
Cyclic Photoisomerization of Azobenzene in Atomistic Simulations: Modeling the Effect of Light on Columnar Aggregates of Azo Stars


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
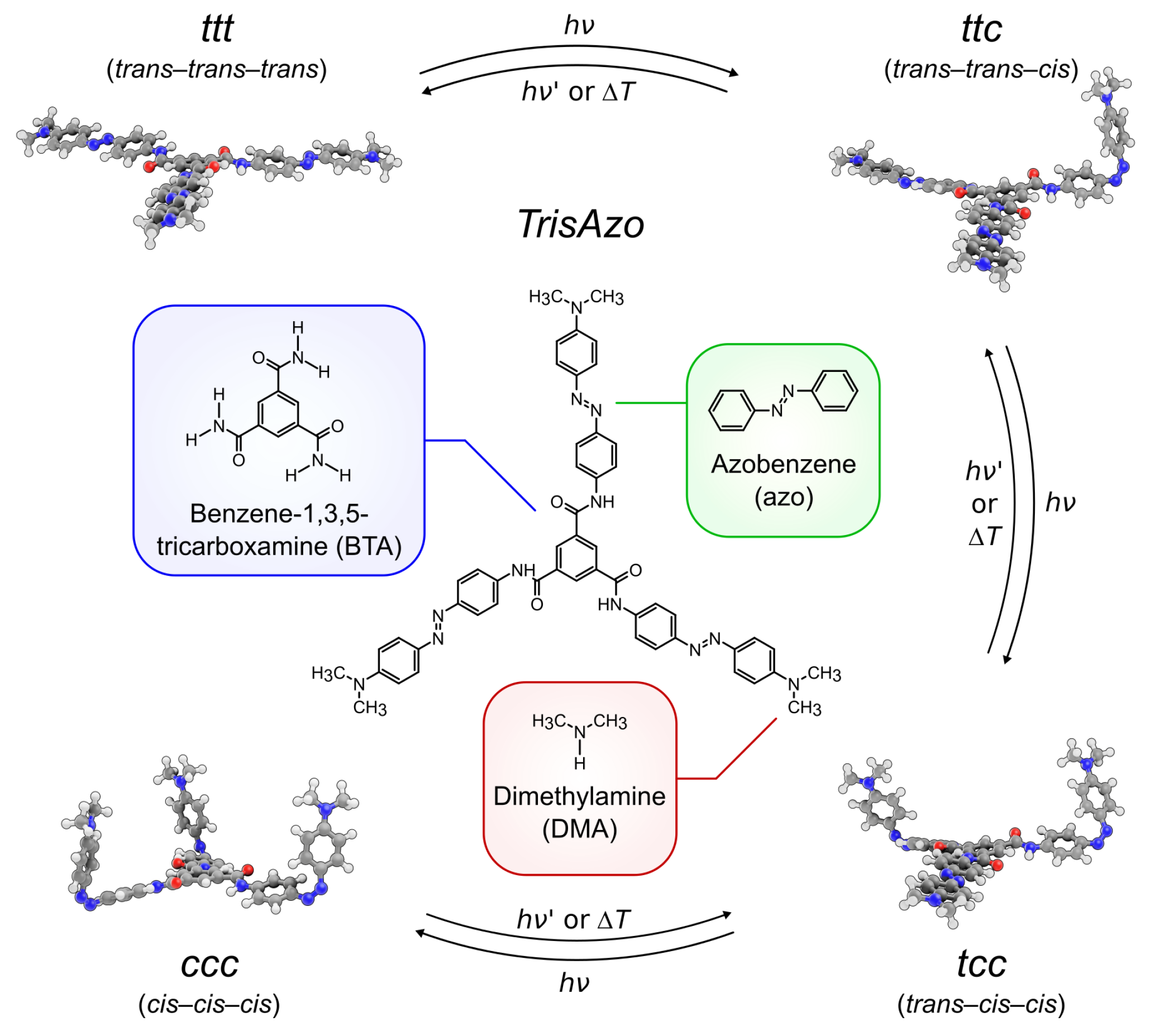
2.1. Object of Study
2.2. Basic Simulation Model
2.3. Modeling the Photoisomerization of Azobenzene
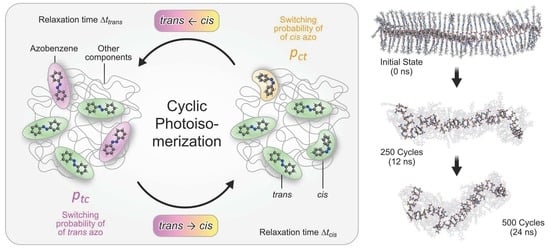
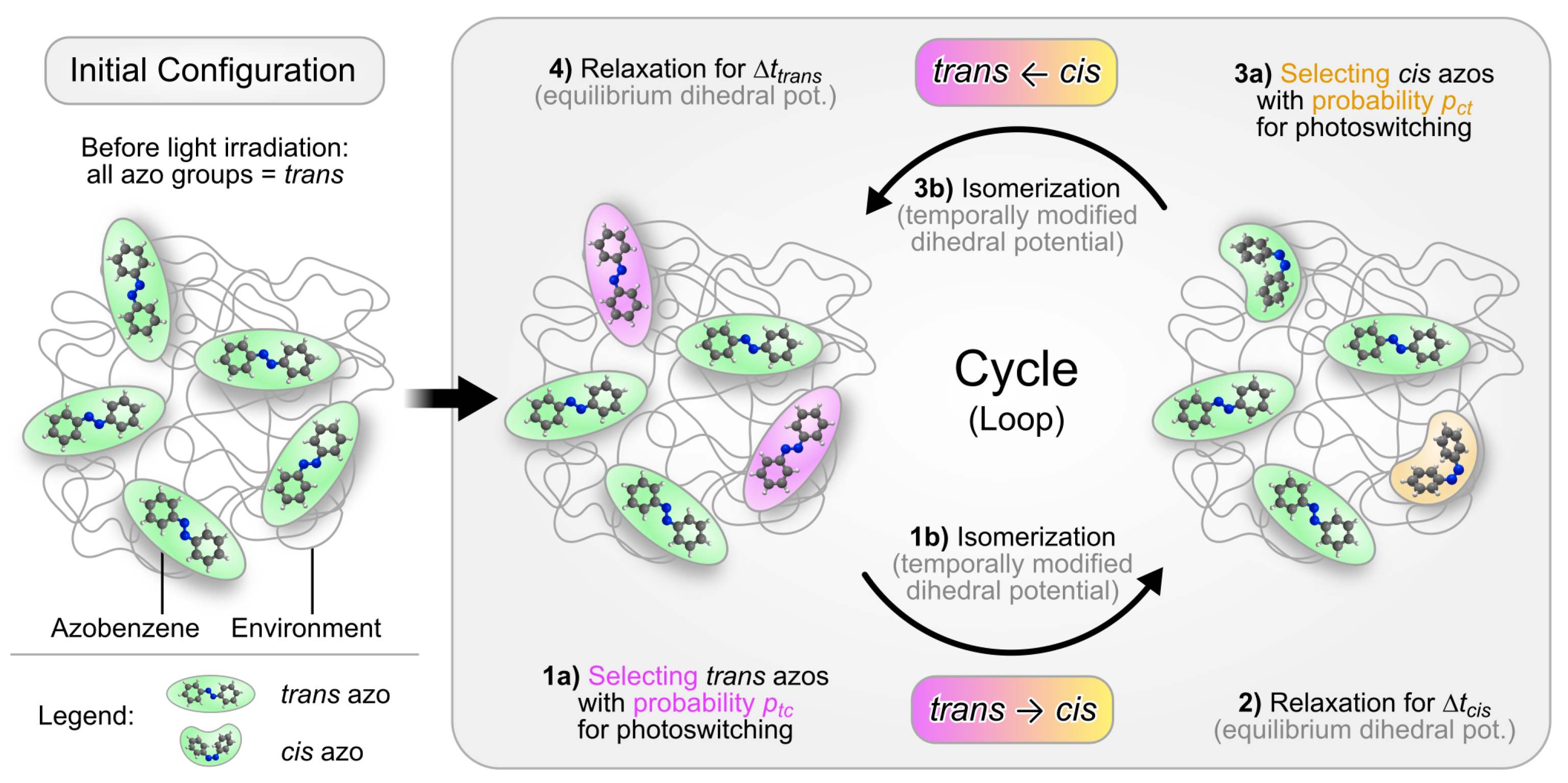
2.4. Modeling the Collective Photoisomerization Kinetics: The Cyclic Photoisomerization Model
2.4.1. Adaptation of the Model by Bedrov et al. (Bedrov Model or CPM-B)
- (a)
- At the beginning of every cycle, a predefined fraction of the trans-azo groups is randomly selected for photoswitching. A trans-azo moiety is selected with the probability This fraction represents those trans isomers, which absorb a photon and subsequently undergo photoisomerization. Note that in the original model, irradiation with linearly polarized light is simulated. To implement this, the switching probability should depend on the angle between the long axis of the azo moiety and the polarization direction of the incoming light. In particular, this probability should be proportional to [79]. The probability for the cistrans back reaction is assumed to remain angle-independent [79]. After implementing such an angle-dependence, the photo-orientation effect of azobenzene can be realized [43,79]. Here, the switching probabilities remain angle-independent since we do not simulate light of a certain polarization.
- (b)
- For the subset of azo groups now selected for switching, the photoisomerization model of Heinz et al. [42] is applied (see Section 2.3). To this end, the C-N=N-C dihedral potential of the selected azobenzene groups is temporally modified. The simulation is then continued for , allowing the isomerization reaction to complete.
- Before the simulation enters the next stage, the dihedral potential parameters of the isomerized azobenzene groups are reverted to their equilibrium values. The cis isomers remain stable because thermal relaxation back to the trans geometry is energetically hindered. The simulation then proceeds for . During this period the system can relax, now containing a mixture of trans and cis isomers.
- (a)
- Next, a fixed fraction of the cis isomers is selected for isomerization back to trans. Cis groups are selected with the probability In this implementation (CPM-B), this probability is = 100%. In other words, all cis isomers will be switched back to trans.
- (b)
- The C-N=N-C dihedral potential of the cis-azobenzene groups is temporally modified again, now using the parameters for the back reaction [42]. The simulation continues for to complete the conversion.
- Finally, the modified dihedral potentials are reset to their equilibrium settings. Now all azobenzene groups are again present as trans isomers, like in the initial stage. They remain like this for , allowing the system to relax before another iteration of the loop begins.
