
Structural Requirements of 1-(2-Pyridinyl)-5-pyrazolones for Disproportionation of Boronic Acids


Abstract
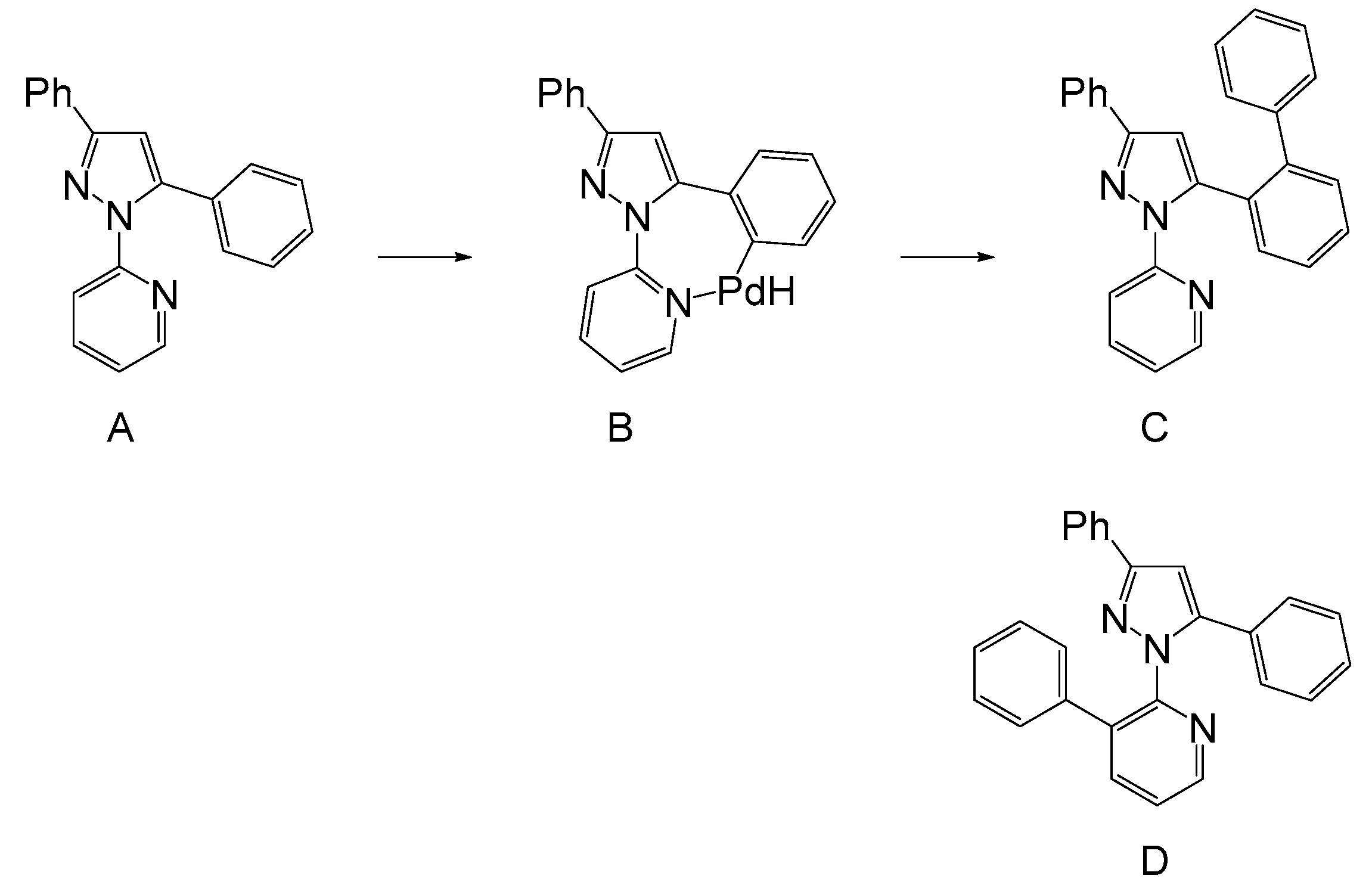
:1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. Generals
3.2. Representative Procedure for the Synthesis of Four-Coordinate Boron Complexes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Karrouchi, K.; Radi, S.; Ramli, Y.; Taoufik, J.; Mabkhot, Y.N.; Al-aizari, F.A.; Ansar, M. Synthesis and Pharmacological Activities of Pyrazole Derivatives: A Review. Molecules 2018, 23, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bin, H.-R.; Bae, Y.S.; Kim, G.; Lee, K.-I. Palladium-Catalyzed Peripheral Arylation of 5-Pyrazolones via Enolizable Bond Protection. Synthesis 2011, 11, 1783–1791. [Google Scholar]
- Joo, J.H.; Huh, J.E.; Lee, J.H.; Park, D.R.; Lee, Y.; Lee, S.G.; Choi, S.; Lee, H.J.; Song, S.W.; Jeong, Y.; et al. A novel pyrazole derivative protects from ovariectomy-induced osteoporosis through the inhibition of NADPH oxidase. Sci. Rep. 2016, 6, 22389. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.G.; Lee, J.; Kim, K.M.; Lee, K.-I.; Bae, Y.S.; Lee, H.J. Pharmacokinetic Study of NADPH Oxidase Inhibitor Ewha-18278, a Pyrazole Derivative. Pharmaceutics 2019, 11, 482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadu, V.S.; Choi, Y.M.; Kamma, K.R.; Kim, C.H.; Bae, Y.S.; Lee, K.-I. Crystal structure determination of N-and O-alkylated tautomers of 1-(2-pyridinyl)-5-hydroxypyrazole. J. Mol. Struct. 2020, 1215, 128272. [Google Scholar] [CrossRef]
- Nagata, Y.; Chujo, Y. Main-chain-type organoboron quinolate polymers: Synthesis and photoluminescence properties. Macromolecules 2007, 40, 6–8. [Google Scholar] [CrossRef]
- Marciasini, L.; Cacciuttolo, B.; Vaultier, M.; Pucheault, M. Synthesis of borinic acids and borinate adducts using diisopropylaminoborane. Org. Lett. 2015, 17, 3532–3535. [Google Scholar] [CrossRef] [PubMed]
- Frath, D.; Massue, J.; Ulrich, G.; Ziessel, R. Luminescent materials: Locking π-conjugated and heterocyclic ligands with boron (III). Angew. Chem. Int. Ed. 2014, 53, 2290–2310. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-Y.; Lin, H.-C.; Cheng, K.-M.; Su, W.-N.; Sung, K.-C.; Lin, T.-P.; Huang, J.-J.; Lin, S.-K.; Wong, F.F. Efficient di-bromination of 5-pyrazolones and 5-hydroxypyrazoles by N-bromobenzamide. Tetrahedron 2009, 65, 9592–9597. [Google Scholar] [CrossRef]
