.; Clarke, S.J.; Halestrap, A.P. Role of Critical Thiol Groups on the Matrix Surface of the Adenine Nucleotide Translocase in the Mechanism of the Mitochondrial Permeability Transition Pore. Biochem. J. 2002, 367, 541–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertholet, A.M.; Chouchani, E.T.; Kazak, L.; Angelin, A.; Fedorenko, A.; Long, J.Z.; Vidoni, S.; Garrity, R.; Cho, J.; Terada, N.; et al. H+ Transport Is an Integral Function of the Mitochondrial ADP/ATP Carrier. Nature 2019, 571, 515–520. [Google Scholar] [CrossRef] [PubMed]
- Beyer, K.; Klingenberg, M. ADP/ATP Carrier Protein from Beef Heart Mitochondria Has High Amounts of Tightly Bound Cardiolipin, as Revealed by 31P Nuclear Magnetic Resonance. Biochemistry 1985, 24, 3821–3826. [Google Scholar] [CrossRef] [PubMed]
- Rottenberg, H.; Marbach, M. Regulation of Ca2+ Transport in Brain Mitochondria. II. The Mechanism of the Adenine Nucleotides Enhancement of Ca2+ Uptake and Retention. BBA-Bioenerg. 1990, 1016, 87–98. [Google Scholar] [CrossRef]
- Karch, J.; Bround, M.J.; Khalil, H.; Sargent, M.A.; Latchman, N.; Terada, N.; Peixoto, P.M.; Molkentin, J.D. Inhibition of Mitochondrial Permeability Transition by Deletion of the ANT Family and CypD. Sci. Adv. 2019, 5, 4597–4625. [Google Scholar] [CrossRef] [Green Version]
- Zhou, A.; Rohou, A.; Schep, D.G.; Bason, J.V.; Montgomery, M.G.; Walker, J.E.; Grigorieffniko, N.; Rubinstein, J.L. Structure and Conformational States of the Bovine Mitochondrial ATP Synthase by Cryo-EM. Elife 2015, 4, e10180. [Google Scholar] [CrossRef]
- Spikes, T.E.; Montgomery, M.G.; Walker, J.E. Structure of the Dimeric ATP Synthase from Bovine Mitochondria. Proc. Natl. Acad. Sci. USA 2020, 117, 23519–23526. [Google Scholar] [CrossRef]
- Davies, K.M.; Anselmi, C.; Wittig, I.; Faraldo-Gómez, J.D.; Kühlbrandt, W. Structure of the Yeast F1FO-ATP Synthase Dimer and Its Role in Shaping the Mitochondrial Cristae. Proc. Natl. Acad. Sci. USA 2012, 109, 13602–13607. [Google Scholar] [CrossRef] [Green Version]
- Giorgio, V.; Von Stockum, S.; Antoniel, M.; Fabbro, A.; Fogolari, F.; Forte, M.; Glick, G.D.; Petronilli, V.; Zoratti, M.; Szabó, I.; et al. Dimers of Mitochondrial ATP Synthase Form the Permeability Transition Pore. Proc. Natl. Acad. Sci. USA 2013, 110, 5887–5892. [Google Scholar] [CrossRef] [Green Version]
- Alavian, K.N.; Beutner, G.; Lazrove, E.; Sacchetti, S.; Park, H.A.; Licznerski, P.; Li, H.; Nabili, P.; Hockensmith, K.; Graham, M.; et al. An Uncoupling Channel within the C-Subunit Ring of the F1FO ATP Synthase Is the Mitochondrial Permeability Transition Pore. Proc. Natl. Acad. Sci. USA 2014, 111, 10580–10585. [Google Scholar] [CrossRef] [Green Version]
- Von Stockum, S.; Giorgio, V.; Trevisan, E.; Lippe, G.; Glick, G.D.; Forte, M.A.; Da-Rè, C.; Checchetto, V.; Mazzotta, G.; Costa, R.; et al. F-ATPase of Drosophila Melanogaster Forms 53-Picosiemen (53-PS) Channels Responsible for Mitochondrial Ca2+-Induced Ca2+ Release. J. Biol. Chem. 2015, 290, 4537–4544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinke, G.; Zhou, L.; Sazanov, L.A. Cryo-EM Structure of the Entire Mammalian F-Type ATP Synthase. Nat. Struct. Mol. Biol. 2020, 27, 1077–1085. [Google Scholar] [CrossRef] [PubMed]
