
Mollusc-Derived Brominated Indoles for the Selective Inhibition of Cyclooxygenase: A Computational Expedition

 , and
, and
Abstract
:1. Introduction
2. Results and Discussion
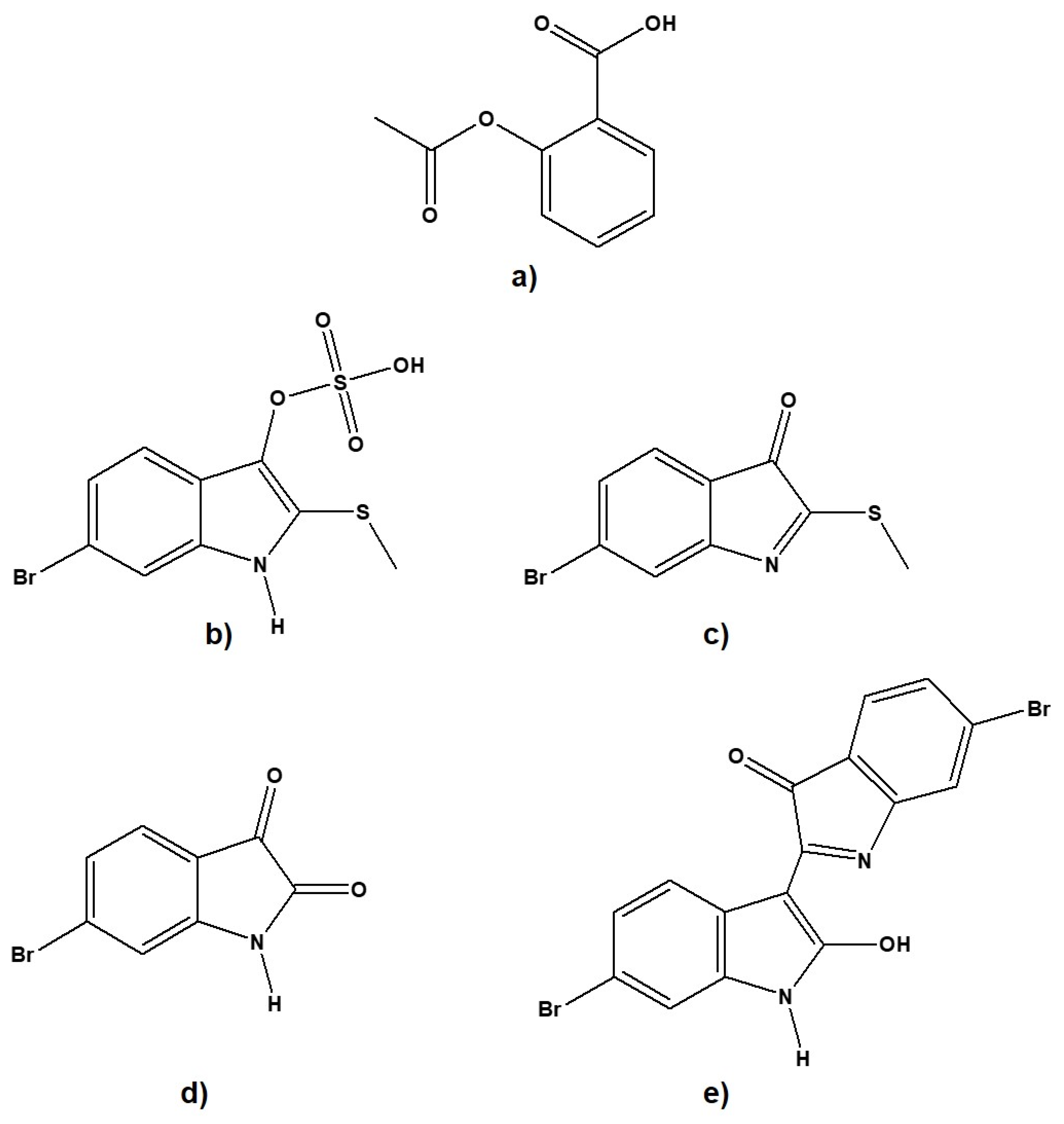
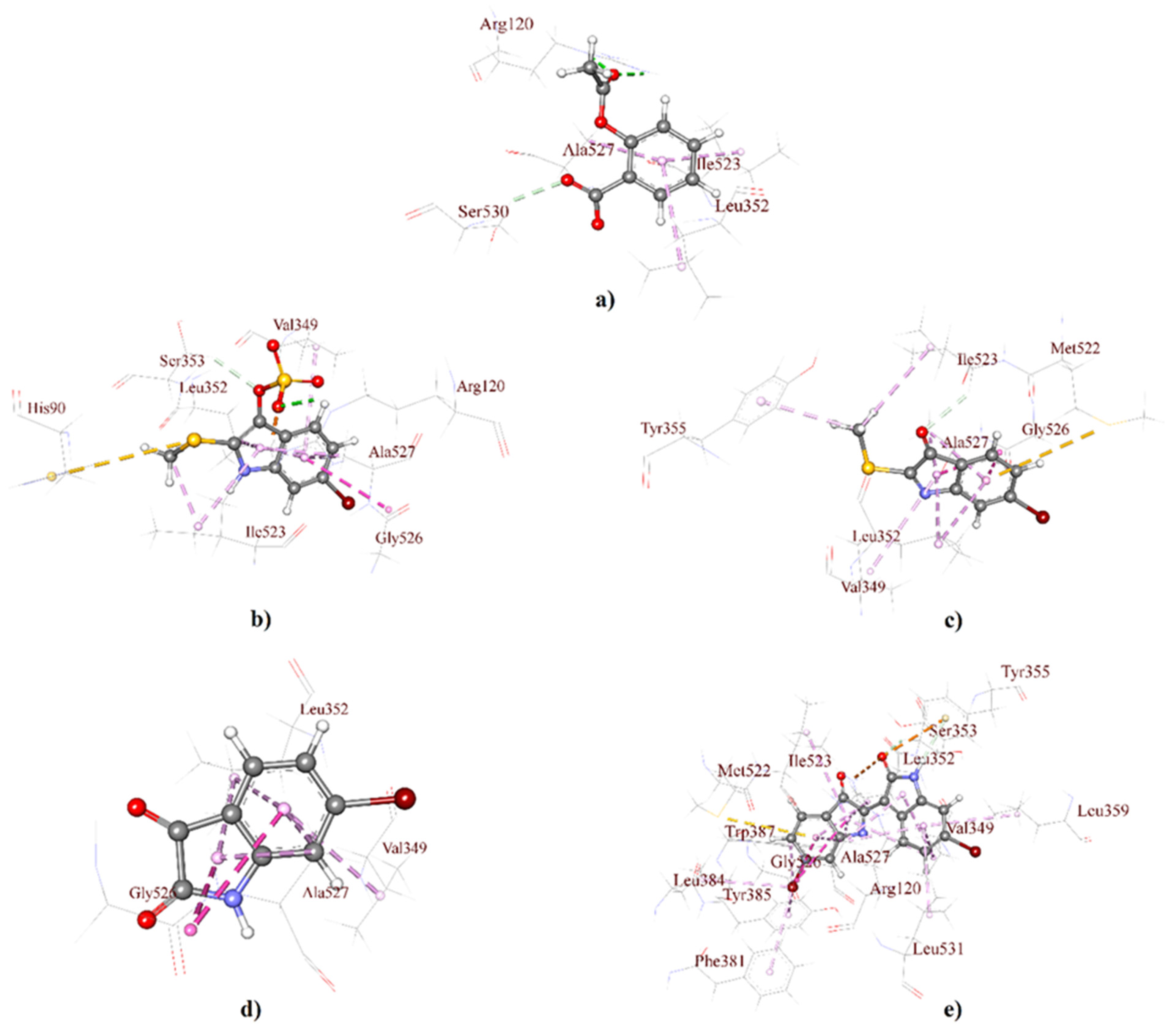
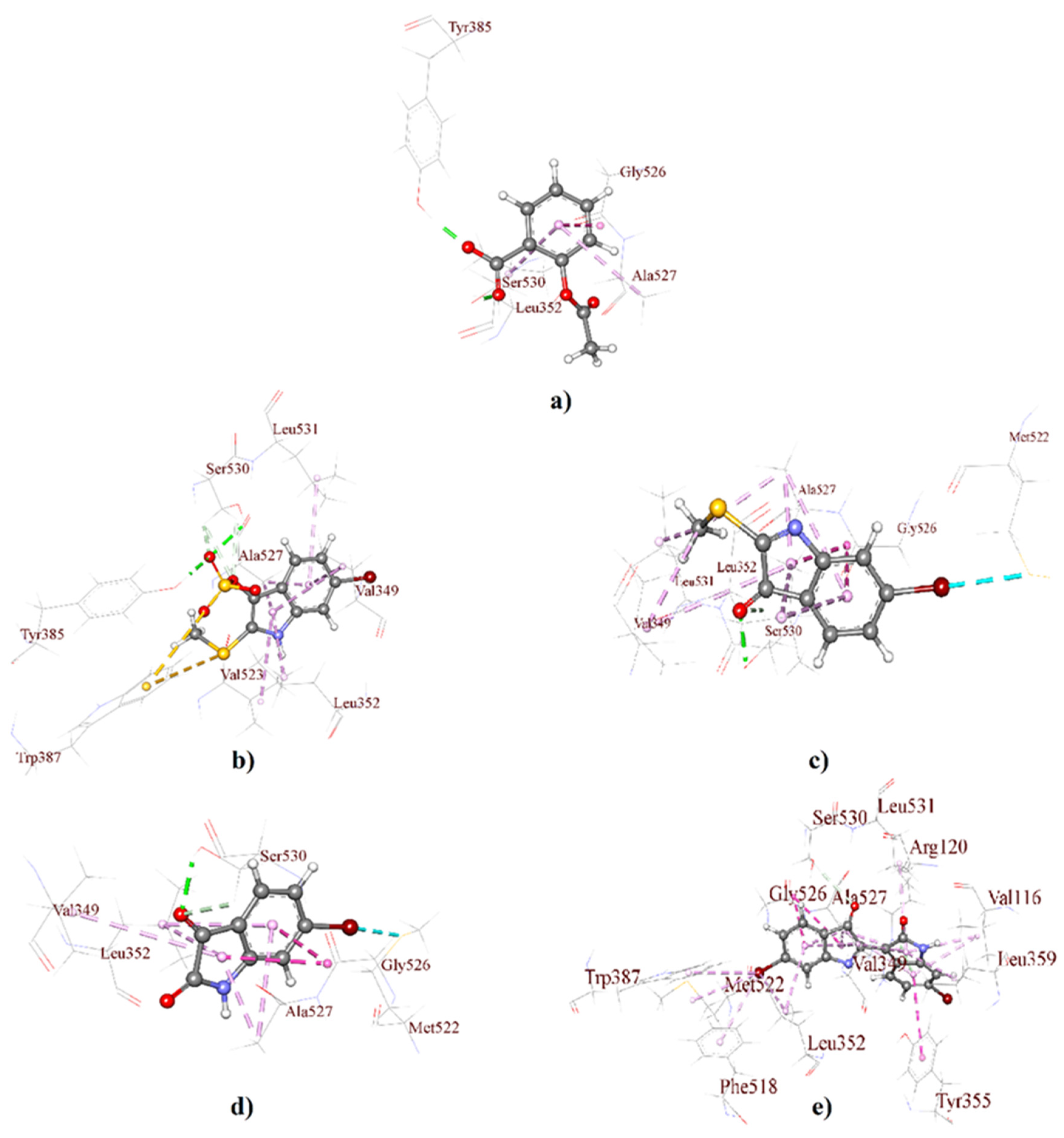
2.1. Molecular Docking Analysis
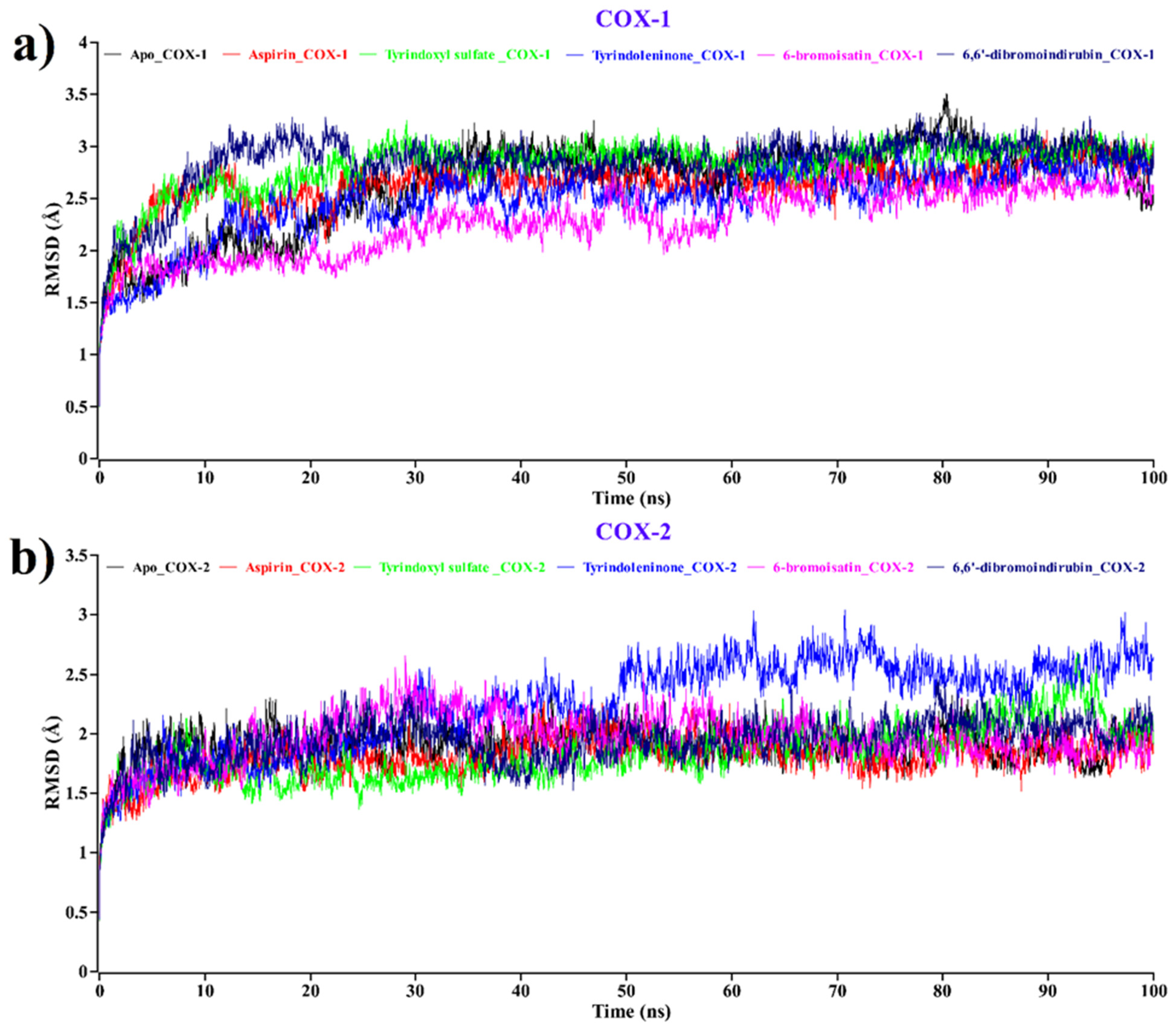
2.2. Molecular Dynamics Simulation Analysis
2.2.1. Root Mean Square Deviation (RMSD)
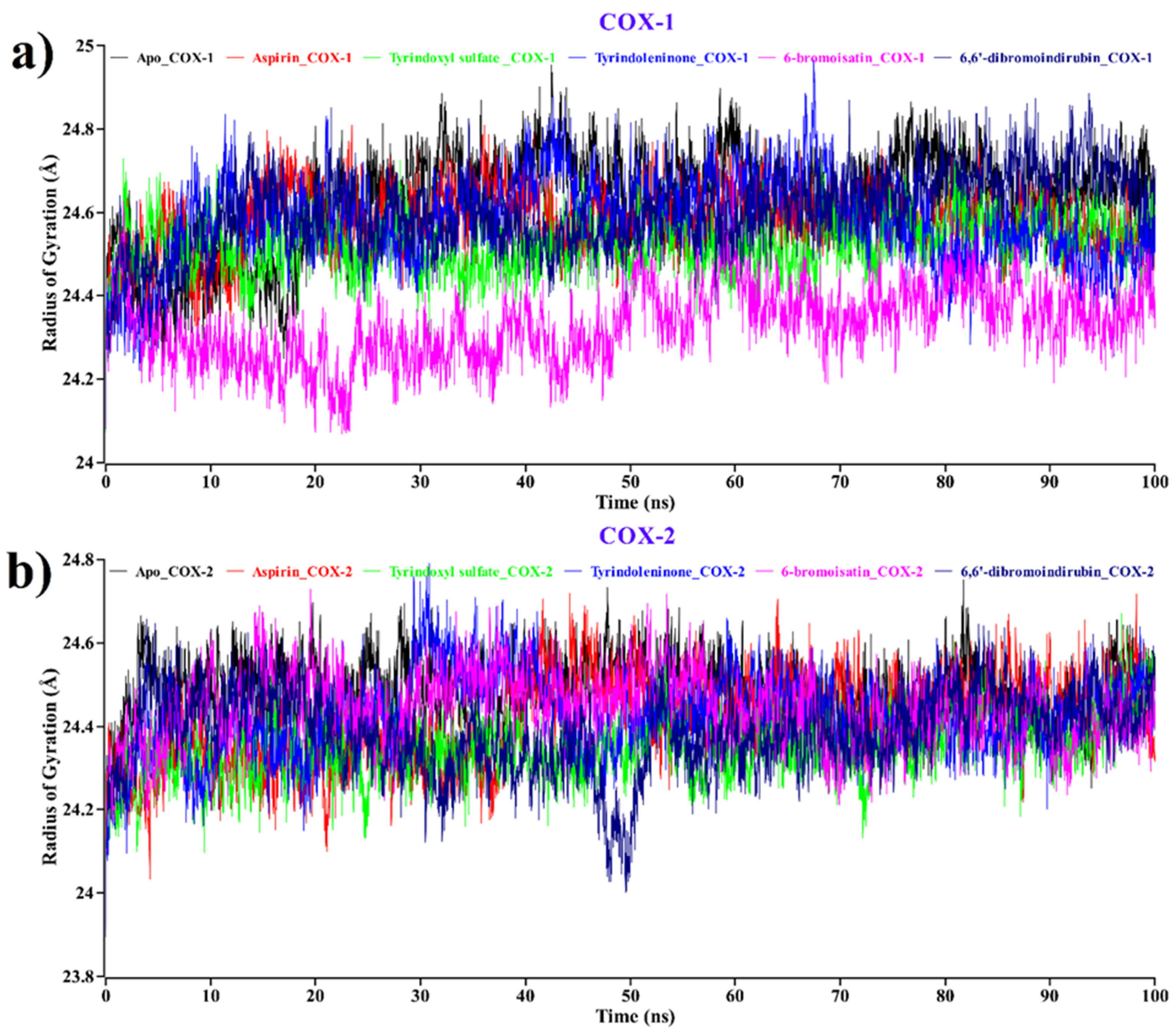
2.2.2. Radius of Gyration (Rg)
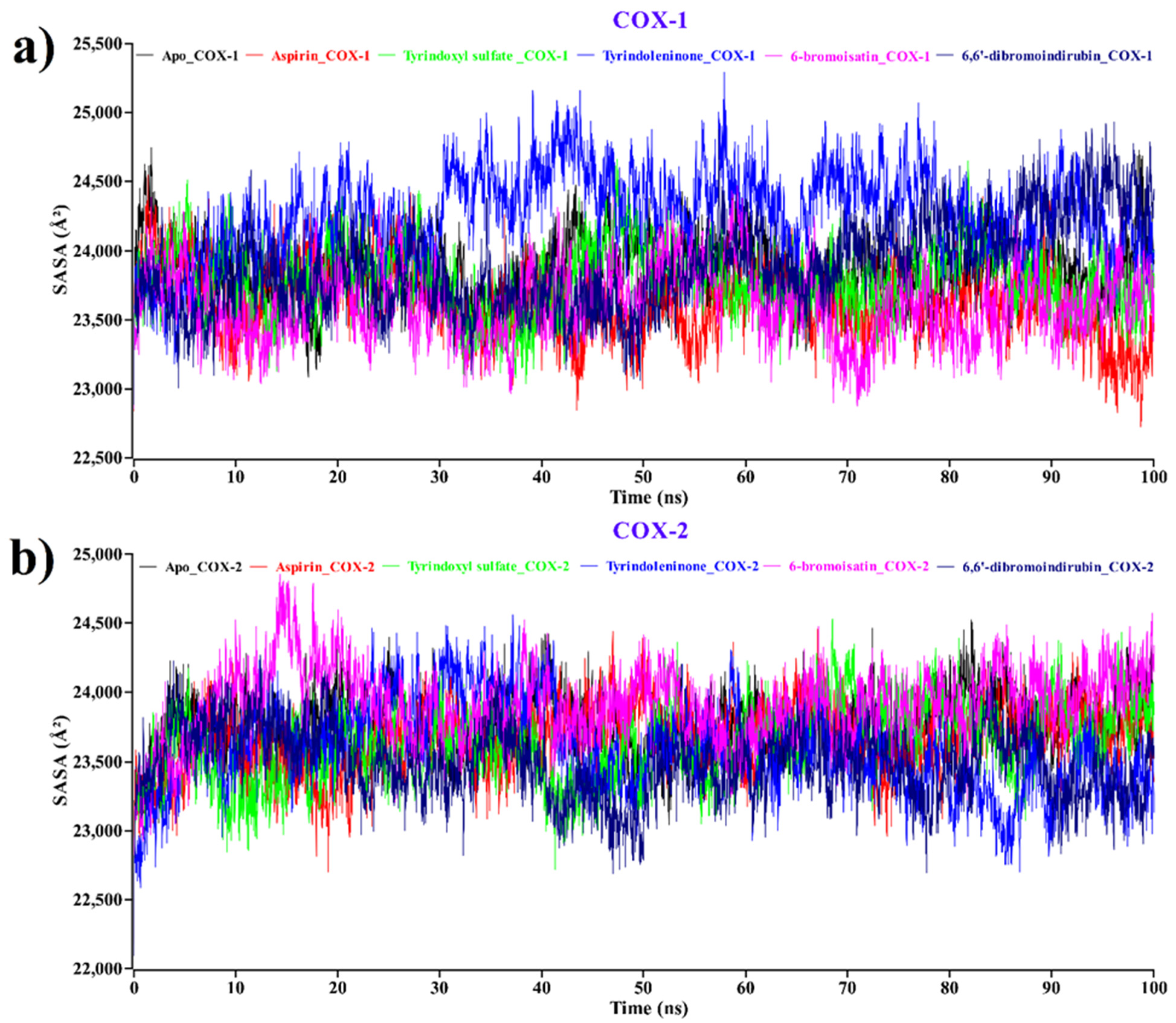
2.2.3. Solvent Accessible Surface Area (SASA)
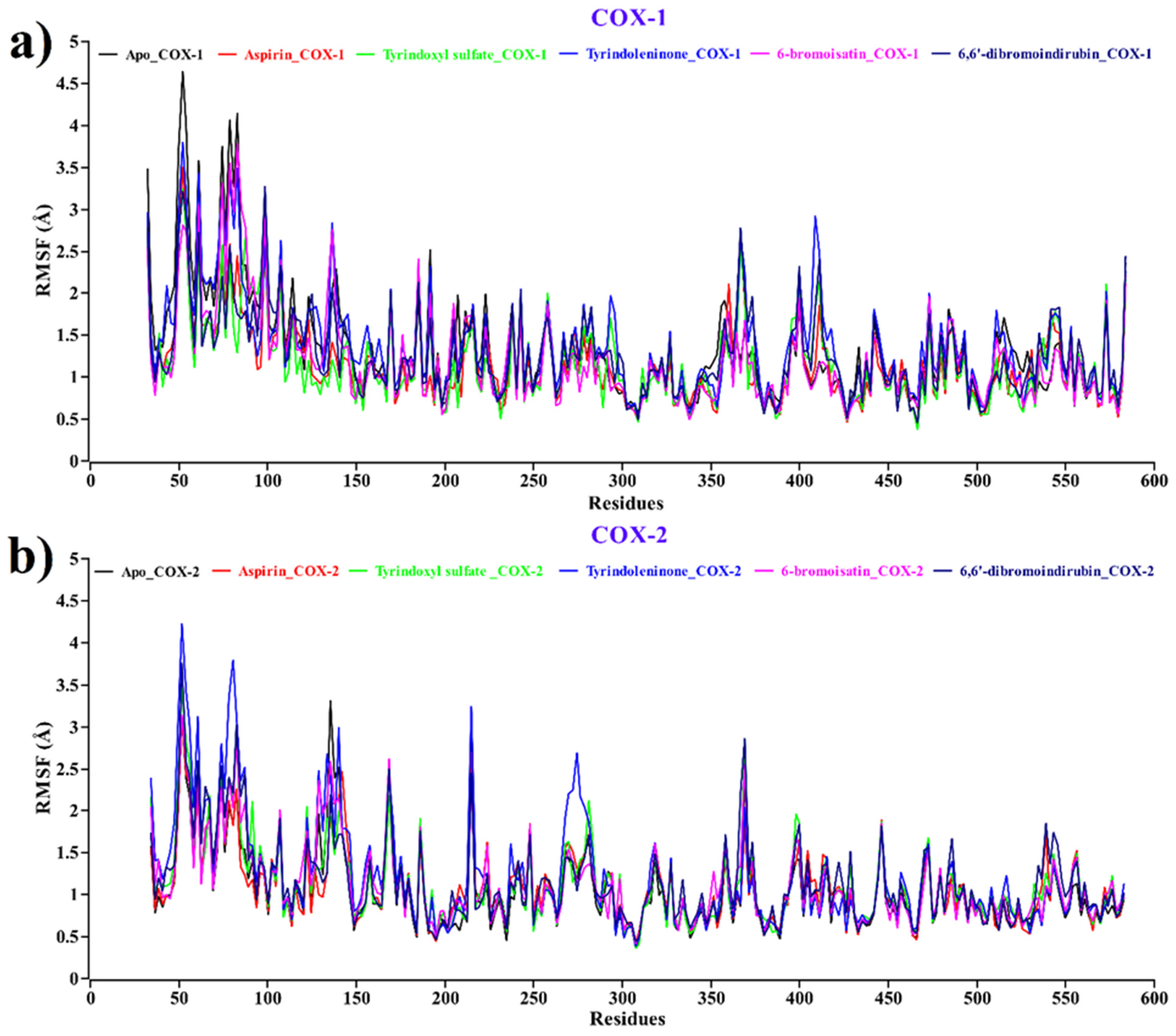
2.2.4. Root Mean Square Fluctuations (RMSFs)
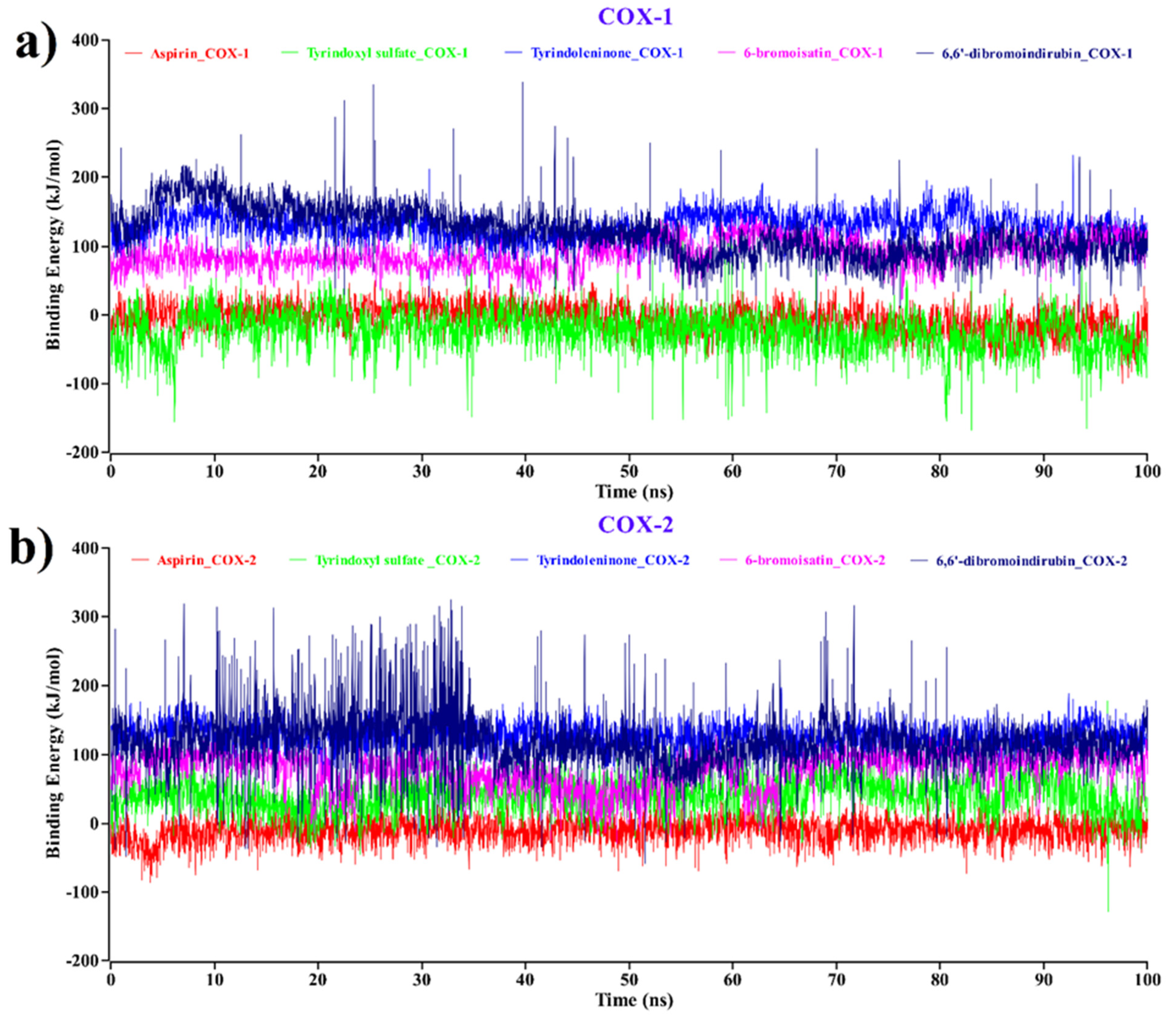
2.3. MM–PBSA Binding Free Energy Analysis
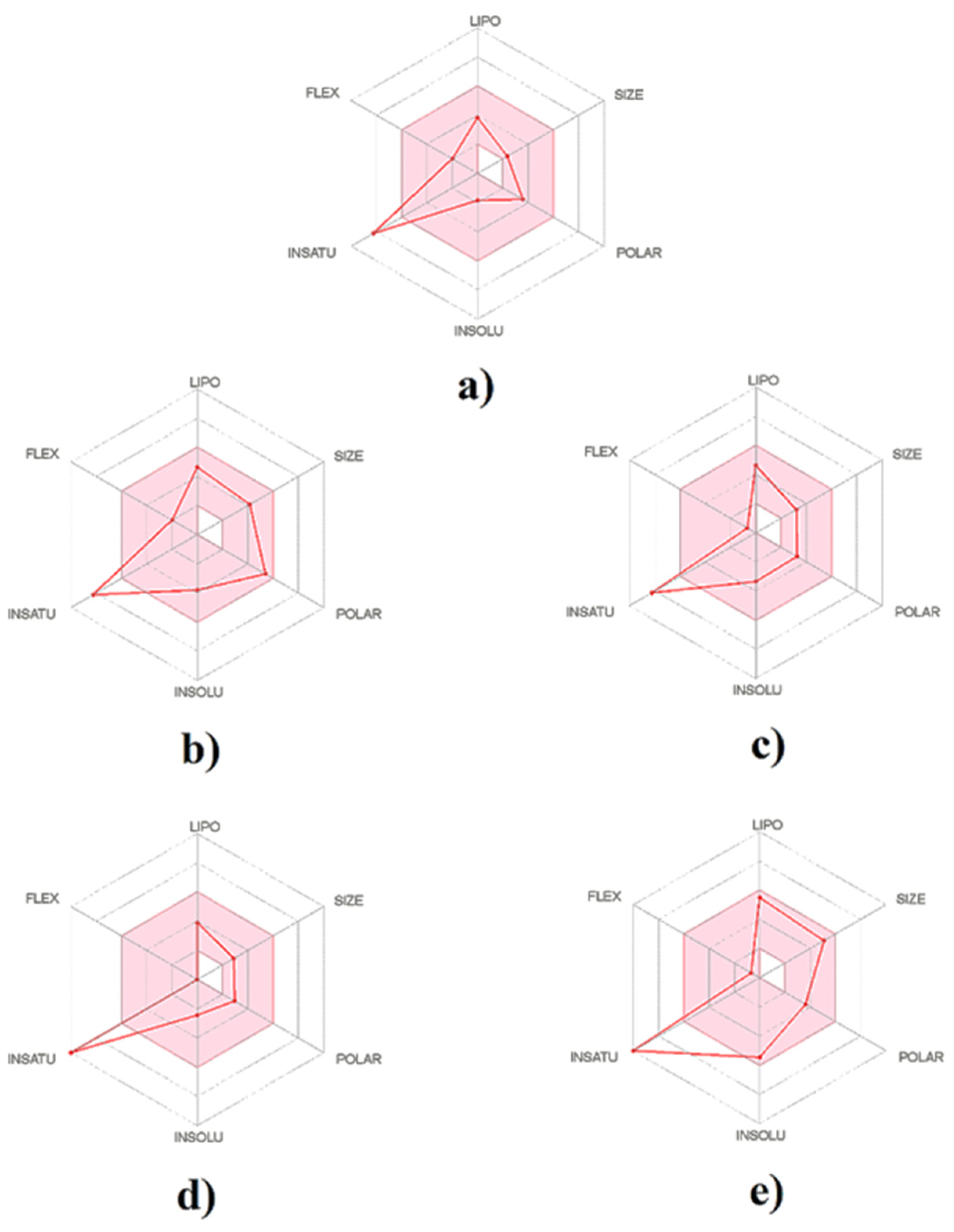
2.4. Physicochemical Properties and Drug-Likeness
2.5. Pharmacokinetics and Toxicological Properties
2.6. Modelling Biological Predictions to Physicochemical Properties
3. Materials and Methods
3.1. Preparation of Ligand
3.2. Preparation of Protein
3.3. Grid Generation
3.4. Molecular Docking Studies
3.5. Molecular Dynamics Simulation
3.6. Binding Free Energy Calculation
3.7. Physicochemical, Drug-Likeness, Pharmacokinetic and Toxicokinetic Properties Prediction
3.8. Distance-Based Linear Modeling of Physicochemical Properties, COX-1 and -2 Binding, Pharmacokinetic and Toxicokinetic Predictions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bittencourt, J.A.; Neto, M.F.; Lacerda, P.S.; Bittencourt, R.C.; Silva, R.C.; Lobato, C.C.; Silva, L.B.; Leite, F.H.; Zuliani, J.P.; Rosa, J. In silico evaluation of ibuprofen and two benzoylpropionic acid derivatives with potential anti-inflammatory activity. Molecules 2019, 24, 1476. [Google Scholar] [CrossRef] [Green Version]
- Cheung, R.C.F.; Ng, T.B.; Wong, J.H.; Chen, Y.; Chan, W.Y. Marine natural products with anti-inflammatory activity. Appl. Microbiol. Biotechnol. 2016, 100, 1645–1666. [Google Scholar] [CrossRef]
- Guo, H.; Callaway, J.B.; Ting, J.P. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef] [Green Version]
- Mota, F.V.B.; de Araújo Neta, M.S.; de Souza Franco, E.; Bastos, I.V.G.A.; da Araújo, L.C.C.; da Silva, S.C.; de Oliveira, T.B.; Souza, E.K.; de Almeida, V.M.; Ximenes, R.M. Evaluation of anti-inflammatory activity and molecular docking study of new aza-bicyclic isoxazoline acylhydrazone derivatives. Medchemcomm 2019, 10, 1916–1925. [Google Scholar] [CrossRef] [PubMed]
- de Morais Lima, G.R.; de Albuquerque Montenegro, C.; de Almeida, C.L.F.; de Athayde-Filho, P.F.; Barbosa-Filho, J.M.; Batista, L.M. Database survey of anti-inflammatory plants in South America: A review. Int. J. Mol. Sci. 2011, 12, 2692–2749. [Google Scholar] [CrossRef] [Green Version]
- Ferrero-Miliani, L.; Nielsen, O.; Andersen, P.; Girardin, S. Chronic inflammation: Importance of NOD2 and NALP3 in interleukin-1β generation. Clin. Exp. Immunol. 2007, 147, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [Green Version]
- McGeer, P.L.; Rogers, J.; McGeer, E.G. Inflammation, anti-inflammatory agents, and Alzheimer’s disease: The last 22 years. J. Alzheimer’s Dis. 2016, 54, 853–857. [Google Scholar] [CrossRef]
- Smolen, J.S.; Aletaha, D.; McInnes, I.B. Rheumatoid arthritis. Lancet 2016, 388, 2023–2038. [Google Scholar] [CrossRef]
- Feng, D.; Mukhopadhyay, P.; Qiu, J.; Wang, H. Inflammation in Liver Diseases. Mediat. Inflamm. 2018, 2018, 3927134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brusselle, G.; Bracke, K. Targeting immune pathways for therapy in asthma and chronic obstructive pulmonary disease. Ann. Am. Thorac. Soc. 2014, 11, S322–S328. [Google Scholar] [CrossRef]
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2018, 9, 7204–7218. [Google Scholar] [CrossRef] [Green Version]
- Miner-Williams, W.M.; Moughan, P.J. Intestinal barrier dysfunction: Implications for chronic inflammatory conditions of the bowel. Nutr. Res. Rev. 2016, 29, 40–59. [Google Scholar] [CrossRef]
- Alessandri, A.L.; Sousa, L.P.; Lucas, C.D.; Rossi, A.G.; Pinho, V.; Teixeira, M.M. Resolution of inflammation: Mechanisms and opportunity for drug development. Pharmacol. Ther. 2013, 139, 189–212. [Google Scholar] [CrossRef] [Green Version]
- De cassia da Silveira e Sa, R.; Andrade, L.N.; de Sousa, D.P. A review on anti-inflammatory activity of monoterpenes. Molecules 2013, 18, 1227–1254. [Google Scholar] [CrossRef]
- Jarapula, R.; Gangarapu, K.; Manda, S.; Rekulapally, S. Synthesis, in vivo anti-inflammatory activity, and molecular docking studies of new isatin derivatives. Int. J. Med. Chem. 2016, 2016, 2181027. [Google Scholar] [PubMed] [Green Version]
- Vane, J. Formation and actions of prostaglandins and inhibition of their synthesis. In Therapeutic Roles of SELECTIVE Cox-2 Inhibitors; Vane, J.R., Botting, R.M., Eds.; William Harvey Press: London, UK, 2001; pp. 1–47. [Google Scholar]
- Mikra, C.; Rossos, G.; Hadjikakou, S.; Kourkoumelis, N. Molecular docking and structure activity relationship studies of nsaids. what do they reveal about IC50? Lett. Drug Des. Discov. 2017, 14, 949–958. [Google Scholar] [CrossRef]
- Simmons, D.; Botting, R.; Hla, T. Cyclooxygenase isozymes: The biology of prostaglandin synthesis and inhibition. Pharmacol. Rev. 2004, 56, 387–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oniga, S.; Pacureanu, L.; Stoica, C.; Palage, M.; Crăciun, A.; Rusu, L.; Crisan, E.-L.; Araniciu, C. COX inhibition profile and molecular docking studies of some 2-(trimethoxyphenyl)-thiazoles. Molecules 2017, 22, 1507. [Google Scholar] [CrossRef] [Green Version]