2.4.2. The Extended Model (CPM-E)
3. Results and Discussion
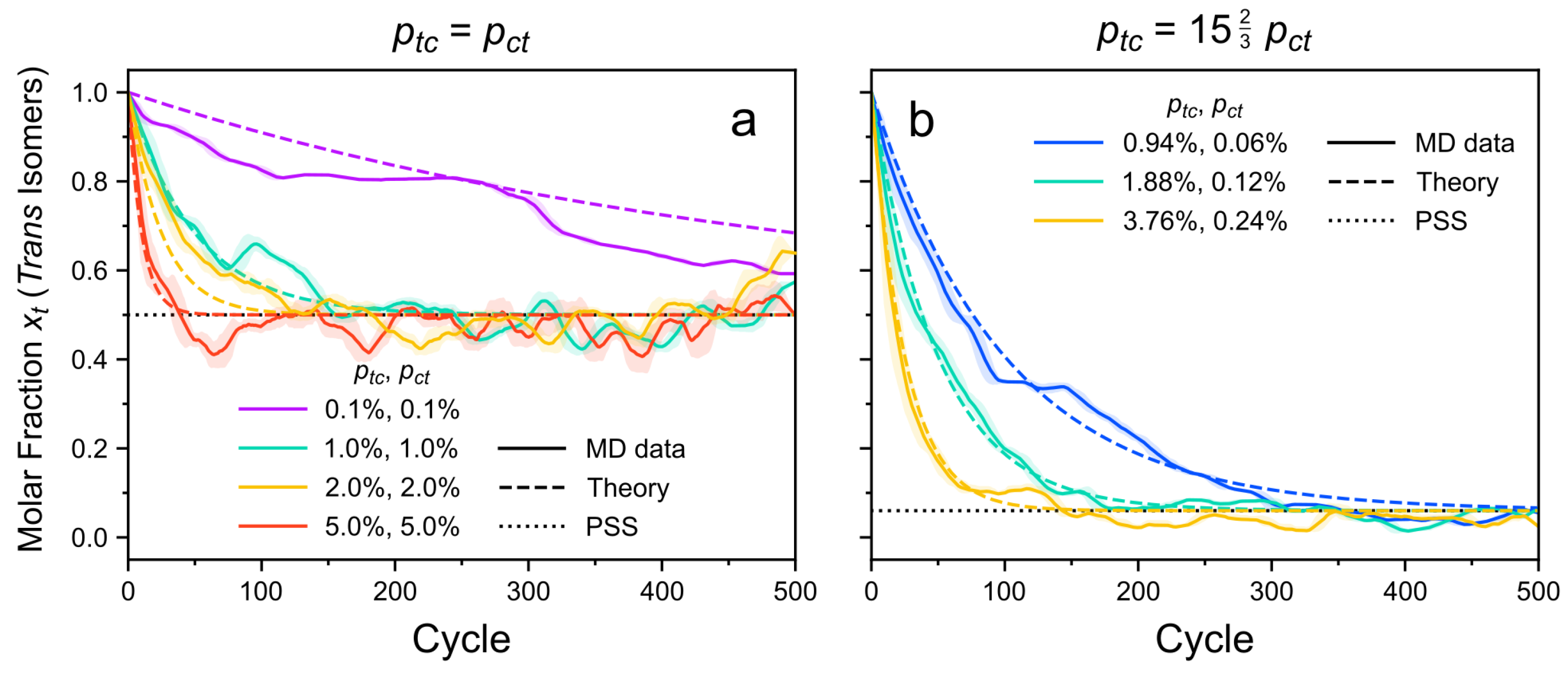
3.1. Photoisomerization Kinetics
3.1.1. Photoisomerization Kinetics in the CPM-B Approach
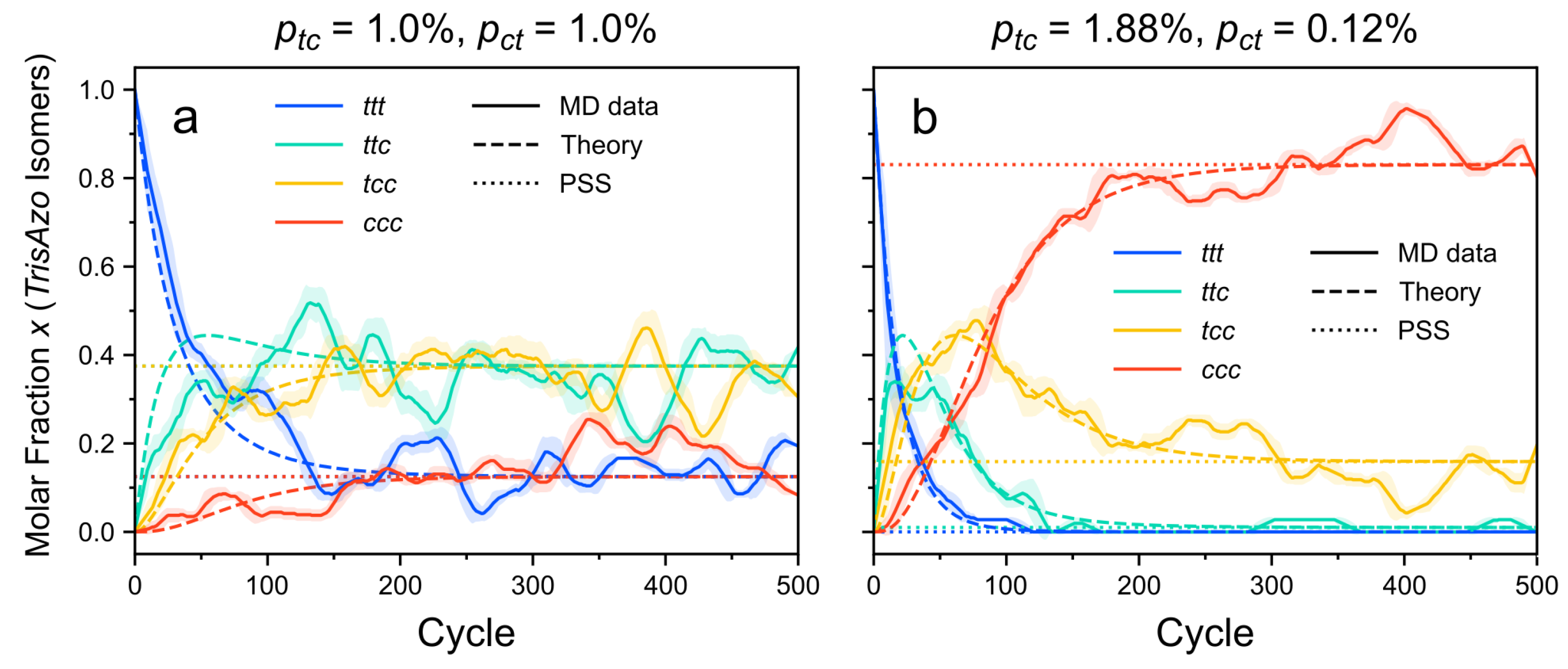
3.1.2. Photoisomerization Kinetics in the CPM-E Approach
3.1.3. Comparison of CPM-E Results with Theory
3.1.4. Comparison of CPM-E Results with Experiments
3.2. Cluster Structure before and during Light Irradiation
3.2.1. Equilibrium Structure of the Cluster (Initial State)
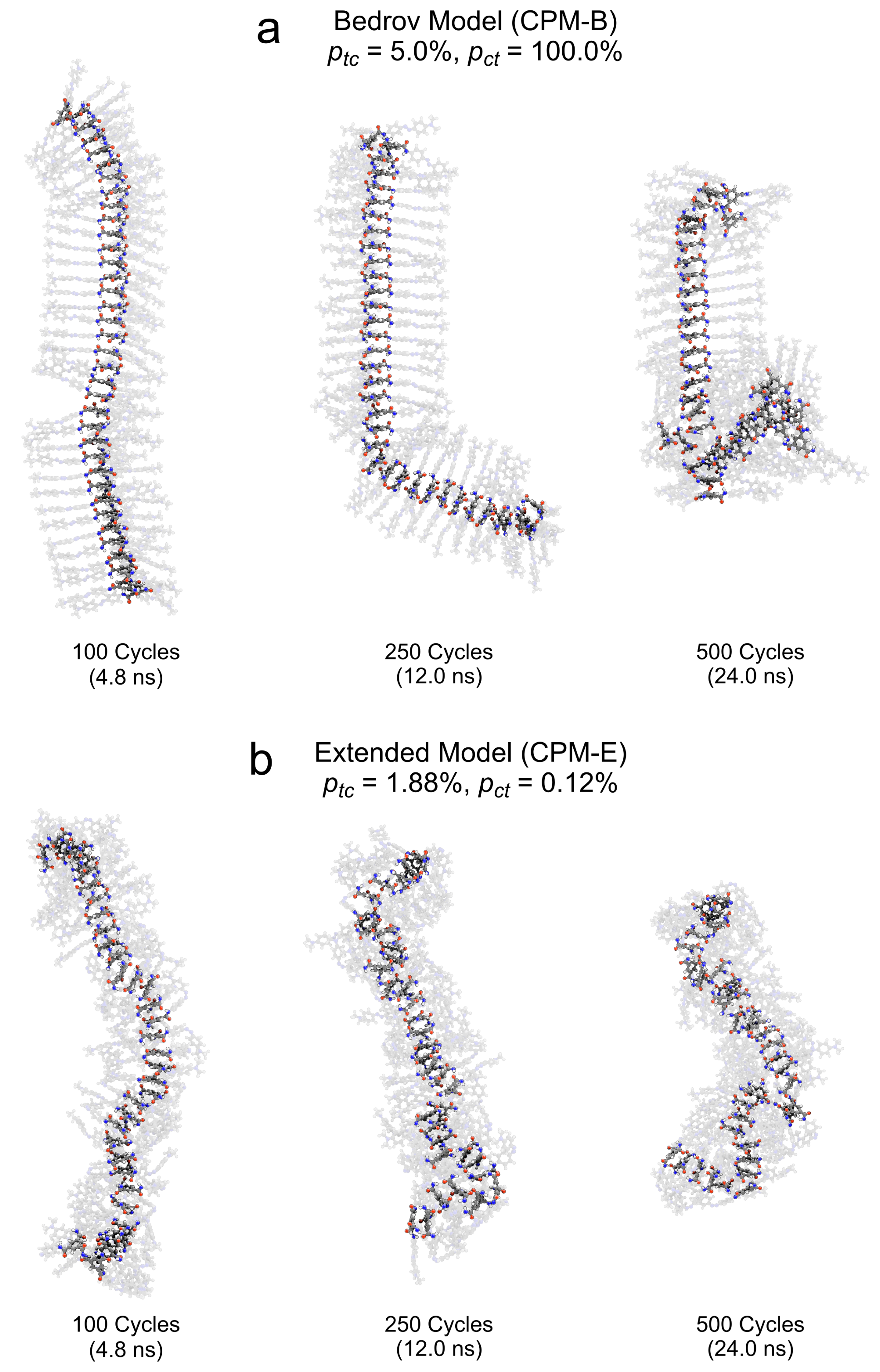
3.2.2. Cluster Structure upon Application of the CPM-B Approach
3.2.3. Cluster Structure upon Application of CPM-E Approach
3.3. Intermolecular Energy of TrisAzo Stacks before and during Light Irradiation
3.3.1. Intermolecular Energy of the Cluster in Equilibrium (Initial State)
3.3.2. Intermolecular Energy upon Application of the CPM-B Approach
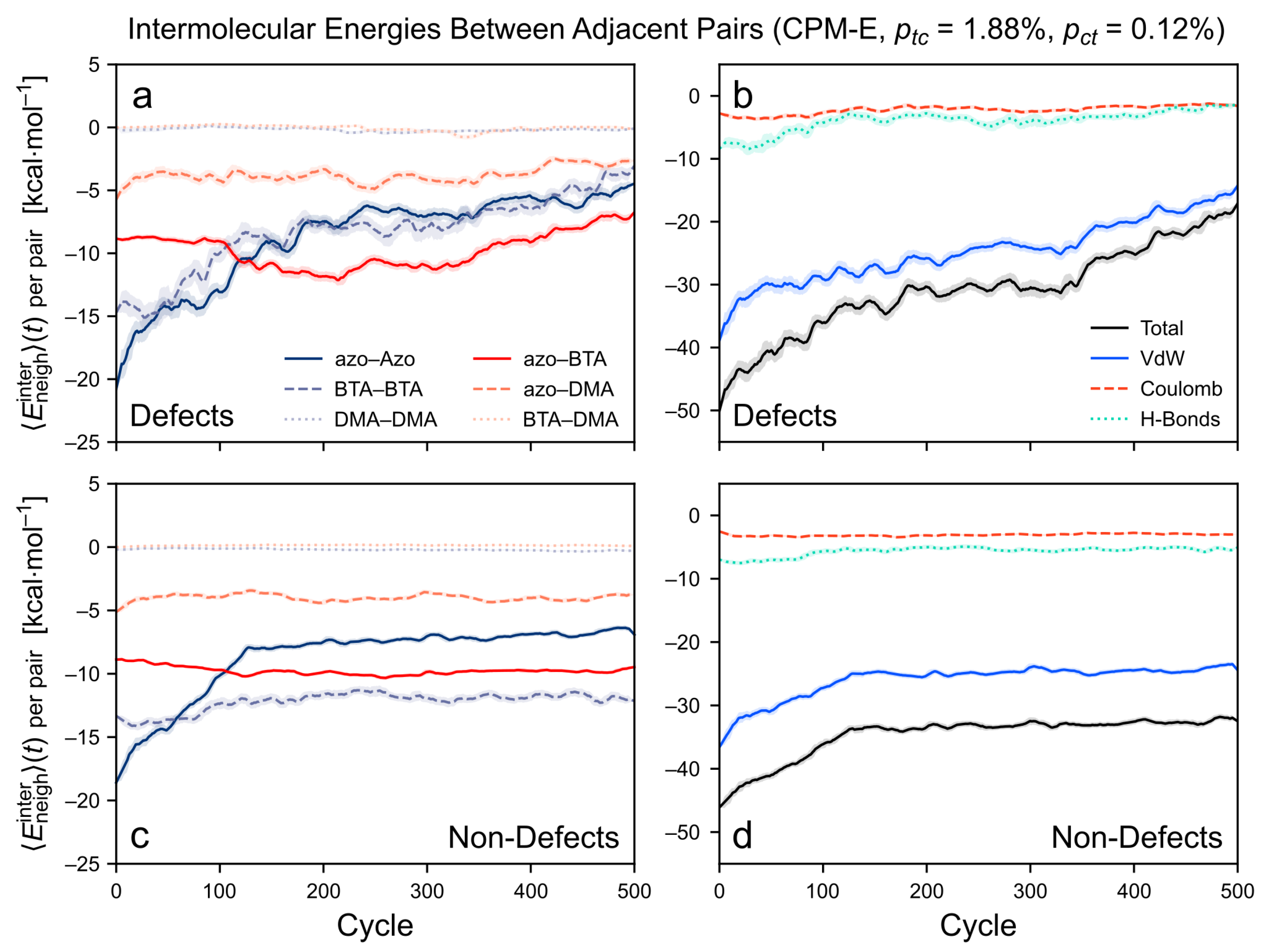
3.3.3. Intermolecular Energy upon Application of the CPM-E Approach
3.4. Mechanism of Defect Formation
The Impact of Cis-Azo Accumulation vs. Energy Dissipation via Photoswitching
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| azo | Azobenzene |
| BTA | Benzene-1,3,5-tricarboxamide |
| COM | Center of mass |
| CPM | Cyclic photoisomerization model |
| DMA | Dimethylamine / Dimethylamino group |
| DMSO | Dimethylsulfoxide |
| LAMMPS | Large-scale atomic/molecular massively parallel simulator |
| LJ | Lennard–Jones |
| MD | Molecular dynamics |
| ODE | Ordinary differential equation |
| PPPM | Particle-particle-particle-mesh Fourier-based Ewald summation method |
| PSS | Photostationary state |
| vdW | Van der Waals |
References
- Zhao, Y.; Ikeda, T. (Eds.) Smart Light-Responsive Materials: Azobenzene-Containing Polymers and Liquid Crystals; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2009. [Google Scholar] [CrossRef]
- Yagai, S.; Kitamura, A. Recent advances in photoresponsive supramolecular self-assemblies. Chem. Soc. Rev. 2008, 37, 1520–1529. [Google Scholar] [CrossRef]
- Yao, X.; Li, T.; Wang, J.; Ma, X.; Tian, H. Recent Progress in Photoswitchable Supramolecular Self-Assembling Systems. Adv. Opt. Mater. 2016, 4, 1322–1349. [Google Scholar] [CrossRef]
- Baroncini, M.; Bergamini, G. Azobenzene: A Photoactive Building Block for Supramolecular Architectures. Chem. Rec. 2017, 17, 700–712. [Google Scholar] [CrossRef]
- Zhu, M.; Zhou, H. Azobenzene-based small molecular photoswitches for protein modulation. Org. Biomol. Chem. 2018, 16, 8434–8445. [Google Scholar] [CrossRef] [PubMed]
- Rochon, P.; Batalla, E.; Natansohn, A. Optically induced surface gratings on azoaromatic polymer films. Appl. Phys. Lett. 1995, 66, 136–138. [Google Scholar] [CrossRef]