- Giorgio, V.; Burchell, V.; Schiavone, M.; Bassot, C.; Minervini, G.; Petronilli, V.; Argenton, F.; Forte, M.; Tosatto, S.; Lippe, G.; et al. Ca2+ Binding to F-ATP Synthase β Subunit Triggers the Mitochondrial Permeability Transition. EMBO Rep. 2017, 18, 1065–1076. [Google Scholar] [CrossRef]
- Antoniel, M.; Jones, K.; Antonucci, S.; Spolaore, B.; Fogolari, F.; Petronilli, V.; Giorgio, V.; Carraro, M.; Di Lisa, F.; Forte, M.; et al. The Unique Histidine in OSCP Subunit of F-ATP Synthase Mediates Inhibition of the Permeability Transition Pore by Acidic PH. EMBO Rep. 2018, 19, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Carraro, M.; Jones, K.; Sartori, G.; Schiavone, M.; Antonucci, S.; Kucharczyk, R.; di Rago, J.-P.; Franchin, C.; Arrigoni, G.; Forte, M.; et al. The Unique Cysteine of F-ATP Synthase OSCP Subunit Participates in Modulation of the Permeability Transition Pore. Cell Rep. 2020, 32, 108095. [Google Scholar] [CrossRef] [PubMed]
- Bonora, M.; Morganti, C.; Morciano, G.; Pedriali, G.; Lebiedzinska-Arciszewska, M.; Aquila, G.; Giorgi, C.; Rizzo, P.; Campo, G.; Ferrari, R.; et al. Mitochondrial Permeability Transition Involves Dissociation of F1FO ATP Synthase Dimers and C-ring Conformation. EMBO Rep. 2017, 18, 1077–1089. [Google Scholar] [CrossRef]
- Morciano, G.; Pedriali, G.; Bonora, M.; Pavasini, R.; Mikus, E.; Calvi, S.; Bovolenta, M.; Lebiedzinska-Arciszewska, M.; Pinotti, M.; Albertini, A.; et al. A Naturally Occurring Mutation in ATP Synthase Subunit c Is Associated with Increased Damage Following Hypoxia/Reoxygenation in STEMI Patients. Cell Rep. 2021, 35. [Google Scholar] [CrossRef]
- Carraro, M.; Checchetto, V.; Sartori, G.; Kucharczyk, R.; Di Rago, J.P.; Minervini, G.; Franchin, C.; Arrigoni, G.; Giorgio, V.; Petronilli, V.; et al. High-Conductance Channel Formation in Yeast Mitochondria Is Mediated by F-ATP Synthase e and g Subunits. Cell. Physiol. Biochem. 2018, 50, 1840–1855. [Google Scholar] [CrossRef] [PubMed]
- Niedzwiecka, K.; Baranowska, E.; Panja, C.; Kucharczyk, R. ATP Synthase Subunit a Supports Permeability Transition in Yeast Lacking Dimerization Subunits and Modulates YPTP Conductance. Cell. Physiol. Biochem. 2020, 54, 211–229. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Carraro, M.; Carrer, A.; Minervini, G.; Urbani, A.; Masgras, I.; Tosatto, S.C.E.; Szabò, I.; Bernardi, P.; Lippe, G. Arg-8 of Yeast Subunit e Contributes to the Stability of F-ATP Synthase Dimers and to the Generation of the Full-Conductance Mitochondrial Megachannel. J. Biol. Chem. 2019, 294, 10987–10997. [Google Scholar] [CrossRef]
- Galber, C.; Minervini, G.; Cannino, G.; Boldrin, F.; Petronilli, V.; Tosatto, S.; Lippe, G.; Giorgio, V. The f Subunit of Human ATP Synthase Is Essential for Normal Mitochondrial Morphology and Permeability Transition. Cell Rep. 2021, 35. [Google Scholar] [CrossRef]
- He, J.; Ford, H.C.; Carroll, J.; Ding, S.; Fearnley, I.M.; Walker, J.E. Persistence of the Mitochondrial Permeability Transition in the Absence of Subunit c of Human ATP Synthase. Proc. Natl. Acad. Sci. USA 2017, 114, 3409–3414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, J.; Carroll, J.; Ding, S.; Fearnley, I.M.; Walker, J.E. Permeability Transition in Human Mitochondria Persists in the Absence of Peripheral Stalk Subunits of ATP Synthase. Proc. Natl. Acad. Sci. USA 2017, 114, 9086–9091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carroll, J.; He, J.; Ding, S.; Fearnley, I.M.; Walker, J.E. Persistence of the Permeability Transition Pore in Human Mitochondria Devoid of an Assembled ATP Synthase. Proc. Natl. Acad. Sci. USA 2019, 116, 12816–12821. [Google Scholar] [CrossRef] [Green Version]
