
Efficacy of Selenourea Organocatalysts in Asymmetric Michael Reactions under Standard and Solvent-Free Conditions


Abstract
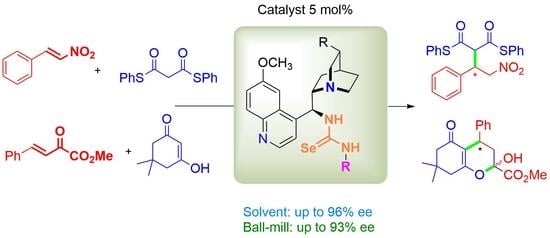
:
1. Introduction
2. Results and Discussion
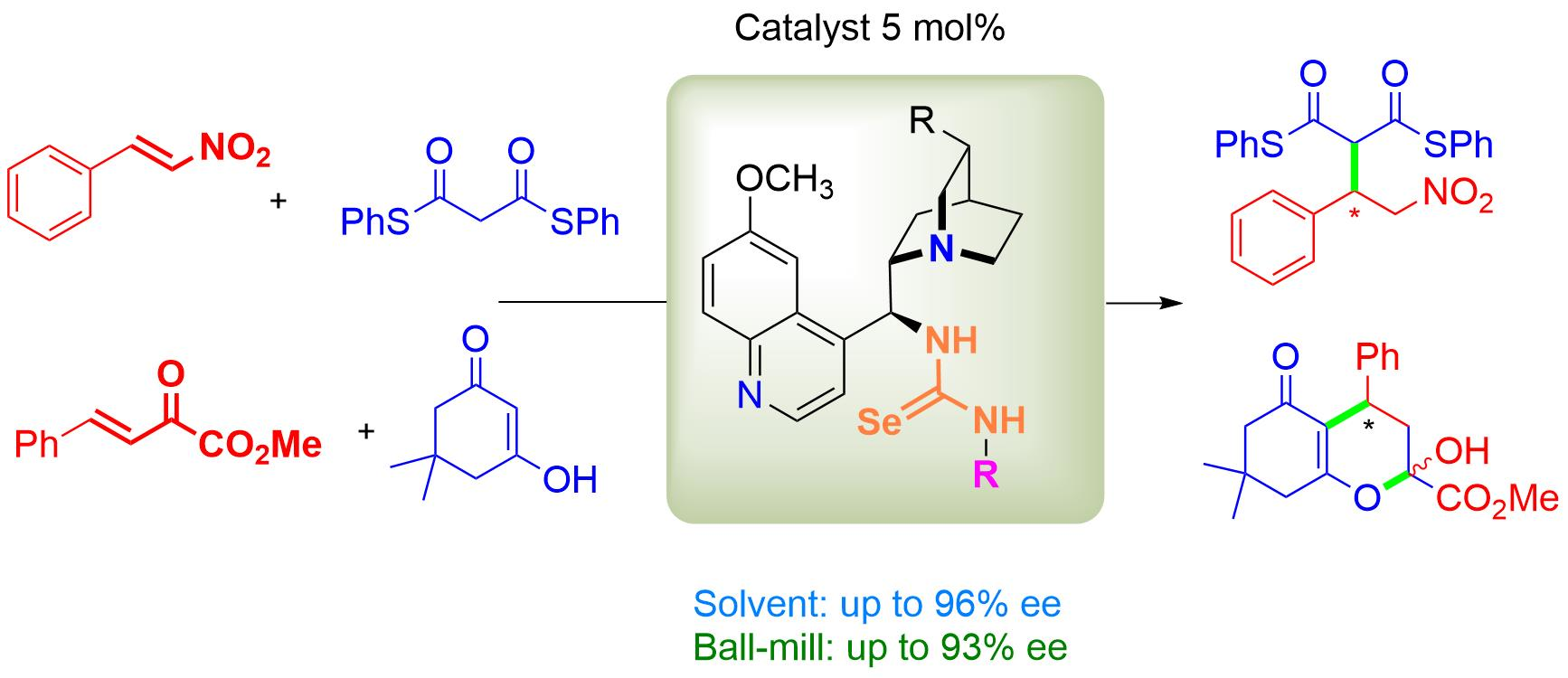
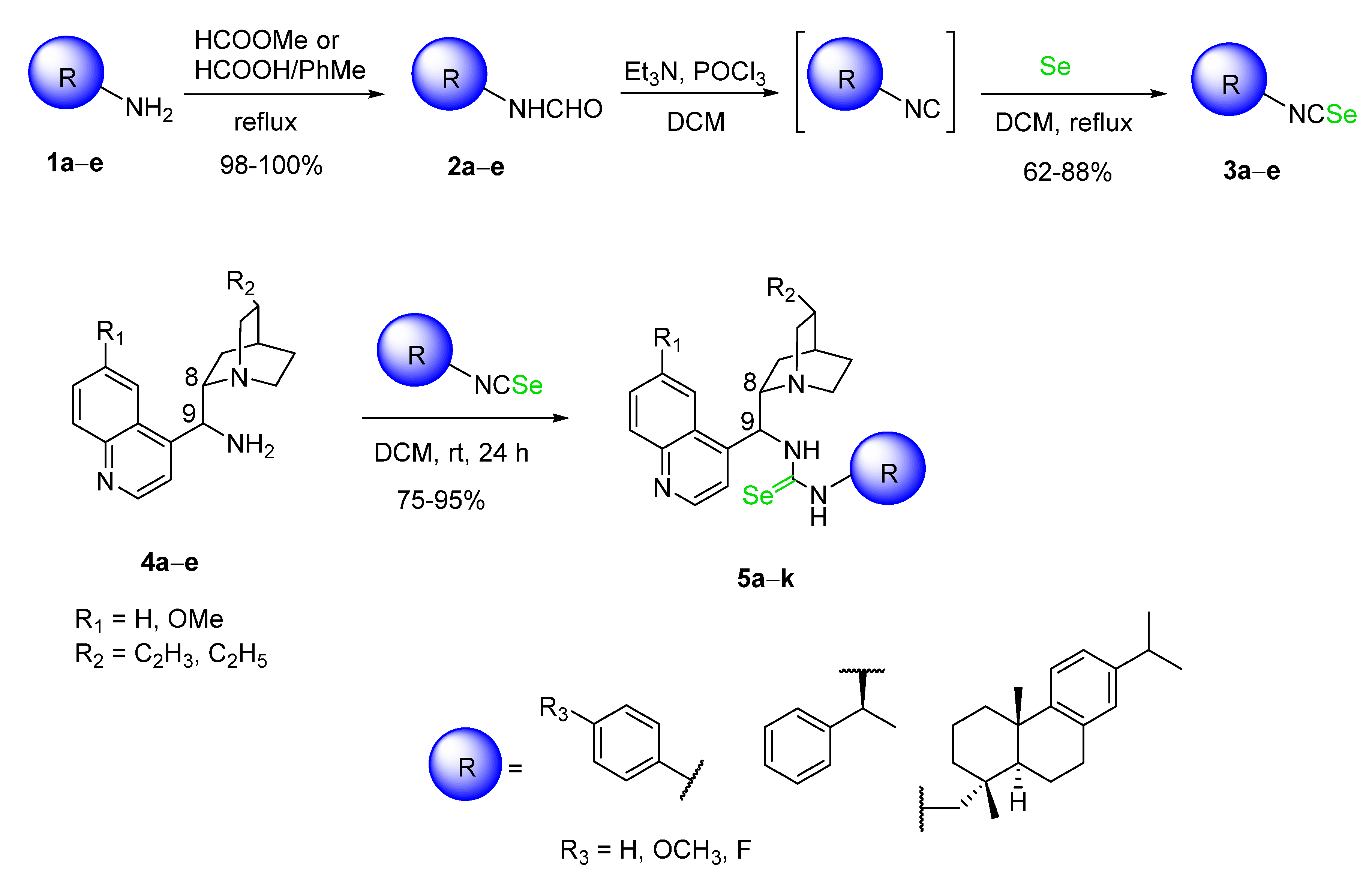
2.1. Synthesis of Cinchona Selenoureas
2.2. Bifunctional Selenourea-Based Catalysts in Various Conjugate Additions
3. Materials and Methods
3.1. General Information
3.2. Preparation of Starting Compounds
3.2.1. Preparation of Formamides 2a–e
- N-Phenylformamide2a. Yield 98%, beige solid. The mixture of two rotamers (ratio 1.1:1) was observed in the NMR spectra. 1H NMR (400 MHz, CDCl3): δ 7.05–7.16 (m, 2H), 7.17–7.25 (m, 2H), 7.29–7.37 (m, 5H), 7.55 (d, J = 8.0 Hz, 2H), 8.18 (s, 1H), 8.34 (br s, 1H), and 8.69 (d, J = 11.3 Hz, 1H) ppm. The spectral data are in agreement with the reported values [34].