- Al-Saeed, A. Gastrointestinal and cardiovascular risk of nonsteroidal anti-inflammatory drugs. Oman Med. J. 2011, 26, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.-J.; Mbonye, U.R.; DeLong, C.J.; Wada, M.; Smith, W.L. Regulation of intracellular cyclooxygenase levels by gene transcription and protein degradation. Prog. Lipid Res. 2007, 46, 108–125. [Google Scholar] [CrossRef] [Green Version]
- Knights, K.M.; Mangoni, A.A.; Miners, J.O. Defining the COX inhibitor selectivity of NSAIDs: Implications for understanding toxicity. Expert Rev. Clin. Pharmacol. 2010, 3, 769–776. [Google Scholar] [CrossRef]
- Hoxha, M. A systematic review on the role of eicosanoid pathways in rheumatoid arthritis. Adv. Med. Sci. 2018, 63, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Crofford, L.J.; Lipsky, P.E.; Brooks, P.; Abramson, S.B.; Simon, L.S.; Van De Putte, L. Basic biology and clinical application of specific cyclooxygenase-2 inhibitors. Arthritis Rheum. 2000, 43, 4–13. [Google Scholar] [CrossRef] [Green Version]
- Perrone, M.; Scilimati, A.; Simone, L.; Vitale, P. Selective COX-1 inhibition: A therapeutic target to be reconsidered. Curr. Med. Chem. 2010, 17, 3769–3805. [Google Scholar] [CrossRef] [PubMed]
- Sever, B.; Altıntop, M.D.; Kus, G.; Ozkurt, M.; Özdemir, A.; Kaplancıklı, Z.A. Indomethacin based new triazolothiadiazine derivatives: Synthesis, evaluation of their anticancer effects on T98 human glioma cell line related to COX-2 inhibition and docking studies. Eur. J. Med. Chem. 2016, 113, 179–186. [Google Scholar] [CrossRef]
- Shaikh, M.M.; Patel, A.P.; Patel, S.P.; Chikhalia, K.H. Synthesis, in vitro COX-1/COX-2 inhibition testing and molecular docking study of novel 1, 4-benzoxazine derivatives. N. J. Chem. 2019, 43, 10305–10317. [Google Scholar] [CrossRef]
- Coy-Barrera, E. Discrimination of naturally-occurring 2-arylbenzofurans as cyclooxygenase-2 inhibitors: Insights into the binding mode and enzymatic inhibitory activity. Biomolecules 2020, 10, 176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vitale, P.; Panella, A.; Scilimati, A.; Perrone, M.G. COX-1 inhibitors: Beyond structure toward therapy. Med. Res. Rev. 2016, 36, 641–671. [Google Scholar] [CrossRef]
- Ahmad, B.T.; Liu, L.; Kotiw, M.; Benkendorff, K. Review of anti-inflammatory, immune-modulatory and wound healing properties of molluscs. J. Ethnopharmacol. 2018, 210, 156–178. [Google Scholar] [CrossRef]
- Joy, M.; Chakraborty, K. Specialised oxygenated heterocyclics from Villorita cyprinoides with cyclooxygenase-2 and 5-lipoxygenase inhibitory properties. Food Res. Int. 2018, 106, 164–172. [Google Scholar] [CrossRef]
- Benkendorff, K. Natural product research in the Australian marine invertebrate Dicathais orbita. Mar. Drugs 2013, 11, 1370–1398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, B.T.; Rudd, D.; Smith, J.; Kotiw, M.; Mouatt, P.; Seymour, L.M.; Liu, L.; Benkendorff, K. Anti-inflammatory activity and structure-activity relationships of brominated indoles from a marine mollusc. Mar. Drugs 2017, 15, 133. [Google Scholar] [CrossRef]
- Ahmad, T.; Rudd, D.; Benkendorff, K.; Mahdi, L.K.; Pratt, K.-A.; Dooley, L.; Wei, C.; Kotiw, M. Brominated indoles from a marine mollusc inhibit inflammation in a murine model of acute lung injury. PLoS ONE 2017, 12, e0186904. [Google Scholar] [CrossRef] [Green Version]
- Meijer, L.; Skaltsounis, A.-L.; Magiatis, P.; Polychronopoulos, P.; Knockaert, M.; Leost, M.; Ryan, X.P.; Vonica, C.A.; Brivanlou, A.; Dajani, R. GSK-3-selective inhibitors derived from Tyrian purple indirubins. Chem. Biol. 2003, 10, 1255–1266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benkendorff, K.; Rudd, D.; Nongmaithem, B.D.; Liu, L.; Young, F.; Edwards, V.; Avila, C.; Abbott, C.A. Are the traditional medical uses of Muricidae molluscs substantiated by their pharmacological properties and bioactive compounds? Mar. Drugs 2015, 13, 5237–5275. [Google Scholar] [CrossRef] [Green Version]
- Nordin, N.A.; Lawai, V.; Ngaini, Z.; Abd Halim, A.N.; Hwang, S.S.; Linton, R.E.; Lee, B.K.; Neilsen, P.M. In vitro cytotoxicity evaluation of thiourea derivatives bearing Salix sp. constituent against HK-1 cell lines. Nat. Prod. Res. 2020, 34, 1505–1514. [Google Scholar] [CrossRef]
- Uzzaman, M.; Mahmud, T. Structural modification of aspirin to design a new potential cyclooxygenase (COX-2) inhibitors. Silico Pharmacol. 2020, 8, 1. [Google Scholar] [CrossRef]
- Ibrahim, I.M.; Abdelmalek, D.H.; Elshahat, M.E.; Elfiky, A.A. COVID-19 Spike-host cell receptor GRP78 binding site prediction. J. Infect. 2020, 80, 554–562. [Google Scholar] [CrossRef]
- Yang, J.; Shen, C.; Huang, N. Predicting or pretending: Artificial intelligence for protein-ligand interactions lack of sufficiently large and unbiased datasets. Front. Pharmacol. 2020, 11, 69. [Google Scholar] [CrossRef] [PubMed]