- Neginskaya, M.A.; Solesio, M.E.; Berezhnaya, E.V.; Amodeo, G.F.; Mnatsakanyan, N.; Jonas, E.A.; Pavlov, E.V. ATP Synthase C-Subunit-Deficient Mitochondria Have a Small Cyclosporine A-Sensitive Channel, but Lack the Permeability Transition Pore. Cell Rep. 2019, 26, 11–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrer, A.; Tommasin, L.; Šileikytė, J.; Ciscato, F.; Filadi, R.; Urbani, A.; Forte, M.; Rasola, A.; Szabò, I.; Carraro, M.; et al. Defining the Molecular Mechanisms of the Mitochondrial Permeability Transition through Genetic Manipulation of F-ATP Synthase. Nat. Commun. 2021, 12, 4835. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Ko, Y.; Delannoy, M.; Ludtke, S.J.; Chiu, W.; Pedersen, P.L. Mitochondrial ATP Synthasome. Three-Dimensional Structure by Electron Microscopy of the ATP Synthase in Complex Formation with Carriers for P i and ADP/ATP. J. Biol. Chem. 2004, 279, 31761–31768. [Google Scholar] [CrossRef] [Green Version]
- Beutner, G.; Alanzalon, R.E.; Porter, G.A. Cyclophilin D Regulates the Dynamic Assembly of Mitochondrial ATP Synthase into Synthasomes. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernardi, P.; von Stockum, S. The Permeability Transition Pore as a Ca2+ Release Channel: New Answers to an Old Question. Cell Calcium 2012, 52, 22–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barsukova, A.; Komarov, A.; Hajnóczky, G.; Bernardi, P.; Bourdette, D.; Forte, M. Activation of the Mitochondrial Permeability Transition Pore Modulates Ca2+ Responses to Physiological Stimuli in Adult Neurons. Eur. J. Neurosci. 2011, 33, 831–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, X.; Kwong, J.Q.; Molkentin, J.D.; Bers, D.M. Individual Cardiac Mitochondria Undergo Rare Transient Permeability Transition Pore Openings. Circ. Res. 2016, 118, 834–841. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, A.; Wu, P.H.; Hughes, E.G.; Fukaya, M.; Tischfield, M.A.; Langseth, A.J.; Wirtz, D.; Bergles, D.E. Transient Opening of the Mitochondrial Permeability Transition Pore Induces Microdomain Calcium Transients in Astrocyte Processes. Neuron 2017, 93, 587–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elrod, J.W.; Wong, R.; Mishra, S.; Vagnozzi, R.J.; Sakthievel, B.; Goonasekera, S.A.; Karch, J.; Gabel, S.; Farber, J.; Force, T.; et al. Cyclophilin D Controls Mitochondrial Pore-Dependent Ca2+ Exchange, Metabolic Flexibility, and Propensity for Heart Failure in Mice. J. Clin. Invest. 2010, 120, 3680–3687. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial Reactive Oxygen Species (ROS) and ROS-Induced ROS Release. Physiological Reviews. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petronilli, V.; Penzo, D.; Scorrano, L.; Bernardi, P.; Di Lisa, F. The Mitochondrial Permeability Transition, Release of Cytochrome c and Cell Death. Correlation with the Duration of Pore Openings in Situ. J. Biol. Chem. 2001, 276, 12030–12034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Lisa, F.; Carpi, A.; Giorgio, V.; Bernardi, P. The Mitochondrial Permeability Transition Pore and Cyclophilin D in Cardioprotection. Biochimica et Biophysica Acta-Mol. Cell Res. 2011, 1813, 1316–1322. [Google Scholar] [CrossRef] [Green Version]