- N-(4-Methoxyphenyl)formamide2b. Yield 99%, yellow solid. The mixture of two rotamers (ratio 1:1) was observed in the NMR spectra. 1H NMR (400 MHz, CDCl3): δ 3.79 (s, 3H), 3.80 (s, 3H), 6.90–6.84 (m, 4H), 7.03 (d, J = 8.9 Hz, 2 H), 7.32 (br s, 1H) 7.44 (d, J = 8.9 Hz, 2H), 7.97 (br s, 1H), 8.32 (s, 1H), and 8.50 (d, J = 11.4 Hz) ppm. The spectral data are in agreement with the reported values [34].
- N-(4-Fluorophenyl)formamide2c. Yield 100%, yellow solid. The mixture of two rotamers (ratio 1.7:1) was observed in the NMR spectra. 1H NMR (400 MHz, CDCl3): δ, 6.99–7.18 (m, 5H), 7.34–7.66 (m, 5 H), 8.34 (m, 1H), and 8.56 (d, J = 12.6 Hz, 1H) ppm. The spectral data are in agreement with the reported values [35].
- N-[(1S)-1-Phenylethyl]formamide2d. Yield 99%, brown oil. The mixture of two rotamers (ratio 5:1) was observed in the NMR spectra. 1H NMR (600 MHz, CDCl3): δ 1.52 (d, J = 6.9 Hz, 3H), 1.56 (d, J = 6.9 Hz, 3H), 4.69 (quint, J = 6.9 Hz, 1H), 5.21 (quint, J = 7.0 Hz, 1H), 5.95 (br s, 1H), 6.09 (br s, 1H), 7.24–7.38 (m, 10 H), and 8.15 (m, 2H) ppm. The spectral data are in agreement with the reported values [36].