- Junaid, M.; Islam, N.; Hossain, M.K.; Ullah, M.O.; Halim, M.A. Metal based donepezil analogues designed to inhibit human acetylcholinesterase for Alzheimer’s disease. PLoS ONE 2019, 14, e0211935. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Bhatia, P.; Alam, O.; Naim, M.J.; Nawaz, F.; Sheikh, A.A.; Jha, M. Recent advancement in the discovery and development of COX-2 inhibitors: Insight into biological activities and SAR studies (2008–2019). Bioorganic Chem. 2019, 89, 103007. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Hermanson, D.J.; Banerjee, S.; Ghebreselasie, K.; Clayton, G.M.; Garavito, R.M.; Marnett, L.J. Oxicams bind in a novel mode to the cyclooxygenase active site via a two-water-mediated H-bonding network. J. Biol. Chem. 2014, 289, 6799–6808. [Google Scholar] [CrossRef] [Green Version]
- Kurumbail, R.G.; Stevens, A.M.; Gierse, J.K.; McDonald, J.J.; Stegeman, R.A.; Pak, J.Y.; Gildehaus, D.; Penning, T.D.; Seibert, K.; Isakson, P.C. Structural basis for selective inhibition of cyclooxygenase-2 by anti-inflammatory agents. Nature 1996, 384, 644–648. [Google Scholar] [CrossRef]
- Blobaum, A.L.; Marnett, L.J. Structural and functional basis of cyclooxygenase inhibition. J. Med. Chem. 2007, 50, 1425–1441. [Google Scholar] [CrossRef] [Green Version]
- Picot, D.; Loll, P.J.; Garavito, R.M. The X-ray crystal structure of the membrane protein prostaglandin H2 synthase-1. Nature 1994, 367, 243–249. [Google Scholar] [CrossRef]
- Goodsell, D.S. The molecular perspective: Cyclooxygenase-2. Oncologist 2000, 5, 169–171. [Google Scholar] [CrossRef] [PubMed]
- Polski, A.; Kaczor, A.A.; Sobotka-Polska, K.; Pitucha, M. From synthesis and spectral analysis to molecular modelling–multidimensional teaching of medicinal chemistry: Aspirin as an example. Indian J. Pharm. Educ. Res. 2015, 49, 82–87. [Google Scholar] [CrossRef] [Green Version]
- Lecomte, M.; Laneuville, O.; Ji, C.; DeWitt, D.L.; Smith, W.L. Acetylation of human prostaglandin endoperoxide synthase-2 (cyclooxygenase-2) by aspirin. J. Biol. Chem. 1994, 269, 13207–13215. [Google Scholar] [CrossRef]
- Shimokawa, T.; Smith, W. Prostaglandin endoperoxide synthase. The aspirin acetylation region. J. Biol. Chem. 1992, 267, 12387–12392. [Google Scholar] [CrossRef]
- Rowlinson, S.W.; Crews, B.C.; Goodwin, D.C.; Schneider, C.; Gierse, J.K.; Marnett, L.J. Spatial requirements for 15-(r)-hydroxy-5z, 8z, 11z, 13e-eicosatetraenoic acid synthesis within the cyclooxygenase active site of murine COX-2 why acetylated COX-1 does not synthesize 15-(r)-hete. J. Biol. Chem. 2000, 275, 6586–6591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prusakiewicz, J.J.; Felts, A.S.; Mackenzie, B.S.; Marnett, L.J. Molecular basis of the time-dependent inhibition of cyclooxygenases by indomethacin. Biochemistry 2004, 43, 15439–15445. [Google Scholar] [CrossRef]
- Walters, M.J.; Blobaum, A.L.; Kingsley, P.J.; Felts, A.S.; Sulikowski, G.A.; Marnett, L.J. The influence of double bond geometry in the inhibition of cyclooxygenases by sulindac derivatives. Bioorganic Med. Chem. Lett. 2009, 19, 3271–3274. [Google Scholar] [CrossRef] [Green Version]
- Schneider, C.; Boeglin, W.E.; Prusakiewicz, J.J.; Rowlinson, S.W.; Marnett, L.J.; Samel, N.; Brash, A.R. Control of prostaglandin stereochemistry at the 15-carbon by cyclooxygenases-1 and-2 a critical role for serine 530 and valine 349. J. Biol. Chem. 2002, 277, 478–485. [Google Scholar] [CrossRef] [Green Version]
- Thuresson, E.D.; Lakkides, K.M.; Rieke, C.J.; Sun, Y.; Wingerd, B.A.; Micielli, R.; Mulichak, A.M.; Malkowski, M.G.; Garavito, R.M.; Smith, W.L. Prostaglandin Endoperoxide H Synthase-1 the functions of cyclooxygenase active site residues in the binding, positioning, and oxygenation of arachidonic acid. J. Biol. Chem. 2001, 276, 10347–10357. [Google Scholar] [CrossRef] [Green Version]
- Thuresson, E.D.; Lakkides, K.M.; Smith, W.L. Different catalytically competent arrangements of arachidonic acid within the cyclooxygenase active site of prostaglandin endoperoxide H synthase-1 lead to the formation of different oxygenated products. J. Biol. Chem. 2000, 275, 8501–8507. [Google Scholar] [CrossRef] [Green Version]
- Schneider, C.; Boeglin, W.E.; Brash, A.R. Identification of two cyclooxygenase active site residues, Leucine 384 and Glycine 526, that control carbon ring cyclisation in prostaglandin biosynthesis. J. Biol. Chem. 2004, 279, 4404–4414. [Google Scholar] [CrossRef] [Green Version]
- Limongelli, V.; Bonomi, M.; Marinelli, L.; Gervasio, F.L.; Cavalli, A.; Novellino, E.; Parrinello, M. Molecular basis of cyclooxygenase enzymes (COXs) selective inhibition. Proc. Natl. Acad. Sci. USA 2010, 107, 5411–5416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhat, M.A.; Al-Omar, M.A.; Raish, M.; Ansari, M.A.; Abuelizz, H.A.; Bakheit, A.H.; Naglah, A.M. Indole derivatives as cyclooxygenase inhibitors: Synthesis, biological evaluation and docking studies. Molecules 2018, 23, 1250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irfan, M. Selective cyclooxygenase-2 inhibitors: A review of recent chemical scaffolds with promising anti-inflammatory and COX-2 inhibitory activities. Med. Chem. Res. 2020, 29, 809–830. [Google Scholar]
- Malkowski, M.; Ginell, S.; Smith, W.; Garavito, R. The productive conformation of arachidonic acid bound to prostaglandin synthase. Science 2000, 289, 1933–1937. [Google Scholar] [CrossRef] [PubMed]
- Rowlinson, S.W.; Crews, B.C.; Lanzo, C.A.; Marnett, L.J. The binding of arachidonic acid in the cyclooxygenase active site of mouse prostaglandin endoperoxide synthase-2 (COX-2) a putative L-shaped binding conformation utilizing the top channel region. J. Biol. Chem. 1999, 274, 23305–23310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vecchio, A.J.; Simmons, D.M.; Malkowski, M.G. Structural basis of fatty acid substrate binding to cyclooxygenase-2. J. Biol. Chem. 2010, 285, 22152–22163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garavito, R.M.; DeWitt, D.L. The cyclooxygenase isoforms: Structural insights into the conversion of arachidonic acid to prostaglandins. Biochim. Biophys. Acta (Bba)-Mol. Cell Biol. Lipids 1999, 1441, 278–287. [Google Scholar] [CrossRef]