- Bond, J.M.; Chacon, E.; Herman, B.; Lemasters, J.J. Intracellular PH and Ca2+ Homeostasis in the PH Paradox of Reperfusion Injury to Neonatal Rat Cardiac Myocytes. Am. J. Physiol.-Cell Physiol. 1993, 265, C129–C137. [Google Scholar] [CrossRef] [PubMed]
- Duchen, M.R.; McGuinness, O.; Brown, L.A.; Crompton, M. On the Involvement of a Cyclosporin A Sensitive Mitochondrial Pore in Myocardial Reperfusion Injury. Cardiovasc. Res. 1993, 27, 1790–1794. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, E.J.; Halestrap, A.P. Protection by Cyclosporin A of Ischemia/Reperfusion-Induced Damage in Isolated Rat Hearts. J. Mol. Cell. Cardiol. 1993, 25, 1461–1469. [Google Scholar] [CrossRef] [PubMed]
- Schinder, A.F.; Olson, E.C.; Spitzer, N.C.; Montal, M. Mitochondrial Dysfunction Is a Primary Event in Glutamate Neurotoxicity. J. Neurosci. 1996, 16, 6125–6133. [Google Scholar] [CrossRef] [Green Version]
- White, R.J.; Reynolds, I.J. Mitochondrial Depolarization in Glutamate-Stimulated Neurons: An Early Signal Specific to Excitotoxin Exposure. J. Neurosci. 1996, 16, 5688–5697. [Google Scholar] [CrossRef] [PubMed]
- Trost, L.C.; Lemasters, J.J. The Mitochondrial Permeability Transition: A New Pathophysiological Mechanism for Reye’s Syndrome and Toxic Liver Injury. J. Pharmacol. Exp. Ther. 1996, 278, 1000–1005. [Google Scholar] [PubMed]
- Forte, M.; Gold, B.G.; Marracci, G.; Chaudhary, P.; Basso, E.; Johnsen, D.; Yu, X.; Fowlkes, J.; Bernardi, P.; Bourdette, D. Cyclophilin D Inactivation Protects Axons in Experimental Autoimmune Encephalomyelitis, an Animal Model of Multiple Sclerosis. Proc. Natl. Acad. Sci. USA 2007, 104, 7558–7563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, L.J. The Mitochondrial Permeability Transition Pore: A Molecular Target for Amyotrophic Lateral Sclerosis Therapy. Biochim. Biophys. Acta-Mol. Basis Dis. 2010, 1802, 186–197. [Google Scholar] [CrossRef]
- Du, H.; Guo, L.; Fang, F.; Chen, D.; Sosunov, A.A.; M McKhann, G.; Yan, Y.; Wang, C.; Zhang, H.; Molkentin, J.D.; et al. Cyclophilin D Deficiency Attenuates Mitochondrial and Neuronal Perturbation and Ameliorates Learning and Memory in Alzheimer’s Disease. Nat. Med. 2008, 14, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Guo, L.; Zhang, W.; Rydzewska, M.; Yan, S. Cyclophilin D Deficiency Improves Mitochondrial Function and Learning/Memory in Aging Alzheimer Disease Mouse Model. Neurobiol. Aging 2011, 32, 398–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irwin, W.A.; Bergamin, N.; Sabatelli, P.; Reggiani, C.; Megighian, A.; Merlini, L.; Braghetta, P.; Columbaro, M.; Volpin, D.; Bressan, G.M.; et al. Mitochondrial Dysfunction and Apoptosis in Myopathic Mice with Collagen VI Deficiency. Nat. Genet. 2003, 35, 367–371. [Google Scholar] [CrossRef] [PubMed]
- Tiepolo, T.; Angelin, A.; Palma, E.; Sabatelli, P.; Merlini, L.; Nicolosi, L.; Finetti, F.; Braghetta, P.; Vuagniaux, G.; Dumont, J.M.; et al. The Cyclophilin Inhibitor Debio 025 Normalizes Mitochondrial Function, Muscle Apoptosis and Ultrastructural Defects in Col6a1-/-Myopathic Mice. Br. J. Pharmacol. 2009, 157, 1045–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sambri, I.; Massa, F.; Gullo, F.; Meneghini, S.; Cassina, L.; Carraro, M.; Dina, G.; Quattrini, A.; Patanella, L.; Carissimo, A.; et al. Impaired Flickering of the Permeability Transition Pore Causes SPG7 Spastic Paraplegia. EBioMedicine 2020, 61, 103050. [Google Scholar] [CrossRef] [PubMed]