- Dvorak, C.A.; Rudolph, D.A.; Ma, S.; Carruthers, N.I. Palladium-Catalyzed Coupling of Pyrazole Triflates with Arylboronic Acids. J. Org. Chem. 2005, 70, 4188–4190. [Google Scholar] [CrossRef] [PubMed]
- Lyons, T.W.; Sanford, M.S. Palladium-Catalyzed Ligand-Directed C–H Functionalization Reactions. Chem. Rev. 2010, 110, 1147–1169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwak, S.H.; Gulia, N.; Daugulis, O. Synthesis of Unsymmetrical 2,6-Diarylanilines by Palladium-Catalyzed C–H Bond Functionalization Methodology. J. Org. Chem. 2018, 83, 5844–5850. [Google Scholar] [CrossRef] [PubMed]
- Crociani, B.; Antonaroli, S.; Marini, A.; Matteoli, U.; Scrivanti, A. Mechanistic study on the coupling reaction of aryl bromides with arylboronic acids catalyzed by (iminophosphine)palladium(0) complexes. Detection of a palladium(ii) intermediate with a coordinated boron anion. Dalton Trans. 2006, 22, 2698–2705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhakal, B.; Bohé, L.; Crich, D. Trifluoromethanesulfonate Anion as Nucleophile in Organic Chemistry. J. Org. Chem. 2017, 82, 9263–9269. [Google Scholar] [CrossRef] [PubMed]
- Hagen, H.; Reinoso, S.; Albrecht, M.; Boersma, J.; Spek, A.L.; van Koten, G. O,N-Chelated boron aminophenolate complexes. Crystal structure of BPh2(OC6H4(CH2NMe2)-2). J. Organomet. Chem. 2000, 608, 27–33. [Google Scholar]
- Aronoff, M.R.; VanVeller, B.; Raines, R.T. Detection of Boronic acids through excited-state intramolecular proton-transfer fluorescence. Org. Lett. 2013, 15, 5382–5385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhari, S.R. Screening and assignment of phenylboronic acid and its anhydride formation by NMR spectroscopy. Chem. Phys. Lett. 2015, 634, 95–97. [Google Scholar] [CrossRef]
- Candeias, N.R.; Montalbano, F.; Cal, P.M.S.D.; Gois, P.M.P. Boronic Acids and Esters in the Petasis-Borono Mannich Multicomponent Reaction. Chem. Rev. 2010, 110, 6169–6193. [Google Scholar] [CrossRef]