- Schreiber, J.; Eling, T.E.; Mason, R.P. The oxidation of arachidonic acid by the cyclooxygenase activity of purified prostaglandin H synthase: Spin trapping of a carbon-centered free radical intermediate. Arch. Biochem. Biophys. 1986, 249, 126–136. [Google Scholar] [CrossRef]
- Tsai, A.-L.; Kulmacz, R.J.; Palmer, G. Spectroscopic evidence for reaction of prostaglandin H synthase-1 tyrosyl radical with arachidonic acid. J. Biol. Chem. 1995, 270, 10503–10508. [Google Scholar] [CrossRef] [Green Version]
- Gierse, J.K.; McDonald, J.J.; Hauser, S.D.; Rangwala, S.H.; Koboldt, C.M.; Seibert, K. A single amino acid difference between cyclooxygenase-1 (COX-1) and −2 (COX-2) reverses the selectivity of COX-2 specific inhibitors. J. Biol. Chem. 1996, 271, 15810–15814. [Google Scholar] [CrossRef] [Green Version]
- Wong, E.; Bayly, C.; Waterman, H.L.; Riendeau, D.; Mancini, J.A. Conversion of prostaglandin G/H synthase-1 into an enzyme sensitive to PGHS-2-selective inhibitors by a double His513→ Arg and Ile523→ Val mutation. J. Biol. Chem. 1997, 272, 9280–9286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, L.W.; DeWitt, D.L.; Garavito, R.M. Cyclooxygenases: Structural, cellular, and molecular biology. Annu. Rev. Biochem. 2000, 69, 145–182. [Google Scholar] [CrossRef] [Green Version]
- Kamaraj, B.; Rajendran, V.; Sethumadhavan, R.; Kumar, C.V.; Purohit, R. Mutational analysis of FUS gene and its structural and functional role in amyotrophic lateral sclerosis 6. J. Biomol. Struct. Dyn. 2015, 33, 834–844. [Google Scholar] [CrossRef]
- Junaid, M.; Alam, M.J.; Hossain, M.K.; Halim, M.A.; Ullah, M.O. Molecular docking and dynamics of Nickel-Schiff base complexes for inhibiting β-lactamase of Mycobacterium tuberculosis. Silico Pharmacol. 2018, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Ausaf Ali, S.; Hassan, I.; Islam, A.; Ahmad, F. A review of methods available to estimate solvent-accessible surface areas of soluble proteins in the folded and unfolded states. Curr. Protein Pept. Sci. 2014, 15, 456–476. [Google Scholar]
- Fogolari, F.; Brigo, A.; Molinari, H. Protocol for MM/PBSA molecular dynamics simulations of proteins. Biophys. J. 2003, 85, 159–166. [Google Scholar] [CrossRef] [Green Version]
- Miller, B.R., III; McGee, T.D., Jr.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA. py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Wang, C.; Greene, D.A.; Xiao, L.; Qi, R.; Luo, R. Recent developments and applications of the MMPBSA method. Front. Mol. Biosci. 2018, 4, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W. Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef]
- Yang, T.; Wu, J.C.; Yan, C.; Wang, Y.; Luo, R.; Gonzales, M.B.; Dalby, K.N.; Ren, P. Virtual screening using molecular simulations. Proteins: Struct. Funct. Bioinform. 2011, 79, 1940–1951. [Google Scholar]
- Srinivasan, E.; Rajasekaran, R. Computational investigation of curcumin, a natural polyphenol that inhibits the destabilisation and the aggregation of human SOD1 mutant (Ala4Val). Rsc. Adv. 2016, 6, 102744–102753. [Google Scholar] [CrossRef]
- Swanson, J.M.; Henchman, R.H.; McCammon, J.A. Revisiting free energy calculations: A theoretical connection to MM/PBSA and direct calculation of the association free energy. Biophys. J. 2004, 86, 67–74. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Ndombera, F.T.; Maiyoh, G.K.; Tuei, V.C. Pharmacokinetic, physicochemical and medicinal properties of n-glycoside anti-cancer agent more potent than 2-deoxy-d-glucose in lung cancer cells. J. Pharm. Pharmacol. 2019, 7, 165–176. [Google Scholar]
- Daina, A.; Blatter, M.-C.; Baillie Gerritsen, V.; Palagi, P.M.; Marek, D.; Xenarios, I.; Schwede, T.; Michielin, O.; Zoete, V. Drug design workshop: A web-based educational tool to introduce computer-aided drug design to the general public. J. Chem. Educ. 2017, 94, 335–344. [Google Scholar] [CrossRef]
- Leo, A.; Hansch, C.; Elkins, D. Partition coefficients and their uses. Chem. Rev. 1971, 71, 525–616. [Google Scholar] [CrossRef]
- Mignani, R.; Petrucci, A.; Cardone, F. Geometrical lorentz violation and quantum mechanical physics. Quantum Matter 2014, 3, 219–225. [Google Scholar] [CrossRef] [Green Version]
- Hansch, C.; Leo, A.; Unger, S.H.; Kim, K.H.; Nikaitani, D.; Lien, E. Aromatic substituent constants for structure-activity correlations. J. Med. Chem. 1973, 16, 1207–1216. [Google Scholar] [CrossRef]
- Ertl, P.; Rohde, B.; Selzer, P. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J. Med. Chem. 2000, 43, 3714–3717. [Google Scholar] [CrossRef]
- Fjodorova, N.; Novic, M.; Venko, K.; Rasulev, B. A Comprehensive Cheminformatics Analysis of Structural Features Affecting the Binding Activity of Fullerene Derivatives. Nanomaterials 2020, 10, 90. [Google Scholar] [CrossRef] [Green Version]
- Pliska, V.; Testa, B.; Van de Waterbeemd, H.; Mannhold, R.; Kubinyi, H.; Timmerman, H. Lipophilicity in drug action and toxicology. J. Med. Chem. 1996, 39, 5287–5288. [Google Scholar]
- Arnott, J.A.; Planey, S.L. The influence of lipophilicity in drug discovery and design. Expert Opin. Drug Discov. 2012, 7, 863–875. [Google Scholar] [CrossRef]