- Blatt, N.B.; Boitano, A.E.; Lyssiotis, C.A.; Opipari, A.W.; Glick, G.D. Bz-423 Superoxide Signals Apoptosis via Selective Activation of JNK, Bak, and Bax. Free Radic. Biol. Med. 2008, 45, 1232–1242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, K.M.; Chen, X.; Boitano, A.; Swenson, L.; Opipari, A.W.; Glick, G.D. Identification and Validation of the Mitochondrial F1FO-ATPase as the Molecular Target of the Immunomodulatory Benzodiazepine Bz-423. Chem. Biol. 2005, 12, 485–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Algieri, C.; Trombetti, F.; Pagliarani, A.; Ventrella, V.; Nesci, S. Phenylglyoxal Inhibition of the Mitochondrial F1FO-ATPase Activated by Mg2+ or by Ca2+ Provides Clues on the Mitochondrial Permeability Transition Pore. Arch. Biochem. Biophys. 2020, 681, 108258. [Google Scholar] [CrossRef] [PubMed]
- Symersky, J.; Osowski, D.; Walters, D.E.; Mueller, D.M. Oligomycin Frames a Common Drug-Binding Site in the ATP Synthase. Proc. Natl. Acad. Sci. USA 2012, 109, 13961–13965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hearne, A.; Chen, H.; Monarchino, A.; Wiseman, J.S. Oligomycin-Induced Proton Uncoupling. Toxicol. Vitr. 2020, 67, 104907. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, S.; Xu, Q.; Velours, J.; Reed, J.C. The Mitochondrial FOF1-ATPase Proton Pump Is Required for Function of the Proapoptotic Protein Bax in Yeast and Mammalian Cells. Mol. Cell 1998, 1, 327–336. [Google Scholar] [CrossRef]
- Narita, M.; Shimizu, S.; Ito, T.; Chittenden, T.; Lutz, R.J.; Matsuda, H.; Tsujimoto, Y. Bax Interacts with the Permeability Transition Pore to Induce Permeability Transition and Cytochrome c Release in Isolated Mitochondria. Proc. Natl. Acad. Sci. USA 1998, 95, 14681–14686. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Xu, H.; Huang, K. Mitochondrial Permeabiltiy Transition and Cytochrome c Release Induced by Selenite. J. Inorg. Biochem. 2002, 90, 43–50. [Google Scholar] [CrossRef]
- Novgorodov, S.A.; Gudz, T.I.; Kushnareva, Y.E.; Zorov, D.B.; Kudrjashov, Y.B. Effect of Cyclosporine A and Oligomycin on Non-Specific Permeability of the Inner Mitochondrial Membrane. FEBS Lett. 1990, 270, 108–110. [Google Scholar] [CrossRef] [Green Version]
- Chávez, E.; Rodríguez, J.S.; García, G.; García, N.; Correa, F. Oligomycin Strengthens the Effect of Cyclosporin A on Mitochondrial Permeability Transition by Inducing Phosphate Uptake. Cell Biol. Int. 2005, 29, 551–558. [Google Scholar] [CrossRef]
- Azzi, A.; Azzone, G.F. Swelling and Shrinkage Phenomena in Liver Mitochondria. III. Irreversible Swelling Induced by Inorganic Phosphate and Ca2+. Biochim. Biophys. Acta 1966, 113, 438–444. [Google Scholar] [CrossRef]
- Carbonera, D.; Azzone, G.F. Permeability of Inner Mitochondrial Membrane and Oxidative Stress. BBA-Biomembr. 1988, 943, 245–255. [Google Scholar] [CrossRef]
- Novgorodov, S.A.; Gudz, T.I.; Brierley, G.P.; Pfeiffer, D.R. Magnesium Ion Modulates the Sensitivity of the Mitochondrial Permeability Transition Pore to Cyclosporine A and ADP. Arch. Biochem. Biophys. 1994, 311, 219–228. [Google Scholar] [CrossRef]