- N-{[(1R,4aS,10aR)-1,4a-Dimethyl-7-isopropyl-1,2,3,4,4a,9,10,10a-octahydrophenanthren-1-yl] methyl}formamide2e. Yield 100%, brown oil. The mixture of two rotamers (ratio 2.8:1) was observed in the NMR spectra. 1H NMR (600 MHz, CDCl3): major rotamer, δ 0.95 (s, 3H), 1.22–1.29 (m, 10H), 1.35–1.44 (m, 3H), 1.66–1.78 (m, 3H), 1.87–1.90 (m, 1H), 2.30 (d, J = 12.8 Hz, 1H), 2.80–2.87 (m, 2H), 2.90–2.94 (m, 1H), 3.13 (dd, J = 6.6, 13.8 Hz, 1H), 3.29 (dd, J = 13.7, 6.7 Hz, 1H), 5.55 ( br s, 1H), 6.89 (dd, J = 1.0, 7.2 Hz, 1H), 6.99 (dd, J = 1.6, 8.2 Hz, 1H), 7.16 (dd, J = 2.5, 8.2 Hz, 1H), and 8.20 (s, 1H) ppm. HRMS (ESI): m/z calculated for [C21H31NO + Na]+: 336.2303, found: 336.2324. The spectral data are in agreement with the reported values [37].
3.2.2. Preparation of Isoselenocyanates 3a–e
- N-Phenylisoselenocyanate3a. Yield 62%, beige oil. 1H NMR (600 MHz, CDCl3): δ 3.81 (s, 3H), 6.86 (d, J = 8.2 Hz, 2H), and 7.23 (d, J = 8.3 Hz, 2H) ppm. The spectral data are in agreement with the reported values [38].
- N-(4-Methoxyphenyl)isoselenocyanate3b. Yield 68%, light yellow solid. 1H NMR (600 MHz, CDCl3): δ 3.81 (s, 3H), 6.86 (d, J = 8.2 Hz, 2H), and 7.23 (d, J = 8.3 Hz, 2H) ppm. The spectral data are in agreement with the reported values [38].
- N-(4-Fluorophenyl)isoselenocyanate3c. Yield 69%, solidifying yellow oil. 1H NMR (600 MHz, CDCl3): δ 6.93–7.01 (m, 2H), and 7.15–7.23 (m, 2H) ppm. The spectral data are in agreement with the reported values [38].
- [(1S)-1-Isoselenocyanatoethyl]benzene3d. Yield 82%, dark yellow oil. 1H NMR (600 MHz, CDCl3): = 1.71 (d, J = 6.8 Hz, 3H), 4.99 (q, J = 6.8 Hz, 1H), 7.31–7.36 (m, 3H), and 7.37–7.42 (m, 2H) ppm. 13C NMR (150 MHz, CDCl3): δ 24.7, 57.7, 125.5 (2C overlapped), 128.6, 129.0, 129.1 (2C overlapped), and 139.1 ppm. HRMS (ESI): m/z calculated for [C9H9NSe + H]+: 211.9973, found: 211.9969.
- (1R,4aS,10aR)-1,4a-Dimethyl-7-isopropyl-1-(isoselenocyanatomethyl)-1,2,3,4,4a,9,10,10a-octahydrophenanthrene3e. Yield 88%, yellow oil. 1H NMR (600 MHz, CDCl3): δ 0.99 (s, 3H), 1.22–1.23 (m, 9H), 1.44–1.46 (m, 3H), 1.56 (td, J = 10.3, 2.2Hz, 2H), 1.65–1.68 (m, 1H), 1.72–1.80 (m, 2H), 2.31 (m, 1H), 2.83 (hept. J = 6.9 Hz, 1H), 2.89–2.92 (m, 1H), 3.09–3.10 (m, 1H), 3.40 (d, J = 14.4 Hz, 1H), 3.51 (d, J = 14.4 Hz, 1H), 6.89 (dd, J = 1.1 Hz, 1H), 7.01 (dd, J = 8.1, 1.8 Hz, 1H), and 7.16 (d, J = 8.2 Hz, 1H) ppm. 13C NMR (150 MHz, CDCl3): δ 18.1, 18.6, 19.2, 24.1, 25.3, 30.2, 33.5, 36.5, 37.7, 38.2, 38.5, 45.9, 57.3, 124.2, 124.4, 126.9, 134.3, 145.9, and 146.3 ppm. HRMS (ESI): m/z calculated for [C21H29NSe + H]+: 376.1544, found: 376.1541.
3.3. General Procedure for the Synthesis of Selenourea Catalysts 5a–k
- N-[(8S,9S)-6′-Methoxycinchonan-9-yl]-N′-[(S)-1-phenylethyl]selenourea eQN-5d
- Yield 94%, pale yellow solid, mp 130−132 °C, Rf = 0.35 (CH2Cl2/MeOH 10:1). = −136.6 (c 0.22, CH2Cl2). 1H NMR (400 MHz, CDCl3): δ 0.91 (s, 1H), 1.25–1.61 (m, 8H), 2.12–3.06 (m, 6H), 3.94 (s, 3H), 4.88–4.93 (m, 2H), 5.33 (br s, 1H), 5.55–5.64 (m, 1H), 7.29–7.40 (m, 9H), 7.56 (br s, 1H), 8.04 (d, J = 6.7 Hz, 1H), and 8.74 (d, J = 4.5 Hz, 1H) ppm. 13C NMR (101 MHz, CDCl3): δ 25.6, 27.3, 27.4, 27.6, 29.8, 39.3, 40.4, 50.8, 55.0, 55.8, 61.2, 102.3, 115.0, 120.0, 122.0, 126.1, 127.9 (2C overlapped), 129.1 (2C overlapped), 132.0, 140.9, 142.0 (2C overlapped), 144.8, 147.7 (2C overlapped), 158.1, and 179.8 ppm. HRMS (ESI): m/z calculated for C29H35N4O80Se [M + H]+: 535.1971, found: 535.1985.