- Delaney, J.S. ESOL: Estimating aqueous solubility directly from molecular structure. J. Chem. Inf. Comput. Sci. 2004, 44, 1000–1005. [Google Scholar] [CrossRef]
- Testa, B.; Carrupt, P.-A.; Gaillard, P.; Billois, F.; Weber, P. Lipophilicity in molecular modelling. Pharm. Res. 1996, 13, 335–343. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. iLOGP: A simple, robust, and efficient description of n-octanol/water partition coefficient for drug design using the GB/SA approach. J. Chem. Inf. Modeling 2014, 54, 3284–3301. [Google Scholar] [CrossRef]
- Cheng, T.; Zhao, Y.; Li, X.; Lin, F.; Xu, Y.; Zhang, X.; Li, Y.; Wang, R.; Lai, L. Computation of octanol—Water partition coefficients by guiding an additive model with knowledge. J. Chem. Inf. Modeling 2007, 47, 2140–2148. [Google Scholar] [CrossRef]
- Moriguchi, I.; Hirono, S.; Liu, Q.; Nakagome, I.; Matsushita, Y. Simple method of calculating octanol/water partition coefficient. Chem. Pharm. Bull. 1992, 40, 127–130. [Google Scholar] [CrossRef] [Green Version]
- Moriguchi, I.; Hirono, S.; Nakagome, I.; Hirano, H. Comparison of reliability of log P values for drugs calculated by several methods. Chem. Pharm. Bull. 1994, 42, 976–978. [Google Scholar] [CrossRef] [Green Version]
- Sugiyama, Y. Druggability: Selecting optimised drug candidates. Drug Discov. Today 2005, 23, 1577–1579. [Google Scholar] [CrossRef]
- Yamashita, F.; Hashida, M. In silico approaches for predicting ADME properties of drugs. Drug Metab. Pharmacokinet. 2004, 19, 327–338. [Google Scholar] [CrossRef] [Green Version]
- Dimasi, J.A.; Grabowski, H.G.; Hansen, R.W. Innovation in the pharmaceutical industry: New estimates of R&D costs. J. Health Econ. 2016, 47, 20–33. [Google Scholar]
- Cummings, J.; Reiber, C.; Kumar, P. The price of progress: Funding and financing Alzheimer’s disease drug development. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2018, 4, 330–343. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.W.; Urade, Y.; Jakobsson, P.-J. Enzymes of the cyclooxygenase pathways of prostanoid biosynthesis. Chem. Rev. 2011, 111, 5821–5865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kar, P.; Lipowsky, R.; Knecht, V. Importance of polar solvation and configurational entropy for design of antiretroviral drugs targeting HIV-1 protease. J. Phys. Chem. B 2013, 117, 5793–5805. [Google Scholar] [CrossRef] [PubMed]
- Cronin, M.T. Prediction of drug toxicity. Il Farm. 2001, 56, 149–151. [Google Scholar] [CrossRef]
- Van de Waterbeemd, H. Improving compound quality through in vitro and in silico physicochemical profiling. Chem. Biodivers. 2009, 6, 1760–1766. [Google Scholar] [CrossRef] [PubMed]
- Yadav, T.C.; Kumar, N.; Raj, U.; Goel, N.; Vardawaj, P.K.; Prasad, R.; Pruthi, V. Exploration of interaction mechanism of tyrosol as a potent anti-inflammatory agent. J. Biomol. Struct. Dyn. 2020, 38, 382–397. [Google Scholar] [CrossRef] [PubMed]
- Chadha, N.; Singh, D.; Milton, M.D.; Mishra, G.; Daniel, J.; Mishra, A.K.; Tiwari, A.K. Computational prediction of interaction and pharmacokinetics profile study for polyamino-polycarboxylic ligands on binding with human serum albumin. N. J. Chem. 2020, 44, 2907–2918. [Google Scholar] [CrossRef]
- Rudd, D.A.; Benkendorff, K.; Chahal, C.; Guinan, T.; Gustafsson, O.J.R.; Esmaeelian, B.; Krysinska, H.; Pogson, L.; Voelcker, N.H.; Abbott, C.A. Mapping insoluble indole metabolites in the gastrointestinal environment of a murine colorectal cancer model using desorption/ionisation on porous silicon imaging. Sci. Rep. 2019, 9, 12342. [Google Scholar] [CrossRef] [PubMed]
- Press, B.; Di Grandi, D. Permeability for intestinal absorption: Caco-2 assay and related issues. Curr. Drug Metab. 2008, 9, 893–900. [Google Scholar] [CrossRef]
- Pires, D.E.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Esmaeelian, B.; Benkendorff, K.; Johnston, M.R.; Abbott, C.A. Purified brominated indole derivatives from Dicathais orbita induce apoptosis and cell cycle arrest in colorectal cancer cell lines. Mar. Drugs 2013, 11, 3802–3822. [Google Scholar] [CrossRef]
- Westley, C.B.; McIver, M.C.; Abbott, A.C.; Le Leu, R.K.; Benkendorff, K. Enhanced acute apoptotic response to azoxymethane-induced DNA damage in the rodent colonic epithelium by Tyrian purple precursors: A potential colorectal cancer chemopreventative. Cancer Biol. Ther. 2010, 9, 371–379. [Google Scholar] [CrossRef] [Green Version]
- Osakwe, O. Preclinical in vitro studies: Development and applicability. Chapter 6. In Social Aspects of Drug Discovery, Development and Commercialisation; Osakwe, O., Rizvi, S., Eds.; Elsevier: London, UK, 2016; pp. 129–148. [Google Scholar]
- Dannenberg, A.J.; Altorki, N.K.; Boyle, J.O.; Dang, C.; Howe, L.R.; Weksler, B.B.; Subbaramaiah, K. Cyclooxygenase 2: A pharmacological target for the prevention of cancer. Lancet Oncol. 2001, 2, 544–551. [Google Scholar] [CrossRef]
- Arber, N.; DuBois, R.N. Nonsteroidal anti-inflammatory drugs and prevention of colorectal cancer. Curr. Gastroenterol. Rep. 1999, 1, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.A.; DuBois, R.N. Colorectal cancer prevention and treatment by inhibition of cyclooxygenase-2. Nat. Rev. Cancer 2001, 1, 11–21. [Google Scholar] [CrossRef]
- Abdallah, H.M.; Al-Abd, A.M.; El-Dine, R.S.; El-Halawany, A.M. P-glycoprotein inhibitors of natural origin as potential tumor chemo-sensitizers: A review. J. Adv. Res. 2015, 6, 45–62. [Google Scholar] [CrossRef]