- Natansohn, A.; Rochon, P.; Ho, M.-S.; Barrett, C. Azo Polymers for Reversible Optical Storage. 6. Poly[4-[2-(methacryloyloxy)ethyl] azobenzene]. Macromolecules 1995, 28, 4179–4183. [Google Scholar] [CrossRef]
- Jelken, J.; Santer, S. Light induced reversible structuring of photosensitive polymer films. RSC Adv. 2019, 9, 20295–20305. [Google Scholar] [CrossRef] [Green Version]
- Jelken, J.; Henkel, C.; Santer, S. Solving an old puzzle: Fine structure of diffraction spots from an azo-polymer surface relief grating. Appl. Phys. B 2019, 125, 218. [Google Scholar] [CrossRef] [Green Version]
- Oscurato, S.L.; Salvatore, M.; Maddalena, P.; Ambrosio, A. From nanoscopic to macroscopic photo-driven motion in azobenzene-containing materials. Nanophotonics 2018, 7, 1387–1422. [Google Scholar] [CrossRef]
- Priimagi, A.; Shevchenko, A. Azopolymer-based micro- and nanopatterning for photonic applications. J. Polym. Sci. Part B: Polym. Phys. 2014, 52, 163–182. [Google Scholar] [CrossRef]
- Yu, Y.; Nakano, M.; Ikeda, T. Directed bending of a polymer film by light. Nature 2003, 425, 145. [Google Scholar] [CrossRef] [PubMed]
- Koerner, H.; White, T.J.; Tabiryan, N.V.; Bunning, T.J.; Vaia, R.A. Photogenerating work from polymers. Mater. Today 2008, 11, 34–42. [Google Scholar] [CrossRef]
- White, T.J.; Broer, D.J. Programmable and adaptive mechanics with liquid crystal polymer networks and elastomers. Nat. Mater. 2015, 14, 1087–1098. [Google Scholar] [CrossRef]
- Rau, H. Photoisomerization of Azobenzenes. In Photochemistry and Photophysics; Rabek, J.F., Ed.; CRC Press: Boca Raton, FL, USA, 1990; Volume II, Chapter 4; pp. 119–141. [Google Scholar]
- Bandara, H.M.D.; Burdette, S.C. Photoisomerization in different classes of azobenzene. Chem. Soc. Rev. 2012, 41, 1809–1825. [Google Scholar] [CrossRef] [PubMed]
- Fihey, A.; Perrier, A.; Browne, W.R.; Jacquemin, D. Multiphotochromic Molecular Systems. Chem. Soc. Rev. 2015, 44, 3719–3759. [Google Scholar] [CrossRef]
- Galanti, A.; Santoro, J.; Mannancherry, R.; Duez, Q.; Diez-Cabanes, V.; Valášek, M.; De Winter, J.; Cornil, J.; Gerbaux, P.; Mayor, M.; et al. A New Class of Rigid Multi(azobenzene) Switches Featuring Electronic Decoupling: Unravelling the Isomerization in Individual Photochromes. J. Am. Chem. Soc. 2019, 141, 9273–9283. [Google Scholar] [CrossRef] [Green Version]
- Gupta, D.; Gaur, A.K.; Kumar, P.; Kumar, H.; Mahadevan, A.; Devi, S.; Roy, S.; Venkataramani, S. Tuning of Bistability, Thermal Stability of the Metastable States, and Application Prospects in the C3-Symmetric Designs of Multiple Azo(hetero)arenes Systems. Chem. Eur. J. 2021, 27, 3463–3472. [Google Scholar] [CrossRef]
- Ichimura, K. Photoalignment of Liquid-Crystal Systems. Chem. Rev. 2000, 100, 1847–1874. [Google Scholar] [CrossRef] [PubMed]
- Loebner, S.; Lomadze, N.; Kopyshev, A.; Koch, M.; Guskova, O.; Saphiannikova, M.; Santer, S. Light-Induced Deformation of Azobenzene-Containing Colloidal Spheres: Calculation and Measurement of Opto-Mechanical Stresses. J. Phys. Chem. B 2018, 122, 2001–2009. [Google Scholar] [CrossRef]