- N-{[(1R,4aS,10aR)-1,4a-Dimethyl-7-izopropyl-1,2,3,4,4a,9,10,10a-octahydrophenanthrene-1-yl]methyl}-N′-[(8S,9S)-6′-methoxycinchonan-9-yl]selenourea eQN-5e
- Yield 92%, pale yellow solid, mp 137−139 °C, Rf = 0.52 (CH2Cl2/MeOH 10:1). = −66.3 (c 0.23, CH2Cl2). 1H NMR (600 MHz, CDCl3): δ 0.68–0.79 (m, 4H), 0.83–1.18 (m, 6H), 1.20–12.00 (m, 16H), 1.96–2.24 (s, 1H), 2.35 (s, 1H), 2.53 (s, 1H), 2.66–3.43 (m, 8H), 3.95–4.00 (m, 4H), 4.83–5.08 (m, 2H), 5.62–5.67 (m, 1H), 6.40 (br s, 1H), 6.83 (s, 1H), 6.93–7.17 (m, 2H), 7.24–7.69 (m, 3H), 7.74–8.20 (m, 2H), and 8.50 (br s, 1H) ppm. 13C NMR (151 MHz, CDCl3): δ 18.4 (2C overlapped), 18.7, 19.1 (2C overlapped), 24.0, 24.1 (2C overlapped), 25.4, 27.2, 29.7, 30.0 (2C overlapped), 33.5 (3C overlapped), 36.2, 37.2, 37.5, 37.6, 55.2, 55.3, 59.2, 100.8, 115.6, 122.3, 123.7 (2C overlapped), 124.1 (2C overlapped), 126.9, 127.1, 132.2, 134.6, 140.1, 145.0, 145.4, 146.8, 147.7, 158.4, and 180.5 ppm. HRMS (ESI): m/z calculated for C41H55N4O80Se [M + H]+: 699.3541, found: 699.3556.
- N-[(8R,9R)-6′-Methoxycinchonan-9-yl]-N′-[(S)-1-phenylethyl]selenourea eQD-5i
- Yield 86%, pale yellow solid, mp 123−125 °C, Rf = 0.33 (CH2Cl2/MeOH 10:1). = +302.5 (c 0.24, CH2Cl2). 1H NMR (600 MHz, CDCl3): δ 0.88 (s, 1H), 1.12–1.65 (m, 8H), 2.32 (d, J = 8.2 Hz, 1H), 2.58–3.24 (m, 5H), 3.95 (s, 3H), 4.72–4.80 (m, 1H), 5.13 (m, 3H), 5.72–5.84 (m, 1H), 6.68–7.76 (m, 10H), 7.96 (s, 1H), and 8.15–8.84 (m, 1H) ppm. 13C NMR (151 MHz, CDCl3): δ 24.8, 26.0, 27.2, 38.6, 47.0, 48.7, 50.7, 53.4, 55.7 (2C overlapped), 57.7, 61.8, 102.1, 115.3, 118.8, 122.4, 126.0 (3C overlapped), 128.3, 128.8, 131.9, 139.4, 141.8, 144.6, 147.6 (2C overlapped), 158.2, and 178.7 ppm. HRMS (ESI): m/z calculated for C29H35N4O80Se [M + H]+: 535.1971, found: 535,1984.
- N-{[(1R,4aS,10aR)-1,4a-Dimethyl-7-izopropyl-1,2,3,4,4a,9,10,10a-octahydrophenanthrene-1-yl]methyl}-N′-[(8R,9R)-6′-methoxycinchonan-9-yl]selenourea eQD-5j
- Yield 89%, pale yellow solid, mp 144−146 °C, Rf = 0.40 (CH2Cl2/MeOH 10:1). = +204.4 (c 0.23, CH2Cl2). 1H NMR (600 MHz, CDCl3): δ 0.48–0.66 (m, 4H), 0.71–1.16 (m, 8H), 1.16–1.68 (m, 13H), 1.68–1.92 (m, 2H), 1,98–2.23 (m, 1H), 2.35 (s, 1H), 2.46–3.43 (m, 9H), 3.57–4.14 (m, 3H), 5.00–5.24 (m, 2H), 5.76–6.00 (m, 1H), 6.37 (br s, 1H), 6.88 (s, 1H), 6.97 (d, J = 8.1 Hz, 1H), 7.08 (d, J = 8.0 Hz, 1H), 7.24–7.79 (m, 3H), 7.89–8.29 (m, 2H), and 8.58–9.00 (br s, 1H) ppm. 13C NMR (151 MHz, CDCl3): δ 18.0, 18.2, 19.0 (2C overlapped), 24.0 (3C overlapped), 25.2, 26.2, 27.2, 30.2, 33.5 (2C overlapped), 36.0, 37.0, 37.3, 37.9, 46.2, 46.8, 49.1 (2C overlapped), 55.7, 60.0, 99.4, 115.1, 122.9, 123.9 (2C overlapped), 124.2 (2C overlapped), 126.8 (2C overlapped), 132.3, 134.4, 139.9, 145.1, 145.6, 146.8, 147.8, 158.6, and 180.2 ppm. HRMS (ESI): m/z calculated for C41H55N4O80Se [M + H]+: 699.3541, found: 699.3564.