- Meyer, B.; Wittig, I.; Trifilieff, E.; Karas, M.; Schägger, H. Identification of Two Proteins Associated with Mammalian ATP Synthase. Mol. Cell. Proteomics 2007, 6, 1690–1699. [Google Scholar] [CrossRef] [Green Version]
- Daniele, C.; Dahamna, S.; Firuzi, O.; Sekfali, N.; Saso, L.; Mazzanti, G. Atractylis Gummifera L. Poisoning: An Ethnopharmacological Review. J. Ethnopharmacol. 2005, 97, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Bruni, A.; Luciani, S.; Contessa, A.R. Inhibition by Atractyloside of the Binding of Adenine-Nucleotides to Rat-Liver Mitochondria. Nature 1964, 201, 1219–1220. [Google Scholar] [CrossRef] [PubMed]
- Klingenberg, M.; Grebe, K.; Scherer, B. The Binding of Atractylate and Carboxy-atractylate to Mitochondria. Eur. J. Biochem. 1975, 52, 351–363. [Google Scholar] [CrossRef] [PubMed]
- Welling, W.; Cohen, J.A.; Berends, W. Disturbance of Oxidative Phosphorylation by an Antibioticum Produced by Pseudomonas Cocovenenans. Biochem. Pharmacol. 1960, 3, 122–135. [Google Scholar] [CrossRef]
- Henderson, P.J.; Lardy, H.A. Bongkrekic Acid. An Inhibitor of the Adenine Nucleotide Translocase of Mitochondria. J. Biol. Chem. 1970, 245, 1319–1326. [Google Scholar] [CrossRef]
- Klingenberg, M.; Grebe, K.; Scherer, B. Opposite Effects of Bongkrekic Acid and Atractyloside on the Adenine Nucleotides Induced Mitochondrial Volume Changes and on the Efflux of Adenine Nucleotides. FEBS Lett. 1971, 16, 253–256. [Google Scholar] [CrossRef] [Green Version]
- Todisco, S.; Di Noia, M.A.; Onofrio, A.; Parisi, G.; Punzi, G.; Redavid, G.; De Grassi, A.; Pierri, C.L. Identification of New Highly Selective Inhibitors of the Human ADP/ATP Carriers by Molecular Docking and in Vitro Transport Assays. Biochem. Pharmacol. 2015, 100, 112–132. [Google Scholar] [CrossRef] [PubMed]
- Ravagnan, L.; Marzo, I.; Costantini, P.; Susin, S.A.; Zamzami, N.; Petit, P.X.; Hirsch, F.; Goulbern, M.; Poupon, M.F.; Miccoli, L.; et al. Lonidamine Triggers Apoptosis via a Direct, Bcl-2-Inhibited Effect on the Mitochondrial Permeability Transition Pore. Oncogene 1999, 18, 2537–2546. [Google Scholar] [CrossRef] [Green Version]
- Fancelli, D.; Abate, A.; Amici, R.; Bernardi, P.; Ballarini, M.; Cappa, A.; Carenzi, G.; Colombo, A.; Contursi, C.; Di Lisa, F.; et al. Cinnamic Anilides as New Mitochondrial Permeability Transition Pore Inhibitors Endowed with Ischemia-Reperfusion Injury Protective Effect in Vivo. J. Med. Chem. 2014, 57, 5333–5347. [Google Scholar] [CrossRef]
- Fang, J.; Chavez-Valdez, R.; Flock, D.L.; Avaritt, O.; Saraswati, M.; Robertson, C.; Martin, L.J.; Northington, F.J. An Inhibitor of the Mitochondrial Permeability Transition Pore Lacks Therapeutic Efficacy Following Neonatal Hypoxia Ischemia in Mice. Neuroscience 2019, 406, 202–211. [Google Scholar] [CrossRef]
- Richardson, A.P.; Halestrap, A.P. Quantification of Active Mitochondrial Permeability Transition Pores Using GNX-4975 Inhibitor Titrations Provides Insights into Molecular Identity. Biochem. J. 2016, 473, 1129–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, S.; Šileikyte, J.; Schiavone, M.; Neuenswander, B.; Argenton, F.; Aubé, J.; Hedrick, M.P.; Chung, T.D.Y.; Forte, M.A.; Bernardi, P.; et al. Discovery, Synthesis, and Optimization of Diarylisoxazole-3-Carboxamides as Potent Inhibitors of the Mitochondrial Permeability Transition Pore. ChemMedChem 2015, 10, 1655–1671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, S.; Šileikyte, J.; Neuenswander, B.; Hedrick, M.P.; Chung, T.D.Y.; Aubé, J.; Schoenen, F.J.; Forte, M.A.; Bernardi, P. N-Phenylbenzamides as Potent Inhibitors of the Mitochondrial Permeability Transition Pore. ChemMedChem 2016, 11, 283–288. [Google Scholar] [CrossRef] [Green Version]