- Yadav, B.; Domurath, J.; Kim, K.; Lee, S.; Saphiannikova, M. Orientation Approach to Directional Photodeformations in Glassy Side-Chain Azopolymers. J. Phys. Chem. B 2019, 123, 3337–3347. [Google Scholar] [CrossRef]
- Yadav, B.; Domurath, J.; Saphiannikova, M. Modeling of Stripe Patterns in Photosensitive Azopolymers. Polymers 2020, 12, 735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Oh, S.; Lee, J.; Malpani, Y.; Jung, Y.-S.; Kang, B.; Lee, J.Y.; Ozasa, K.; Isoshima, T.; Lee, S.Y.; et al. Stimulus-Responsive Azobenzene Supramolecules: Fibers, Gels, and Hollow Spheres. Langmuir 2013, 29, 5869–5877. [Google Scholar] [CrossRef]
- Malpani, Y.R.; Oh, S.; Lee, S.; Jung, Y.-S.; Kim, J.-M. Photoinduced Phase Transition of Azobenzene-Coupled Benzenetricarboxamide. Bull. Korean Chem. Soc. 2014, 35, 2563–2566. [Google Scholar] [CrossRef] [Green Version]
- Natansohn, A.; Rochon, P. Photoinduced Motions in Azo-Containing Polymers. Chem. Rev. 2002, 102, 4139–4176. [Google Scholar] [CrossRef]
- Yagai, S.; Karatsu, T.; Kitamura, A. Photocontrollable Self-Assembly. Chem. Eur. J. 2005, 11, 4054–4063. [Google Scholar] [CrossRef]
- Bochicchio, D.; Pavan, G.M. Molecular modelling of supramolecular polymers. Adv. Phys.: X 2018, 3, 1436408. [Google Scholar] [CrossRef] [Green Version]
- Koch, M.; Saphiannikova, M.; Guskova, O. Do Columns of Azobenzene Stars Disassemble under Light Illumination? Langmuir 2019, 35, 14659–14669. [Google Scholar] [CrossRef] [PubMed]
- Bejagam, K.K.; Fiorin, G.; Klein, M.L.; Balasubramanian, S. Supramolecular Polymerization of Benzene-1,3,5-tricarboxamide: A Molecular Dynamics Simulation Study. J. Phys. Chem. B 2014, 118, 5218–5228. [Google Scholar] [CrossRef]
- Bejagam, K.K.; Kulkarni, C.; George, S.J.; Balasubramanian, S. External electric field reverses helical handedness of a supramolecular columnar stack. Chem. Commun. 2015, 51, 16049–16052. [Google Scholar] [CrossRef]
- Cantekin, S.; de Greef, T.F.A.; Palmans, A.R.A. Benzene-1,3,5-tricarboxamide: A versatile ordering moiety for supramolecular chemistry. Chem. Soc. Rev. 2012, 41, 6125–6137. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.-J.; Kim, D.-Y.; Park, M.; Yoon, W.-J.; Lee, Y.; Hwang, J.-K.; Chiang, Y.-W.; Kuo, S.-W.; Hsu, C.-H.; Jeong, K.U. Self-Assembled Hierarchical Superstructures from the Benzene-1,3,5-Tricarboxamide Supramolecules for the Fabrication of Remote-Controllable Actuating and Rewritable Films. ACS Appl. Mater. Interfaces 2016, 8, 9490–9498. [Google Scholar] [CrossRef] [PubMed]
- Garzoni, M.; Baker, M.B.; Leenders, C.M.A.; Voets, I.K.; Albertazzi, L.; Palmans, A.R.A.; Meijer, E.W.; Pavan, G.M. Effect of H-Bonding on Order Amplification in the Growth of a Supramolecular Polymer in Water. J. Am. Chem. Soc. 2016, 138, 13985–13995. [Google Scholar] [CrossRef] [PubMed]