3.4. Preparation of Thiourea Catalysts 6a and 6b
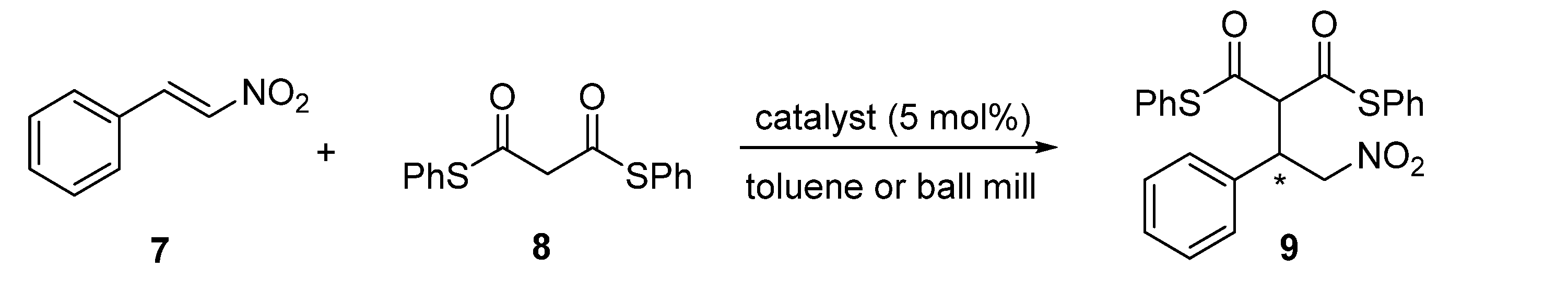
3.5. General Procedure for Michael Addition of S,S′-Diphenyl Dithiomalonate 8 to trans-β-Nitrostyrene 7 in a Solution
- (S)-2-(2-Nitro-1-phenylethyl)-1,3-bis(phenylsulfanyl)propane-1,3-dione9. The following product was obtained as an off-white solid, 108 mg, 99%, = +123.0 (c 1.0, DCM), 95% ee. Mp 160.0–161.5 °C. 1H NMR (CDCl3, 400 MHz): δ 4.42–4.37 (m, 1H), 4.49 (d, J = 9.8 Hz, 1H), 4.89–4.80 (m, 2H), 7.16–7.13 (m, 2H), 7.28–7.26 (m, 2H), 7.39–7.31 (m, 6H), and 7.47–7.40 (m, 5H). 13C NMR (CDCl3, 125 MHz): δ 44.5, 69.4, 77.2, 126.2, 128.5, 128.7, 129.2, 129.5, 129.7, 130.2, 130.4, 134.3, 134.4, 135.3, 189.7, 190.5 ppm. HRMS (ESI) m/z calcd for C23H19O4S2 [M − H]+ 436.0677, found 436.0661. HPLC (Chiralcel IB-3, n-hexane/2-propanol, 9:1, flow rate 1 mL/min, λ = 205 nm): tR = 17.1 min (major), tR = 20.1 min (minor). The spectral data are in agreement with those reported in the literature [24].
3.6. General Procedure for Michael Addition of S,S′-Diphenyl Dithiomalonate 8 to trans-β-Nitrostyrene 7 Using Planetary Ball-Mill
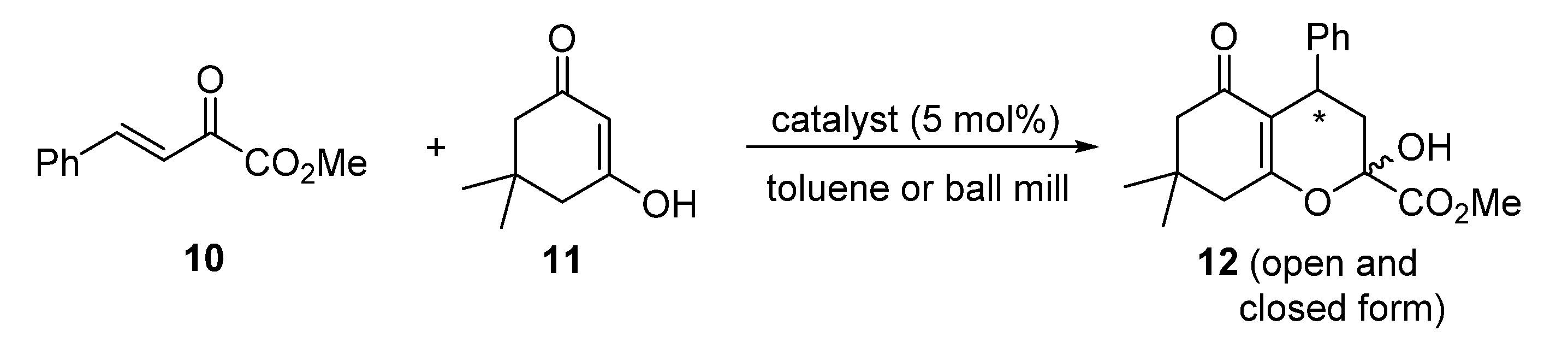
3.7. General Procedure for the Asymmetric Michael-Hemiacetalization Reaction of Benzylidene Pyruvate 10 and Dimedone 11 in a Solution
- (4R)-4-Methyl-2-hydroxy-7,7-dimethyl-5-oxo-4-phenyl-3,4,5,6,7,8-hexahydro-2H-chromene-2-carboxylate12. The following 12 product was obtained as a colorless oil, 33 mg, 99%, 94%ee. 1H NMR (CDCl3, 400 MHz): δ 1.10 (d, J = 8.9 Hz, 3H), 1.19 (d, J = 9.5 Hz, 3H), 2.24–2.21 (m, 2H), 2.58–2.26 (m, 4H), 3.73 (d, J = 2.1 Hz, 2H), 3.84 (s, 1H), 3.91–3.86 (m, 1H), 4.65 (bs, 1H), 7.18–7.13 (m, 3H), 7.28–7.23 (m, 2H) ppm. HPLC (Chiralcel IA-3, n-hexane/2-propanol, 7:3, flow rate 1 mL/min, λ = 254 nm): tR = 4.6 min (major), tR = 6.0 min (minor). The spectral data are in agreement with those reported in the literature [25].