- Šileikytė, J.; Devereaux, J.; de Jong, J.; Schiavone, M.; Jones, K.; Nilsen, A.; Bernardi, P.; Forte, M.; Cohen, M.S. Second-Generation Inhibitors of the Mitochondrial Permeability Transition Pore with Improved Plasma Stability. ChemMedChem 2019, 14, 1771–1782. [Google Scholar] [CrossRef]
- Wieckowski, M.R.; Wojtczak, L. Fatty Acid-Induced Uncoupling of Oxidative Phosphorylation Is Partly Due to Opening of the Mitochondrial Permeability Transition Pore. FEBS Lett. 1998, 423, 339–342. [Google Scholar] [CrossRef]
- Lehninger, A.L.; Remmert, L.F. An Endogenous Uncoupling and Swelling Agent in Liver Mitochondria and Its Enzymic Formation. J. Biol. Chem. 1959, 234, 2459–2464. [Google Scholar] [CrossRef]
- Hülsmann, W.C.; Elliott, W.B.; Slater, E.C. The Nature and Mechanism of Action of Uncoupling Agents Present in Mitochrome Preparations. BBA-Biochim. Biophys. Acta 1960, 39, 267–276. [Google Scholar] [CrossRef]
- Kamp, F.; Hamilton, J.A. PH Gradients across Phospholipid Membranes Caused by Fast Flip-Flop of Un-Ionized Fatty Acids. Proc. Natl. Acad. Sci. USA 1992, 89, 11367–11370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skulachev, V.P. Fatty Acid Circuit as a Physiological Mechanism of Uncoupling of Oxidative Phosphorylation. FEBS 1991, 294, 158–162. [Google Scholar] [CrossRef] [Green Version]
- Schönfeld, P. Does the Function of Adenine Nucleotide Translocase in Fatty Acid Uncoupling Depend on the Type of Mitochondria? FEBS Lett. 1990, 264, 246–248. [Google Scholar] [CrossRef] [Green Version]
- Andreyev, A.Y.; Bondareva, T.O.; Ddedukhova, V.I.; Mokhova, E.N.; Skulachev, V.P.; Tsofina, L.M.; Volkov, N.I.; Vygodina, T.V. The ATP/ADP-antiporter Is Involved in the Uncoupling Effect of Fatty Acids on Mitochondria. Eur. J. Biochem. 1989, 182, 585–592. [Google Scholar] [CrossRef]
- Dedukhova, V.I.; Mokhova, E.N.; Skulachev, V.P.; Starkov, A.A.; Arrigoni-Martelli, E.; Bobyleva, V.A. Uncoupling Effect of Fatty Acids on Heart Muscle Mitochondria and Submitochondrial Particles. FEBS Lett. 1991, 295, 51–54. [Google Scholar] [CrossRef] [Green Version]
- Brustovetsky, N.; Klingenberg, M. The Reconstituted ADP/ATP Carrier Can Mediate H+ Transport by Free Fatty Acids, Which Is Further Stimulated by Mersalyl. J. Biol. Chem. 1994, 269, 27329–27336. [Google Scholar] [CrossRef]
- Penzo, D.; Tagliapietra, C.; Colonna, R.; Petronilli, V.; Bernardi, P. Effects of Fatty Acids on Mitochondria: Implications for Cell Death. Biochim. Biophys. Acta-Bioenerg. 2002, 1555, 160–165. [Google Scholar] [CrossRef] [Green Version]
- Broekemeier, K.M.; Pfeiffer, D.R. Inhibition of the Mitochondrial Permeability Transition by Cyclosporin A during Long Time Frame Experiments: Relationship between Pore Opening and the Activity of Mitochondrial Phospholipases. Biochemistry 1995, 34, 16440–16449. [Google Scholar] [CrossRef] [PubMed]