- Banach, E.; Invernizzi, C.; Baudin, M.; Neier, R.; Carnevale, D. Columnar self-assembly of N,N′,N″-trihexylbenzene-1,3,5-tricarboxamides investigated by means of NMR spectroscopy and computational methods in solution and the solid state. Phys. Chem. Chem. Phys. 2017, 19, 5525–5539. [Google Scholar] [CrossRef] [Green Version]
- Bochicchio, D.; Pavan, G.M. From Cooperative Self-Assembly to Water-Soluble Supramolecular Polymers Using Coarse-Grained Simulations. ACS Nano 2017, 11, 1000–1011. [Google Scholar] [CrossRef]
- Lee, J.; Oh, S.; Pyo, J.; Kim, J.-M.; Je, J.H. A light-driven supramolecular nanowire actuator. Nanoscale 2015, 7, 6457–6461. [Google Scholar] [CrossRef] [Green Version]
- Devi, S.; Bala, I.; Gupta, S.P.; Kumar, P.; Pal, S.K.; Venkataramani, S. Reversibly photoswitchable alkoxy azobenzenes connected benzenetricarboxamide discotic liquid crystals with perpetual long range columnar assembly. Org. Biomol. Chem. 2019, 17, 1947–1954. [Google Scholar] [CrossRef]
- Bochicchio, D.; Kwangmettatam, S.; Kudernac, T.; Pavan, G.M. How Defects Control the Out-of-Equilibrium Dissipative Evolution of a Supramolecular Tubule. ACS Nano 2019, 13, 4322–4334. [Google Scholar] [CrossRef]
- Savchenko, V.; Koch, M.; Pavlov, A.S.; Saphiannikova, M.; Guskova, O. Stacks of Azobenzene Stars: Self-Assembly Scenario and Stabilising Forces Quantified in Computer Modelling. Molecules 2019, 24, 4387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koch, M.; Saphiannikova, M.; Guskova, O. Columnar Aggregates of Azobenzene Stars: Exploring Intermolecular Interactions, Structure, and Stability in Atomistic Simulations. Molecules 2021, 26, 7598. [Google Scholar] [CrossRef]
- Heinz, H.; Vaia, R.A.; Koerner, H.; Farmer, B.L. Photoisomerization of Azobenzene Grafted to Layered Silicates: Simulation and Experimental Challenges. Chem. Mater. 2008, 20, 6444–6456. [Google Scholar] [CrossRef]
- Bedrov, D.; Hooper, J.B.; Glaser, M.A.; Clark, N.A. Photoinduced and Thermal Relaxation in Surface-Grafted Azobenzene-Based Monolayers: A Molecular Dynamics Simulation Study. Langmuir 2016, 32, 4004–4015. [Google Scholar] [CrossRef] [PubMed]
- Teboul, V.; Saiddine, M.; Nunzi, J.-M. Isomerization-Induced Dynamic Heterogeneity in a Glass Former below and above Tg. Phys. Rev. Lett. 2009, 103, 265701. [Google Scholar] [CrossRef] [Green Version]
- Teboul, V.; Saiddine, M.; Nunzi, J.-M.; Accary, J.-B. An isomerization-induced cage-breaking process in a molecular glass former below Tg. J. Chem. Phys. 2011, 134, 114517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teboul, V. Stimuli Thresholds for Isomerization-Induced Molecular Motions in Azobenzene-Containing Materials. J. Phys. Chem. B 2015, 119, 3854–3859. [Google Scholar] [CrossRef] [Green Version]
- Koch, M. The Influence of Light on a Three-Arm Azobenzene Star: A Computational Study. Ph.D. Dissertation, Technische Universität Dresden, Dresden, Germany, 2022. [Google Scholar]