3.8. General Procedure for the Asymmetric Michael-Hemiacetalization Reaction of Benzylidene Pyruvate 10 and Dimedone 11 Using Planetary Ball-Mill
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Tsogoeva, S.B. Recent advances in asymmetric organocatalytic 1,4-conjugate additions. Eur. J. Org. Chem. 2007, 2007, 1701–1716. [Google Scholar] [CrossRef]
- Sigman, M.S.; Jacobsen, E.N. Schiff base catalysts for the asymmetric Strecker reaction identified and optimized from parallel synthetic libraries. J. Am. Chem. Soc. 1998, 120, 4901–4902. [Google Scholar] [CrossRef]
- Okino, T.; Hoashi, Y.; Takemoto, Y. Enantioselective Michael reaction of malonates to nitroolefins catalyzed by bifunctional organocatalysts. J. Am. Chem. Soc. 2003, 125, 12672–12673. [Google Scholar] [CrossRef]
- Connon, S.J. Asymmetric catalysis with bifunctional cinchona alkaloid-based urea and thiourea organocatalysts. Chem. Commun. 2008, 2499–2510. [Google Scholar] [CrossRef] [Green Version]
- Song, C.E. Cinchona Alkaloids in Synthesis and Catalysis: Ligands, Immobilization and Organocatalysis; John Wiley and Sons: Hoboken, NJ, USA, 2009; ISBN 9783527324163. [Google Scholar]
- Jörres, M.; Schiffers, I.; Atodiresei, I.; Bolm, C. Asymmetric Michael additions of α-nitrocyclohexanone to aryl nitroalkenes catalyzed by natural amino acid-derived bifunctional thioureas. Org. Lett. 2012, 14, 4518–4521. [Google Scholar] [CrossRef]
- Zielińska-Błajet, M.; Najdek, J. Novel selenoureas based on Cinchona alkaloid skeleton: Synthesis and catalytic investigations. Materials 2021, 14, 600. [Google Scholar] [CrossRef]
- Obst, M.; König, B. Organic synthesis without conventional solvents. Eur. J. Org. Chem. 2018, 2018, 4213–4232. [Google Scholar] [CrossRef]
- Tan, D.; Friščić, T. Mechanochemistry for organic chemists: An update. Eur. J. Org. Chem. 2018, 2018, 18–33. [Google Scholar] [CrossRef]
- Howard, J.L.; Cao, Q.; Browne, D.L. Mechanochemistry as an emerging tool for molecular synthesis: What can it offer? Chem. Sci. 2018, 9, 3080–3094. [Google Scholar] [CrossRef] [Green Version]
- Avila-Ortiz, C.G.; Juaristi, E. Novel methodologies for chemical activation in organic synthesis under solvent-free reaction conditions. Molecules 2020, 25, 3579. [Google Scholar] [CrossRef]
- Schöbel, J.H.; Liang, W.; Wöll, D.; Bolm, C. Mechanochemical synthesis of 1,2,6-thiadiazine 1-oxides from sulfonimidamides and the fluorescence properties of the products. J. Org. Chem. 2020, 85, 15760–15766. [Google Scholar] [CrossRef]
- Rodríguez, B.; Bruckmann, A.; Bolm, C. A highly efficient asymmetric organocatalytic aldol reaction in a ball mill. Chem. Eur. J. 2007, 13, 4710–4722. [Google Scholar] [CrossRef]
- Veverková, E.; Poláčková, V.; Liptáková, L.; Kázmerová, E.; Mečiarová, M.; Toma, Š.; Šebesta, R. Organocatalyst efficiency in the Michael additions of aldehydes to nitroalkenes in water and in a ball-mill. ChemCatChem 2012, 4, 1013–1018. [Google Scholar] [CrossRef]
- Jörres, M.; Mersmann, S.; Raabe, G.; Bolm, C. Organocatalytic solvent-free hydrogen bonding-mediated asymmetric Michael additions under ball milling conditions. Green Chem. 2013, 15, 612–616. [Google Scholar] [CrossRef]
- Machuca, E.; Rojas, Y.; Juaristi, E. Synthesis and evaluation of (S)-proline-containing α,β-dipeptides as organocatalysts in solvent-free asymmetric aldol reactions under ball-milling conditions. Asian J. Org. Chem. 2015, 4, 46–53. [Google Scholar] [CrossRef]
- Egorov, I.N.; Santra, S.; Kopchuk, D.S.; Kovalev, I.S.; Zyryanov, G.V.; Majee, A.; Ranu, B.C.; Rusinov, V.L.; Chupakhin, O.N. Ball milling: An efficient and green approach for asymmetric organic syntheses. Green Chem. 2020, 22, 302–315. [Google Scholar] [CrossRef]
- Wang, Y.F.; Chen, R.X.; Wang, K.; Zhang, B.B.; Li, Z.B.; Xu, D.Q. Fast, solvent-free and hydrogen-bonding-mediated asymmetric Michael addition in a ball mill. Green Chem. 2012, 14, 893–895. [Google Scholar] [CrossRef]
- Ignatiuk, Ż.A.; Janicki, M.J.; Góra, R.W.; Konieczny, K.; Kowalczyk, R. Applications of thermal activation, ball-milling and aqueous medium in stereoselective Michael addition of nitromethane to enynones catalyzed by chiral squaramides. Adv. Synth. Catal. 2019, 361, 1108–1116. [Google Scholar] [CrossRef]