- Penzo, D.; Petronilli, V.; Angelin, A.; Cusan, C.; Colonna, R.; Scorrano, L.; Pagano, F.; Prato, M.; Di Lisa, F.; Bernardi, P. Arachidonic Acid Released by Phospholipase A2 Activation Triggers Ca2+-Dependent Apoptosis through the Mitochondrial Pathway. J. Biol. Chem. 2004, 279, 25219–25225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scarpa, A.; Lindsay, J.G. Maintenance of Energy-Linked Functions in Rat-Liver Mitochondria Aged in the Presence of Nupercaine. Eur. J. Biochem. 1972, 27, 401–407. [Google Scholar] [CrossRef]


{kind=link}
| Compound Name | Target | Effect on the Permeability Transition (PT) | Mechanism of Action | Comments |
|---|---|---|---|---|
| cyclosporin A (CsA) | cyclophilins | inhibition | CyPD sequestration | CsA/CyPA complex inhibits calcineurin [64,65,66] |
| NIM811 | effective in ischemia/reperfusion injury, dystrophic models, traumatic brain injury and pancreatitis [71,72,73,74] | |||
| Debio025 (or Alisporivir) | effective in dystrophic models [36,79,80] | |||
| sanglifehrin A (SfA) | effective in ischemia-reperfusion injury [76] | |||
| compound 31 | effective in hepatic ischemia/reperfusion injury [78] | |||
| Benzodiazepine-423 (Bz-423) | F-ATP synthase (OSCP subunit) | activation | possible conformational change of the F-ATP synthase | induction of cell death in lymphocytes [81]; effective on channel activity of reconstituted F-ATP synthase [82] |
| phenylglyoxals (PGO) | F-ATP synthase (subunit g) | activation or inhibition (species-specific) | arginine adducts | [25,26,83,84,85,86] |
| oligomycin | F-ATP synthase (subunit c and a) | inhibition (controversial) | ND | no alterations of PT occurrence [13,24,87]; inhibition of channel activity of DDM-extracted F-ATP synthase [88] |
| compound 10 | F-ATP synthase (subunit c) | inhibition | ND | protection in an ex vivo model of myocardial infarction [89] |
| atractylate (ATR) | adenine nucleotide translocator (ANT) | activation | blockage of ANT in the c-state | [90,91,92] |
| bongkrekic acid (BKA) | inhibition | blockage of ANT in the m-state | [90,91,92] | |
| suramin | activation | oxidation of critical thiols | induction of mitochondrial swelling [93]; FDA approved [94] | |
| lonidamine | activation | ND | activation of ANT channel activity [95] | |
| GNX-4728 and GNX-4975 (cinnamic anilides) | adenine nucleotide translocator (ANT)? | inhibition | stabilization of ANT and phosphate carrier (PiC) interaction | effective in amyotrophic lateral sclerosis [96] |
| TR001 and TR002 | ND | ND | effective in dystrophic models and in ischemia/reperfusion injury [97,98] | |
| nupercaine and tetracaine | phospholipases (PLAs) | prevention of fatty acid release | [1,20] | |
| ER-000444793 | ND | ND | [99] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carrer, A.; Laquatra, C.; Tommasin, L.; Carraro, M. Modulation and Pharmacology of the Mitochondrial Permeability Transition: A Journey from F-ATP Synthase to ANT. Molecules 2021, 26, 6463. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26216463
Carrer A, Laquatra C, Tommasin L, Carraro M. Modulation and Pharmacology of the Mitochondrial Permeability Transition: A Journey from F-ATP Synthase to ANT. Molecules. 2021; 26(21):6463. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26216463
Chicago/Turabian StyleCarrer, Andrea, Claudio Laquatra, Ludovica Tommasin, and Michela Carraro. 2021. "Modulation and Pharmacology of the Mitochondrial Permeability Transition: A Journey from F-ATP Synthase to ANT" Molecules 26, no. 21: 6463. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26216463