- Lin, J.H. Yamazaki, Role of P-glycoprotein in pharmacokinetics. Clin. Pharmacokinet. 2003, 42, 59–98. [Google Scholar] [CrossRef] [PubMed]
- Thiebaut, F.; Tsuruo, T.; Hamada, H.; Gottesman, M.M.; Pastan, I.; Willingham, M.C. Cellular localisation of the multidrug-resistance gene product P-glycoprotein in normal human tissues. Proc. Natl. Acad. Sci. USA 1987, 84, 7735–7738. [Google Scholar] [CrossRef] [Green Version]
- Cordon-Cardo, C.; O’brien, J.; Boccia, J.; Casals, D.; Bertino, J.; Melamed, M. Expression of the multidrug resistance gene product (P-glycoprotein) in human normal and tumor tissues. J. Histochem. Cytochem. 1990, 38, 1277–1287. [Google Scholar] [CrossRef] [PubMed]
- Kruger, A.; Maltarollo, V.G.; Wrenger, C.; Kronenberger, T. ADME profiling in drug discovery and a new path paved on silica. In Drug Discovery and Development-New Advances; Gaitonde, V., Karmakar, P., Trivedi, A., Eds.; IntechOpen: London, UK, 2019; pp. 1–31. [Google Scholar]
- Preedy, V.R.; Watson, R.R. Nuts and Seeds in Health and Disease Prevention; Elsevier Science Publishing Co, Inc.: Cambridge, MA, USA, 2020. [Google Scholar]
- Kratz, F.; Elsadek, B. Clinical impact of serum proteins on drug delivery. J. Control. Release 2012, 161, 429–445. [Google Scholar] [CrossRef]
- Buxton, I.L.; Benet, L.Z. Pharmacokinetics: The Dynamics of Drug Absorption, Distribution, Metabolism, and Elimination. Goodman & Gilman’s the Pharmacological Basis of Therapeutics, 12th ed.; Mcgraw-Hill: New York, NY, USA, 2011; pp. 17–39. [Google Scholar]
- Nisha, C.M.; Kumar, A.; Vimal, A.; Bai, B.M.; Pal, D.; Kumar, A. Docking and ADMET prediction of few GSK-3 inhibitors divulges 6-bromoindirubin-3-oxime as a potential inhibitor. J. Mol. Graph. Model. 2016, 65, 100–107. [Google Scholar] [CrossRef]
- Glover, V.; Bhattacharya, S.; Chakrabarti, A.; Sandler, M. The psychopharmacology of isatin: A brief review. Stress Med. 1998, 14, 225–229. [Google Scholar] [CrossRef]
- Hou, L.; Ju, C.; Zhang, J.; Song, J.; Ge, Y.; Yue, W. Antitumor effects of isatin on human neuroblastoma cell line (SH-SY5Y) and the related mechanism. Eur. J. Pharmacol. 2008, 589, 27–31. [Google Scholar] [CrossRef]
- Minghetti, L.; Levi, G. Microglia as effector cells in brain damage and repair: Focus on prostanoids and nitric oxide. Prog. Neurobiol. 1998, 54, 99–125. [Google Scholar] [CrossRef]
- Ajmone-Cat, M.A.; Bernardo, A.; Greco, A.; Minghetti, L. Non-steroidal anti-inflammatory drugs and brain inflammation: Effects on microglial functions. Pharmaceuticals 2010, 3, 1949–1965. [Google Scholar] [CrossRef]
- Bell, R.D.; Zlokovic, B.V. Neurovascular mechanisms and blood–brain barrier disorder in Alzheimer’s disease. Acta Neuropathol. 2009, 118, 103–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thal, S.C.; Schaible, E.-V.; Neuhaus, W.; Scheffer, D.; Brandstetter, M.; Engelhard, K.; Wunder, C.; Förster, C.Y. Inhibition of proteasomal glucocorticoid receptor degradation restores dexamethasone-mediated stabilisation of the blood–brain barrier after traumatic brain injury. Crit. Care Med. 2013, 41, 1305–1315. [Google Scholar] [CrossRef]
- Dokmeci, D. Ibuprofen and Alzheimer’s disease. Folia Med. 2004, 46, 5–10. [Google Scholar]
- Hakan, T.; Toklu, H.Z.; Biber, N.; Ozevren, H.; Solakoglu, S.; Demirturk, P.; Aker, F.V. Effect of COX-2 inhibitor meloxicam against traumatic brain injury-induced biochemical, histopathological changes and blood–brain barrier permeability. Neurol. Res. 2010, 32, 629–635. [Google Scholar] [CrossRef]
- Schlichtiger, J.; Pekcec, A.; Bartmann, H.; Winter, P.; Fuest, C.; Soerensen, J.; Potschka, H. Celecoxib treatment restores pharmacosensitivity in a rat model of pharmacoresistant epilepsy. Br. J. Pharmacol. 2010, 160, 1062–1071. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.-Y.; Duan, Z.J.; Liu, Z.; Tang, S.X.; Li, Y.; He, S.C.; Wang, Q.M.; Chang, Q.Y. Inhibition of P-glycoprotein, multidrug resistance-associated protein 2 and cytochrome P450 3A4 improves the oral absorption of octreotide in rats with portal hypertension. Exp. Ther. Med. 2016, 12, 3716–3722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Y.; Zhang, J.; Hu, C.Q.; Zhang, X.; Ma, B.; Zhang, P. In silico ADME and toxicity prediction of ceftazidime and its impurities. Front. Pharmacol. 2019, 10, 434. [Google Scholar] [CrossRef]
- Lagorce, D.; Douguet, D.; Miteva, M.A.; Villoutreix, B.O. Computational analysis of calculated physicochemical and ADMET properties of protein-protein interaction inhibitors. Sci. Rep. 2017, 7, 46277. [Google Scholar] [CrossRef] [PubMed]
- Koepsell, H. Polyspecific organic cation transporters: Their functions and interactions with drugs. Trends Pharmacol. Sci. 2004, 25, 375–381. [Google Scholar] [CrossRef]
- Ivanyuk, A.; Livio, F.; Biollaz, J.; Buclin, T. Renal drug transporters and drug interactions. Clin. Pharmacokinet. 2017, 56, 825–892. [Google Scholar] [CrossRef] [PubMed]
- Esmaeelian, B. Preclinical In Vitro and In Vivo Effects of Purified and Synthetic Bioactive Compounds from Marine Mollusc Dicathais Orbita on Colorectal Cancer: Cancer Prevention and Toxicity Study; Flinders University: Adelaide, Australia, 2013. [Google Scholar]
- Blomme, E.A.; Will, Y. Toxicology strategies for drug discovery: Present and future. Chem. Res. Toxicol. 2016, 29, 473–504. [Google Scholar] [CrossRef]