- Hestericová, M.; Šebesta, R. Higher enantioselectivities in thiourea-catalyzed Michael additions under solvent-free conditions. Tetrahedron 2014, 70, 901–905. [Google Scholar] [CrossRef]
- Brunner, H.; Bügler, J.; Nuber, B. Preparation of 9-amino(9-deoxy)cinchona alkaloids. Tetrahedron Asymmetry 1995, 6, 1699–1702. [Google Scholar] [CrossRef]
- Kacprzak, K.; Gierczyk, B. Clickable 9-azido-(9-deoxy)-Cinchona alkaloids: Synthesis and conformation. Tetrahedron Asymmetry 2010, 21, 2740–2745. [Google Scholar] [CrossRef]
- Sharma, S.; Maurya, R.A.; Min, K.; Jeong, G.; Kim, P. Odorless isocyanide chemistry: An integrated microfluidic system for a multistep reaction sequence. Angew. Chem. Int. Ed. 2013, 52, 7564–7568. [Google Scholar] [CrossRef]
- Jin, H.; Kim, S.T.; Hwang, G.S.; Ryu, D.H. L -Proline derived bifunctional organocatalysts: Enantioselective Michael addition of dithiomalonates to trans-β-nitroolefins. J. Org. Chem. 2016, 81, 3263–3274. [Google Scholar] [CrossRef]
- Chen, X.K.; Zheng, C.W.; Zhao, S.L.; Chai, Z.; Yang, Y.Q.; Zhao, G.; Cao, W.G. Highly enantioselective Michael addition of cyclic 1,3-dicarbonyl compounds to β,γ-unsaturated α-keto esters. Adv. Synth. Catal. 2010, 352, 1648–1652. [Google Scholar] [CrossRef]
- Wang, Y.F.; Wang, K.; Zhang, W.; Zhang, B.B.; Zhang, C.X.; Xu, D.Q. Enantioselective asymmetric Michael addition of cyclic diketones to β,γ-unsaturated α-keto esters. Eur. J. Org. Chem. 2012, 2012, 3691–3696. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, X.; Wang, M.; He, P.; Lin, L.; Feng, X. Enantioselective synthesis of 3,4-dihydropyran derivatives via organocatalytic Michael reaction of α,β-unsaturated enones. J. Org. Chem. 2012, 77, 4136–4142. [Google Scholar] [CrossRef] [PubMed]
- Boratyński, P.J.; Kowalczyk, R. Click-dimerized Cinchona alkaloids. J. Org. Chem. 2016, 81, 8029–8034. [Google Scholar] [CrossRef]
- Ding, X.; Zhang, X.-D.; Dong, C.-L.; Guan, Z.; He, Y.-H. Enzyme-catalyzed cascade Michael/cyclization reaction for the synthesis of 3,4-dihydropyran derivatives by using a protease. Catal. Lett. 2018, 148, 757–763. [Google Scholar] [CrossRef]
- Dajek, M.; Kowalczyk, R.; Boratyński, P.J. trans-1,2-Diaminocyclohexane-based sulfonamides as effective hydrogen-bonding organocatalysts for asymmetric Michael-hemiacetalization reaction. Catal. Sci. Technol. 2018, 8, 4358–4363. [Google Scholar] [CrossRef]
- Tukhvatshin, R.S.; Kucherenko, A.S.; Nelyubina, Y.V.; Zlotin, S.G. Conjugate addition of carbon acids to β,γ-unsaturated α-keto esters: Product tautomerism and applications for asymmetric synthesis of benzo[α]phenazin-5-ol derivatives. J. Org. Chem. 2019, 84, 13824–13831. [Google Scholar] [CrossRef]
- Dajek, M.; Pruszczyńska, A.; Konieczny, K.A.; Kowalczyk, R. Cinchona squaramide-catalyzed intermolecular desymmetrization of 1,3-diketones leading to chiral 1,4-dihydropyridines. Adv. Synth. Catal. 2020, 362, 3613–3620. [Google Scholar] [CrossRef]
- Zielińska-Błajet, M.; Kucharska, M.; Skarżewski, J. Simple enantiospecific synthesis of sulfides of Cinchona alkaloids. Synthesis 2006, 2006, 1176–1182. [Google Scholar] [CrossRef]
- Nishikawa, Y.; Nakamura, H.; Ukai, N.; Adachi, W.; Hara, O. Tetraethylorthosilicate as a mild dehydrating reagent for the synthesis of N-formamides with formic acid. Tetrahedron Lett. 2017, 58, 860–863. [Google Scholar] [CrossRef]
- Wei, Y.; Wu, J.; Xue, D.; Wang, C.; Liu, Z.; Zhang, Z.; Chen, G.; Xiao, J. Highly efficient rhodium-catalysed transfer hydrogenation of nitroarenes into amines and formanilides. Synlett 2014, 25, 1295–1298. [Google Scholar]
- Patre, R.E.; Mal, S.; Nilkanth, P.R.; Ghorai, S.K.; Deshpande, S.H.; Qacemi, M.E.; Smejkal, T.; Pal, S.; Manjunath, B.N. First report of bio-catalytic N-formylation of amines using ethylformate. Chem. Commun. 2017, 53, 2382–2385. [Google Scholar] [CrossRef] [PubMed]
- Mori, N.; Kuzuya, K.; Watanabe, H. Synthesis of (−)-chamobtusin A from (+)-dehydroabietylamine. J. Org. Chem. 2016, 81, 11866–11870. [Google Scholar] [CrossRef]