- Koch, M.; Saphiannikova, M.; Santer, S.; Guskova, O. Photoisomers of Azobenzene Star with a Flat Core: Theoretical Insights into Multiple States from DFT and MD Perspective. J. Phys. Chem. B 2017, 121, 8854–8867. [Google Scholar] [CrossRef]
- Blender Online Community. Blender—A 3D Modelling and Rendering Package; Blender Foundation, Stichting Blender Foundation: Amsterdam, The Netherlands, 2018. [Google Scholar]
- Durrant, J.D. BlendMol: Advanced macromolecular visualization in Blender. Bioinformatics 2018, 35, 2323–2325. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Thompson, A.P.; Aktulga, H.M.; Berger, R.; Bolintineanu, D.S.; Brown, W.M.; Crozier, P.S.; in’t Veld, P.J.; Kohlmeyer, A.; Moore, S.G.; Nguyen, T.D.; et al. LAMMPS—A flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales. Comput. Phys. Commun. 2022, 271, 108171. [Google Scholar] [CrossRef]
- Mayo, S.L.; Olafson, B.D.; Goddard, W.A. DREIDING: A Generic Force Field for Molecular Simulations. J. Phys. Chem. 1990, 94, 8897–8909. [Google Scholar] [CrossRef]
- Gasteiger, J.; Marsili, M. Iterative partial equalization of orbital electronegativity—A rapid access to atomic charges. Tetrahedron 1980, 36, 3219–3228. [Google Scholar] [CrossRef]
- BIOVIA, Dassault Systèmes. BIOVIA Materials Studio 8.0.100.21; Dassault Systèmes: San Diego, CA, USA, 2014. [Google Scholar]
- Berendsen, H.J.C.; Grigera, J.R.; Straatsma, T.P. The Missing Term in Effective Pair Potentials. J. Phys. Chem. 1987, 91, 6269–6271. [Google Scholar] [CrossRef]
- Jewett, A.I.; Stelter, D.; Lambert, J.; Saladi, S.M.; Roscioni, O.M.; Ricci, M.; Autin, L.; Maritan, M.; Bashusqeh, S.M.; Keyes, T.; et al. Moltemplate: A Tool for Coarse-Grained Modeling of Complex Biological Matter and Soft Condensed Matter Physics. J. Mol. Biol. 2021, 433, 166841. [Google Scholar] [CrossRef]
- Martínez, J.M.; Martínez, L. Packing Optimization for Automated Generation of Complex System’s Initial Configurations for Molecular Dynamics and Docking. J. Comput. Chem. 2003, 24, 819–825. [Google Scholar] [CrossRef]
- Martínez, L.; Andrade, R.; Birgin, E.G.; Martínez, J.M. PACKMOL: A Package for Building Initial Configurations for Molecular Dynamics Simulations. J. Comput. Chem. 2009, 30, 2157–2164. [Google Scholar] [CrossRef]
- Lorentz, H.A. Ueber die Anwendung des Satzes vom Virial in der kinetischen Theorie der Gase. Ann. Phys. 1881, 248, 127–136. [Google Scholar] [CrossRef] [Green Version]
- Berthelot, D. Sur le mélange des gaz. C. R. Hebd. Séances Acad. Sci. 1898, 126, 1703–1706. [Google Scholar]
- Eastwood, J.W.; Hockney, R.W.; Lawrence, D.N. P3M3DP—The three-dimensional periodic particle-particle/particle-mesh program. Comput. Phys. Commun. 1980, 19, 215–261. [Google Scholar] [CrossRef]
- Hockney, R.W.; Eastwood, J.W. Computer Simulation Using Particles; CRC Press: Boca Raton, FL, USA, 1988. [Google Scholar]