- Abraham, D.J.; Rotella, D.P. Burger’s Medicinal Chemistry, Drug Discovery and Development; Wiley: Hoboken, NJ, USA, 2010. [Google Scholar]
- Westley, C.B.; Benkendorff, K.; McIver, C.M.; Le Leu, R.K.; Abbott, C.A. Gastrointestinal and hepatotoxicity assessment of an anticancer extract from muricid molluscs. Evid.-Based Complementary Altern. Med. 2013, 2013, 837370. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Xing, J.; Xu, Y.; Zhou, N.; Peng, J.; Xiong, Z.; Liu, X.; Luo, X.; Luo, C.; Chen, K. In silico ADME/T modelling for rational drug design. Q. Rev. Biophys. 2015, 48, 488–515. [Google Scholar] [CrossRef] [Green Version]
- Asirvatham, S.; Dhokchawle, B.V.; Tauro, S.J. Quantitative structure activity relationships studies of non-steroidal anti-inflammatory drugs: A review. Arab. J. Chem. 2019, 12, 3948–3962. [Google Scholar] [CrossRef]
- Ballard, P.; Brassil, P.; Bui, K.; Dolgos, H.; Petersson, C.; Tunek, A.; Webborn, P. Metabolism and pharmacokinetic optimization strategies in drug discovery. In Drug Discovery and Development-E-Book: Technology in Transition, 2nd ed.; Hill, R.G., Richards, D.B., Eds.; Elsevier: Online, 2012; pp. 135–155. [Google Scholar]
- de Oliveira Moraes, A.D.T.; de Miranda, M.D.S.; Jacob, Í.T.T.; da Cruz Amorim, C.A.; de Moura, R.O.; da Silva, S.Â.S.; Soares, M.B.P.; de Almeida, S.M.V.; de Lima Souza, T.R.C.; de Oliveira, J.F. Synthesis, in vitro and in vivo biological evaluation, COX-1/2 inhibition and molecular docking study of indole-N-acylhydrazone derivatives. Bioorganic Med. Chem. 2018, 26, 5388–5396. [Google Scholar] [CrossRef] [PubMed]
- Schrodinger, L. Schrodinger Release 2018-1: Maestro; Schrodinger LLC: New York, NY, USA, 2018. [Google Scholar]
- Sidhu, R.S.; Lee, J.Y.; Yuan, C.; Smith, W.L. Comparison of cyclooxygenase-1 crystal structures: Cross-talk between monomers comprising cyclooxygenase-1 homodimers. Biochemistry 2010, 49, 7069–7079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orlando, B.J.; Malkowski, M.G. Substrate-selective inhibition of cyclooxygeanse-2 by fenamic acid derivatives is dependent on peroxide tone. J. Biol. Chem. 2016, 291, 15069–15081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Krieger, E.; Darden, T.; Nabuurs, S.B.; Finkelstein, A.; Vriend, G. Making optimal use of empirical energy functions: Force-field parameterisation in crystal space. Proteins Struct. Funct. Bioinform. 2004, 57, 678–683. [Google Scholar] [CrossRef] [PubMed]
- Uzzaman, M.; Junaid, M.; Uddin, M.N. Evaluation of anti-tuberculosis activity of some oxotitanium (IV) Schiff base complexes; molecular docking, dynamics simulation and ADMET studies. SN Appl. Sci. 2020, 2, 880. [Google Scholar] [CrossRef] [Green Version]
- Case, D.A.; Cheatham, T.E., III; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [Green Version]
- Krieger, E.; Nielsen, J.E.; Spronk, C.A.; Vriend, G. Fast empirical pKa prediction by Ewald summation. J. Mol. Graph. Model. 2006, 25, 481–486. [Google Scholar] [CrossRef]
- Krieger, E.; Vriend, G. New ways to boost molecular dynamics simulations. J. Comput. Chem. 2015, 36, 996–1007. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Koraimann, G.; Vriend, G. Increasing the precision of comparative models with YASARA NOVA—A self-parameterizing force field. Proteins 2002, 47, 393–402. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Dash, R.; Ali, M.; Dash, N.; Azad, M.; Kalam, A.; Hosen, S.; Hannan, M.; Moon, I.S. Structural and dynamic characterizations highlight the deleterious role of SULT1A1 R213H polymorphism in substrate binding. Int. J. Mol. Sci. 2019, 20, 6256. [Google Scholar] [CrossRef] [Green Version]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2012, 64, 4–17. [Google Scholar] [CrossRef]
- Joshi, T.; Sharma, P.; Joshi, T.; Chandra, S. In silico screening of anti-inflammatory compounds from Lichen by targeting cyclooxygenase-2. J. Biomol. Struct. Dyn. 2019, 38, 3544–3562. [Google Scholar] [CrossRef] [PubMed]
- Jang, C.; Yadav, D.K.; Subedi, L.; Venkatesan, R.; Venkanna, A.; Afzal, S.; Lee, E.; Yoo, J.; Ji, E.; Kim, S.Y. Identification of novel acetylcholinesterase inhibitors designed by pharmacophore-based virtual screening, molecular docking and bioassay. Sci. Rep. 2018, 8, 14921. [Google Scholar] [CrossRef] [PubMed] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand Name | XP Docking Score (kcal mol−1) | GLIDE Energy (kcal mol−1) | GLIDE Model (kcal mol−1) | GLIDE Ligand Efficiency |
|---|---|---|---|---|
| Aspirin | −2.80 | −26.25 | −33.12 | −0.21 |
| Tyrindoxyl sulfate | −6.17 | −33.26 | −37.64 | −0.36 |
| Tyrindoleninone | −6.85 | −32.49 | −37.17 | −0.52 |
| 6-Bromoisatin | −6.06 | −27.95 | −36.96 | −0.50 |
| 6,6′-Dibromoindirubin | −7.25 | −36.23 | 2.69 | −0.33 |
| Ligand Name | XP Docking Score (kcal mol−1) | GLIDE Energy (kcal mol−1) | GLIDE Model (kcal mol−1) | GLIDE Ligand Efficiency |
|---|---|---|---|---|
| Aspirin | −6.87 | −31.43 | −41.06 | −0.52 |
| Tyrindoxyl sulfate | −6.34 | −34.58 | −44.53 | −0.37 |
| Tyrindoleninone | −7.17 | −29.27 | −30.7 | −0.55 |
| 6-Bromoisatin | −6.19 | −26.1 | −32.51 | −0.51 |
| 6,6′-Dibromoindirubin | −3.14 | −15.27 | 1.96 | −0.14 |
| Parameters | Aspirin | Tyrindoxyl Sulfate | Tyrindoleninone | 6-Bromoisatin | 6,6′-Dibromoindirubin |
|---|---|---|---|---|---|
| IUPAC Name | 2-acetyloxybenzoic acid | (6-bromo-2-methylsulfanyl- 1H-indol-3-yl) hydrogen sulfate | 6-bromo-2-methylsulfanylindol-3-one | 6-bromo-1H-indole-2,3-dione | 6-bromo-2-(6-bromo-2-hydroxy-1H-indol-3-yl) indol-3-one |