- Noonan, G.; Leach, A.G. A mechanistic proposal for the protodeboronation of neat boronic acids: Boronic acid mediated reaction in the solid state. Org. Biomol. Chem. 2015, 13, 2555–2560. [Google Scholar] [CrossRef] [Green Version]
- Voth, S.; Hollett, J.W.; McCubbin, J.A. Transition-Metal-Free Access to Primary Anilines from Boronic Acids and a Common +NH2 Equivalent. J. Org. Chem. 2015, 80, 2545–2553. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Substrate a | Reaction Conditions b | Product (Yield, %) |
|---|---|---|---|
| 1 | 2b | PhB(OH)2, K3PO4, dioxane | 2c (29) |
| 2 | 2a | PhB(OH)2, K3PO4, dioxane | 2c (37) |
| 3 | 1a | PhB(OH)2, K3PO4, dioxane | - c |
| 4 | 2a | PhB(OCMe2CMe2O), K3PO4, dioxane | - c |
| 5 | 2a | PhB(OH)2, dioxane | - c |
| 6 | - d | PhB(OH)2, K3PO4, dioxane |  3a (4%) 3b (26%) e |
| 7 | 2a | 3a, EtOH | 2c (81%) |
 |
 |
| Crystal Data | ||
|---|---|---|
| C26H20BN3O | c = 11.2162 (1) Å | Z = 2 |
| Mr = 401.26 | α = 78.966 (1)o | Mo Kα radiation |
| Triclinic, P1 | β = 81.795 (1)o | μ = 0.08 mm−1 |
| a = 9.7309 (1) Å | γ = 79.993 (1)o | T = 296 K |
| b = 9.8830 (1) Å | V = 1035.91 (2) Å3 | 0.44 × 0.31 × 0.23 mm |
| Data Collection | ||
| Bruker APEXII CCD diffractometer | 5044 independent reflections | |
| 19,866 measured reflections | 3923 reflections with I > 2σ(I) | |
| Refinement | ||
| R[F2 > 2σ(F2)] = 0.041 | 280 parameters | |
| wR(F2) = 0.106 | H-atom parameters constrained | |
| S = 1.03 | Δρmax = 0.21 e Å−3 | |
| 5044 reflections | Δρmin = −0.19 e Å−3 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cho, J.; Sadu, V.S.; Han, Y.; Bae, Y.; Lee, H.; Lee, K.-I. Structural Requirements of 1-(2-Pyridinyl)-5-pyrazolones for Disproportionation of Boronic Acids. Molecules 2021, 26, 6814. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26226814
Cho J, Sadu VS, Han Y, Bae Y, Lee H, Lee K-I. Structural Requirements of 1-(2-Pyridinyl)-5-pyrazolones for Disproportionation of Boronic Acids. Molecules. 2021; 26(22):6814. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26226814
Chicago/Turabian StyleCho, Joungmo, Venkata Subbaiah Sadu, Yohan Han, Yunsoo Bae, Hwajeong Lee, and Kee-In Lee. 2021. "Structural Requirements of 1-(2-Pyridinyl)-5-pyrazolones for Disproportionation of Boronic Acids" Molecules 26, no. 22: 6814. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26226814