- Ryckaert, J.P.; Ciccotti, G.; Berendsen, H.J.C. Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of n-Alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Tiberio, G.; Muccioli, L.; Berardi, R.; Zannoni, C. How Does the Trans–Cis Photoisomerization of Azobenzene Take Place in Organic Solvents? ChemPhysChem 2010, 11, 1018–1028. [Google Scholar] [CrossRef]
- Li, Y.; Hartke, B. Approximate photochemical dynamics of azobenzene with reactive force fields. J. Chem. Phys. 2013, 139, 224303. [Google Scholar] [CrossRef]
- Liu, Z.; Ma, J. Effects of External Electric Field and Self-Aggregations on Conformational Transition and Optical Properties of Azobenzene-Based D-π-A Type Chromophore in THF Solution. J. Phys. Chem. A 2011, 115, 10136–10145. [Google Scholar] [CrossRef]
- Böckmann, M.; Marx, D.; Peter, C.; Site, L.D.; Kremer, K.; Doltsinis, N.L. Multiscale modelling of mesoscopic phenomena triggered by quantum events: Light-driven azo-materials and beyond. Phys. Chem. Chem. Phys. 2011, 13, 7604–7621. [Google Scholar] [CrossRef]
- Böckmann, M.; Braun, S.; Doltsinis, N.L.; Marx, D. Mimicking photoisomerisation of azo-materials by a force field switch derived from nonadiabatic ab initio simulations: Application to photoswitchable helical foldamers in solution. J. Chem. Phys. 2013, 139, 084108. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.; Wen, J.; Ma, J. Reactive molecular dynamics simulations of switching processes of azobenzene-based monolayer on surface. J. Chem. Phys. 2013, 139, 014706. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.; Wen, J.; Ma, J. Dynamic simulations of stimuli-responsive switching of azobenzene derivatives in self-assembled monolayers: Reactive rotation potential and switching functions. Mol. Simul. 2015, 41, 28–42. [Google Scholar] [CrossRef]
- Choi, J.; Chung, H.; Yun, J.-H.; Cho, M. Molecular Dynamics Study on the Photothermal Actuation of a Glassy Photoresponsive Polymer Reinforced with Gold Nanoparticles with Size Effect. ACS Appl. Mater. Interfaces 2016, 8, 24008–24024. [Google Scholar] [CrossRef]
- Xue, X.-G.; Zhao, L.; Lu, Z.-Y.; Li, M.-H.; Li, Z.-S. Molecular dynamics simulation study on the isomerization and molecular orientation of liquid crystals formed by azobenzene and (1-cyclohexenyl)phenyldiazene. Phys. Chem. Chem. Phys. 2011, 13, 11951–11957. [Google Scholar] [CrossRef]
- Choi, J.; Chung, H.; Yun, J.-H.; Cho, M. Photo-isomerization effect of the azobenzene chain on the opto-mechanical behavior of nematic polymer: A molecular dynamics study. Appl. Phys. Lett. 2014, 105, 221906. [Google Scholar] [CrossRef]
- Chung, H.; Choi, J.; Yun, J.-H.; Cho, M. Nonlinear photomechanics of nematic networks: Upscaling microscopic behaviour to macroscopic deformation. Sci. Rep. 2016, 6, 20026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moon, J.; Kim, B.; Choi, J.; Cho, M. Multiscale Study of the Relationship between Photoisomerization and Mechanical Behavior of Azo-Polymer Based on the Coarse-Grained Molecular Dynamics Simulation. Macromolecules 2019, 52, 2033–2049. [Google Scholar] [CrossRef]