| Canonical SMILES | CC(=O)OC1=CC=CC= C1C(=O)O | CSC1=C(C2=C(N1)C= C(C=C2)Br)OS (=O)(=O)O | CSC1=NC2=C(C1=O)C= CC(=C2)Br | C1=CC2=C(C=C1Br) NC(=O)C2=O | C1=CC2=C(C=C1Br)NC(= C2C3=NC4=C(C3=O)C=CC (=C4)Br)O |
| Physicochemical properties | |||||
| Molecular formula | C9H8O4 | C9H8BrNO4S2 | C9H6BrNOS | C8H4BrNO2 | C16H8Br2N2O2 |
| Molecular weight | 180.16 g/mol | 338.20 g/mol | 256.12 g/mol | 226.03 g/mol | 420.05 g/mol |
| Fraction Csp3 | 0.11 | 0.11 | 0.11 | 0.00 | 0.00 |
| Heavy atoms | 13 | 17 | 13 | 12 | 22 |
| Aromatic heavy atoms | 6 | 9 | 6 | 6 | 15 |
| Molar refractivity (MR) | 44.90 | 69.94 | 62.35 | 49.86 | 96.02 |
| Topological polar surface area (TPSA) | 63.60Ų | 113.07 Ų | 54.73 Ų | 46.17 Ų | 65.45 Ų |
| Lipinski violations | 0 | 0 | 0 | 0 | 0 |
| Lipophilicity | |||||
| iLOGP | 1.3- | 1.25 | 2.26 | 1.14 | 2.73 |
| XLOGP3 | 1.19 | 2.64 | 2.60 | 1,33 | 0.91 |
| MLOGP | 1.51 | 1.52 | 1.68 | 0.91 | 2.95 |
| SILICOS-IT | 1.10 | 1.63 | 3.69 | 2.19 | 5.42 |
| Water Solubility | |||||
| Log S (ESOL) | −1.85 | −3.79 | −3.34 | −2.45 | −5.47 |
| Qualitative solubility | Very soluble | Soluble | Soluble | Soluble | Moderately soluble |
| Parameters | Aspirin | Tyrindoxyl Sulfate | Tyrindoleninone | 6-Bromoisatin | 6,6′-Dibromoindirubin |
|---|---|---|---|---|---|
| Absorption | |||||
| Human intestinal absorption | 76.93% | 90.56% | 94.99% | 92.49% | 90.08% |
| CaCo-2 permeability | 0.09 | 0.94 | 1.29 | 1.23 | 0.54 |
| P-glycoprotein I inhibitor | No | No | No | No | No |
| P-glycoprotein II inhibitor | No | No | No | No | No |
| Distribution | |||||
| Plasma protein binding (QPlogKhsa) | −0.75 | −0.41 | −0.45 | −0.61 | 0.33 |
| VDss (human) | −1.71 | −1.85 | 0.21 | −0.03 | 0.40 |
| Fraction unbound (human) | 0.48 | 0.49 | 0.30 | 0.44 | 0.04 |
| Blood brain barrier (BBB) permeability | −0.33 | −0.77 | −0.04 | 0.36 | −0.15 |
| Metabolism | |||||
| CYP 2D6 Substrate | No | No | No | No | No |
| CYP 2D6 Inhibitor | No | No | No | No | No |
| Excretion | |||||
| Total clearance | 0.72 | 0.17 | 0.26 | 0.10 | 0.23 |
| Renal OCT2 substrate | No | No | No | No | Yes |
| Toxicity Assays | |||||
| AMES toxicity | No | No | No | No | No |
| Hepato toxicity | No | No | No | No | No |
| hERG I inhibitor | No | No | No | No | No |
| Oral rat acute toxicity LD50 (mol/kg) | 2.28 | 1.33 | 2.47 | 2.42 | 2.48 |
| (A) | Marginal Tests, p-Value | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Physico-Chemical Parameter | Cox 1 | Cox 2 | Intestinal Absorption | CaCo-2 Permeability | QPlog Khsa | VDss | Unbound Fraction | BBB Permeability | Total Clearance | Oral LD50 |
| iLOGP | 0.146 | 0.155 | 0.716 | 0.945 | 0.069 | 0.193 | 0.042 | 0.876 | 0.986 | 0.373 |
| XLOGP3 | 0.056 | 0.115 | 0.485 | 0.895 | 0.011 | 0.364 | 0.103 | 0.713 | 0.518 | 0.989 |
| MLOGP | 0.194 | 0.181 | 0.945 | 0.585 | 0.088 | 0.596 | 0.1 | 0.715 | 0.923 | 0.5 |
| SILICOS-IT | 0.062 | 0.065 | 0.353 | 0.791 | 0.04 | 0.128 | 0.013 | 0.667 | 0.576 | 0.362 |
| Log S (ESOL) | 0.041 | 0.091 | 0.419 | 0.874 | 0.009 | 0.417 | 0.083 | 0.792 | 0.409 | 0.965 |
| Molecular weight | 0.049 | 0.093 | 0.525 | 0.943 | 0.006 | 0.576 | 0.149 | 0.631 | 0.377 | 0.876 |
| Fraction Csp3 | 0.813 | 0.094 | 0.885 | 1 | 0.415 | 0.289 | 0.498 | 0.273 | 0.507 | 0.628 |
| Heavy atoms | 0.142 | 0.125 | 0.916 | 0.636 | 0.03 | 0.811 | 0.158 | 0.526 | 0.792 | 0.967 |
| Aromatic heavy atoms | 0.149 | 0.087 | 0.953 | 0.728 | 0.056 | 0.813 | 0.209 | 0.608 | 0.589 | 1 |
| Molar refractivity | 0.044 | 0.109 | 0.464 | 0.981 | 0.009 | 0.476 | 0.091 | 0.719 | 0.495 | 0.987 |
| Topological polar surface area | 0.889 | 0.558 | 0.906 | 0.883 | 0.833 | 0.161 | 0.642 | 0.02 | 0.783 | 0.114 |
| Human Intestinal Absorption | MLOGP, Log S, Molecular weight | 0.2 | ||||||||
| CaCo-2 Permeability | iLOGP, MLOGP, Molecular weight | 1 | ||||||||
| Plasma protein binding (QPlogKhsa) | iLOGP, SILICOS-IT, Aromatic heavy atoms | 1 | ||||||||
| VDss (human) | XLOGP3, Heavy atoms, Aromatic heavy atoms | 1 | ||||||||
| Unbound fraction (human) | iLOGP, MLOGP, Fraction Csp3 | 0.1 | ||||||||
| Blood-brain barrier (BBB) permeability | iLOGP, Fraction Csp3, Heavy atoms | 0.95516 | ||||||||
| Total clearance | iLOGP, SILICOS-IT, Fraction Csp3 | 0.999697 | ||||||||
| (B) | BEST model | R2 | ||||||||
| COX-1 | Log S, Molecular refractivity, Total polar surface area | 0.98852 | ||||||||
| COX-2 | iLOGP, Heavy atoms, Aromatic heavy atoms | 0.99933 | ||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rahman, M.M.; Junaid, M.; Hosen, S.M.Z.; Mostafa, M.; Liu, L.; Benkendorff, K. Mollusc-Derived Brominated Indoles for the Selective Inhibition of Cyclooxygenase: A Computational Expedition. Molecules 2021, 26, 6538. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26216538
Rahman MM, Junaid M, Hosen SMZ, Mostafa M, Liu L, Benkendorff K. Mollusc-Derived Brominated Indoles for the Selective Inhibition of Cyclooxygenase: A Computational Expedition. Molecules. 2021; 26(21):6538. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26216538
Chicago/Turabian StyleRahman, Md. Mominur, Md. Junaid, S. M. Zahid Hosen, Mohammad Mostafa, Lei Liu, and Kirsten Benkendorff. 2021. "Mollusc-Derived Brominated Indoles for the Selective Inhibition of Cyclooxygenase: A Computational Expedition" Molecules 26, no. 21: 6538. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26216538