- Barton, D.H.R.; Parekh, S.I.; Tajbakhsh, M.; Theodorakis, E.A.; Chi-Lam, T. A convenient and high yielding procedure for the preparation of isoselenocyanates. Synthesis and reactivity of O-alkylselenocarbamates. Tetrahedron 1994, 50, 639–654. [Google Scholar] [CrossRef]
- Peérez-Labrada, K.; Brouard, I.; Méndez, I.; Rivera, D.G. Multicomponent synthesis of Ugi-type ceramide analogues and neoglycolipids from lipidic isocyanides. J. Org. Chem. 2012, 77, 4660–4670. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, Y.; Zhang, G.; Fu, D.; Zhang, F.; Kai, M.; Wang, R. Enantioselective synthesis of cyclic thioureas via Mannich reaction and concise synthesis of highly optically active methylthioimidazolines: Discovery of a more potent antipyretic agent. Adv. Synth. Catal. 2011, 353, 1787–1796. [Google Scholar] [CrossRef]
- Zhang, G.; Zhang, Y.; Yan, J.; Chen, R.; Wang, S.; Ma, Y.; Wang, R. One-pot enantioselective synthesis of functionalized pyranocoumarins and 2-amino-4H-chromenes: Discovery of a type of potent antibacterial agent. J. Org. Chem. 2012, 77, 878–888. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Catalyst | Time (min) | Conv. (%) b | ee (%) c | Config. d |
|---|---|---|---|---|---|
| 1 | eQN-5a | 15 | >99 | 87 | S |
| 2 | eQN-5b | 15 | 99 (94) | 96 | S |
| 3 | eQN-5c | 30 | 99 (96) | 95 (72) e | S |
| 4 | eQN-5d | 5 | >99 (95) | 96 (78) f | S |
| 5 | eQN-5e | 30 | >99 (97) | 93 (69) e | S |
| 6 | eCD-5f | 30 | >99 | 85 | S |
| 7 | eDHQN-5g | 15 | 99 | 86 | S |
| 8 | eQD-5h | 5 | >99 (98) | 96 (87) f | R |
| 9 | eQD-5i | 5 | 99 (95) | 94 (89) f | R |
| 10 | eQD-5j | 90 | 94 | 87 (72) e | R |
| 11 | eDHQD-5k | 15 | 89 (83) | 94 | R |
| 12 | eQN-6a | 5 | >99 (93) | 95 | S |
| 13 | eQN-6b | 90 | 96 | 76 | S |
| Entry | Catalyst | Conv. (%) b | ee (%) c | Config. d |
|---|---|---|---|---|
| 1 | eQN-5a | 96 | 73 | S |
| 2 | eQN-5b | 94 | 71 | S |
| 3 | eQN-5c | 98 | 71 | S |
| 4 | eQN-5d | >99 (96) | 77 | S |
| 5 | eQN-5e | >99 | 69 | S |
| 6 | eCD-5f | 94 | 76 | S |
| 7 | eDHQN-5g | 93 (89) | 90 | S |
| 8 | eQD-5h | 95 (95) | 84 | R |
| 9 | eQD-5i | 98 (95) | 93 | R |
| 10 | eQD-5j | 91 (87) | 87 | R |
| 11 | eDHQD-5k | 95 (92) | 93 | R |
| 12 | eQN-6a | 99 | 77 | S |
| 13 | eQN-6b | >99 | 59 | S |
| Entry | Catalyst | Conv. (%) b | ee (%) c | Config. d |
|---|---|---|---|---|
| 1 | eQN-5a | >99 | 67 | R |
| 2 | eQN-5b | >99 | 64 | R |
| 3 | eQN-5c | >99 | 65 (69) e | R |
| 4 | eQN-5d | >99 (95) | 84 (76) e | R |
| 5 | eQN-5e | >99 (97) | 94 (89) e | R |
| 6 | eCD-5f | >99 | 66 | R |
| 7 | eDHQN-5g | >99 | 69 | R |
| 8 | eQD-5h | >99 | 73 (80) e | S |
| 9 | eQD-5i | >99 (94) | 85 (80) f | S |
| 10 | eQD-5j | >99 (96) | 93 (85) f | S |
| 11 | eDHQD-5k | >99 (93) | 82 | S |
| 12 | eQN-6a | >99 (94) | 91 | R |
| 13 | eQN-6b | >99 | 76 | R |
| Entry | Catalyst | Conv. (%) b | ee (%) c | Config. d |
|---|---|---|---|---|
| 1 | eQN-5a | 82 | 50 | R |
| 2 | eQN-5b | 82 | 35 | R |
| 3 | eQN-5c | 88 | 34 | R |
| 4 | eQN-5d | 67 (65) | 58 | R |
| 5 | eQN-5e | 78 | 53 | R |
| 6 | eCD-5f | 92 | 50 | R |
| 7 | eDHQN-5g | 98 | 53 | R |
| 8 | eQD-5h | 32 | 39 | S |
| 9 | eQD-5i | 96 (94) | 64 | S |
| 10 | eQD-5j | 78 (74) | 73 | S |
| 11 | eDHQD-5k | 36 | 39 | S |
| 12 | eQN-6a | 38 | 48 | R |
| 13 | eQN-6b | 49 (46) | 61 | R |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zielińska-Błajet, M.; Mała, Ż.A.; Kowalczyk, R. Efficacy of Selenourea Organocatalysts in Asymmetric Michael Reactions under Standard and Solvent-Free Conditions. Molecules 2021, 26, 7303. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26237303
Zielińska-Błajet M, Mała ŻA, Kowalczyk R. Efficacy of Selenourea Organocatalysts in Asymmetric Michael Reactions under Standard and Solvent-Free Conditions. Molecules. 2021; 26(23):7303. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26237303
Chicago/Turabian StyleZielińska-Błajet, Mariola, Żaneta A. Mała, and Rafał Kowalczyk. 2021. "Efficacy of Selenourea Organocatalysts in Asymmetric Michael Reactions under Standard and Solvent-Free Conditions" Molecules 26, no. 23: 7303. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26237303