- Mechau, N.; Saphiannikova, M.; Neher, D. Dielectric and Mechanical Properties of Azobenzene Polymer Layers under Visible and Ultraviolet Irradiation. Macromolecules 2005, 38, 3894–3902. [Google Scholar] [CrossRef]
- Ilnytskyi, J.M.; Toshchevikov, V.; Saphiannikova, M. Modeling of the photo-induced stress in azobenzene polymers by combining theory and computer simulations. Soft Matter 2019, 15, 9894–9908. [Google Scholar] [CrossRef] [PubMed]
- Ilnytskyi, J.M.; Saphiannikova, M. Reorientation Dynamics of Chromophores in Photosensitive Polymers by Means of Coarse-Grained Modeling. ChemPhysChem 2015, 16, 3180–3189. [Google Scholar] [CrossRef]
- Böckmann, M.; Doltsinis, N.L. Towards understanding photomigration: Insights from atomistic simulations of azopolymer films explicitly including light-induced isomerization dynamics. J. Chem. Phys. 2016, 145, 154701. [Google Scholar] [CrossRef]
- Teboul, V.; Accary, J.B. Formation of Surface Relief Gratings: Effect of the Density of the Host Material. J. Phys. Chem. B 2012, 116, 12621–12625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kind, J.; Kaltschnee, L.; Leyendecker, M.; Thiele, C.M. Distinction of trans–cis photoisomers with comparable optical properties in multiple-state photochromic systems – examining a molecule with three azobenzenes via in situ irradiation NMR spectroscopy. Chem. Commun. 2016, 52, 12506–12509. [Google Scholar] [CrossRef]
- Toshchevikov, V.; Ilnytskyi, J.; Saphiannikova, M. Photoisomerization Kinetics and Mechanical Stress in Azobenzene-Containing Materials. J. Phys. Chem. Lett. 2017, 8, 1094–1098. [Google Scholar] [CrossRef]
- Gerth, M.; Berrocal, J.A.; Bochicchio, D.; Pavan, G.M.; Voets, I.K. Discordant Supramolecular Fibres Reversibly Depolymerised by Temperature and Light. Chem. Eur. J. 2021, 27, 1829–1838. [Google Scholar] [CrossRef]












Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koch, M.; Saphiannikova, M.; Guskova, O. Cyclic Photoisomerization of Azobenzene in Atomistic Simulations: Modeling the Effect of Light on Columnar Aggregates of Azo Stars. Molecules 2021, 26, 7674. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26247674
Koch M, Saphiannikova M, Guskova O. Cyclic Photoisomerization of Azobenzene in Atomistic Simulations: Modeling the Effect of Light on Columnar Aggregates of Azo Stars. Molecules. 2021; 26(24):7674. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26247674
Chicago/Turabian StyleKoch, Markus, Marina Saphiannikova, and Olga Guskova. 2021. "Cyclic Photoisomerization of Azobenzene in Atomistic Simulations: Modeling the Effect of Light on Columnar Aggregates of Azo Stars" Molecules 26, no. 24: 7674. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26247674