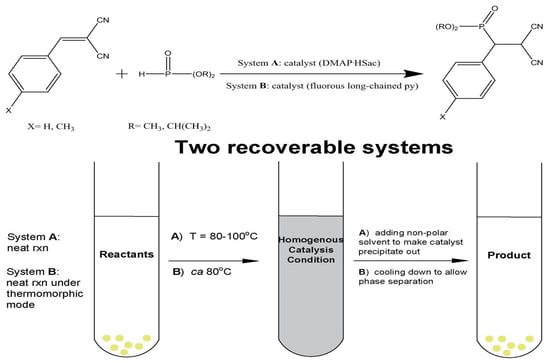
2.1.2. Recoverable DMAP·HSac-Catalyzed Phospha-Michael Addition Reaction
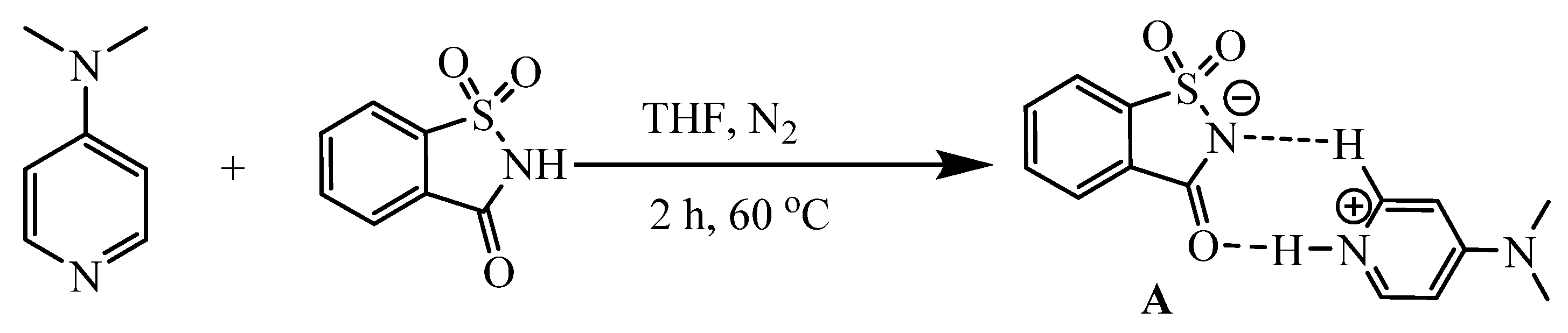
The DMAP·HSac adduct
A was then examined for the following neat phospha-Michel addition reactions, where
A catalyzed addition of alkyl (R group) substituted diorganophosphite compounds [
1a (R= isopropyl);
1b (R= methyl)] and tris(organo)phosphite compound [
2 (R = ethyl)] with 2-benzylidinemelanonitrile-type substrates [
3 (X = H);
4 (X = CH
3)] as indicated in
Scheme 2 and
Scheme 3.
- a.
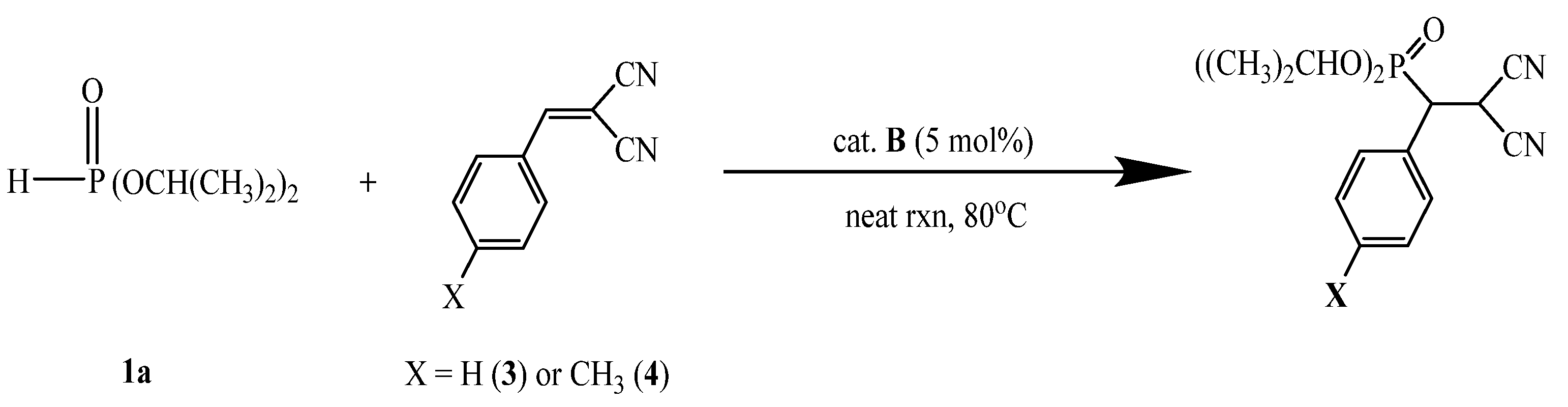
A-catalyzed phospha-Michael addition of diisopropyl phosphite (1a) and benzylidenemalononitrile (3)
As shown in
Scheme 2, the DMAP·HSac (
A)-catalyzed phospha-Michael addition reaction of diisopropyl phosphite (
1a) with 2-benzylidenemalononitrile (
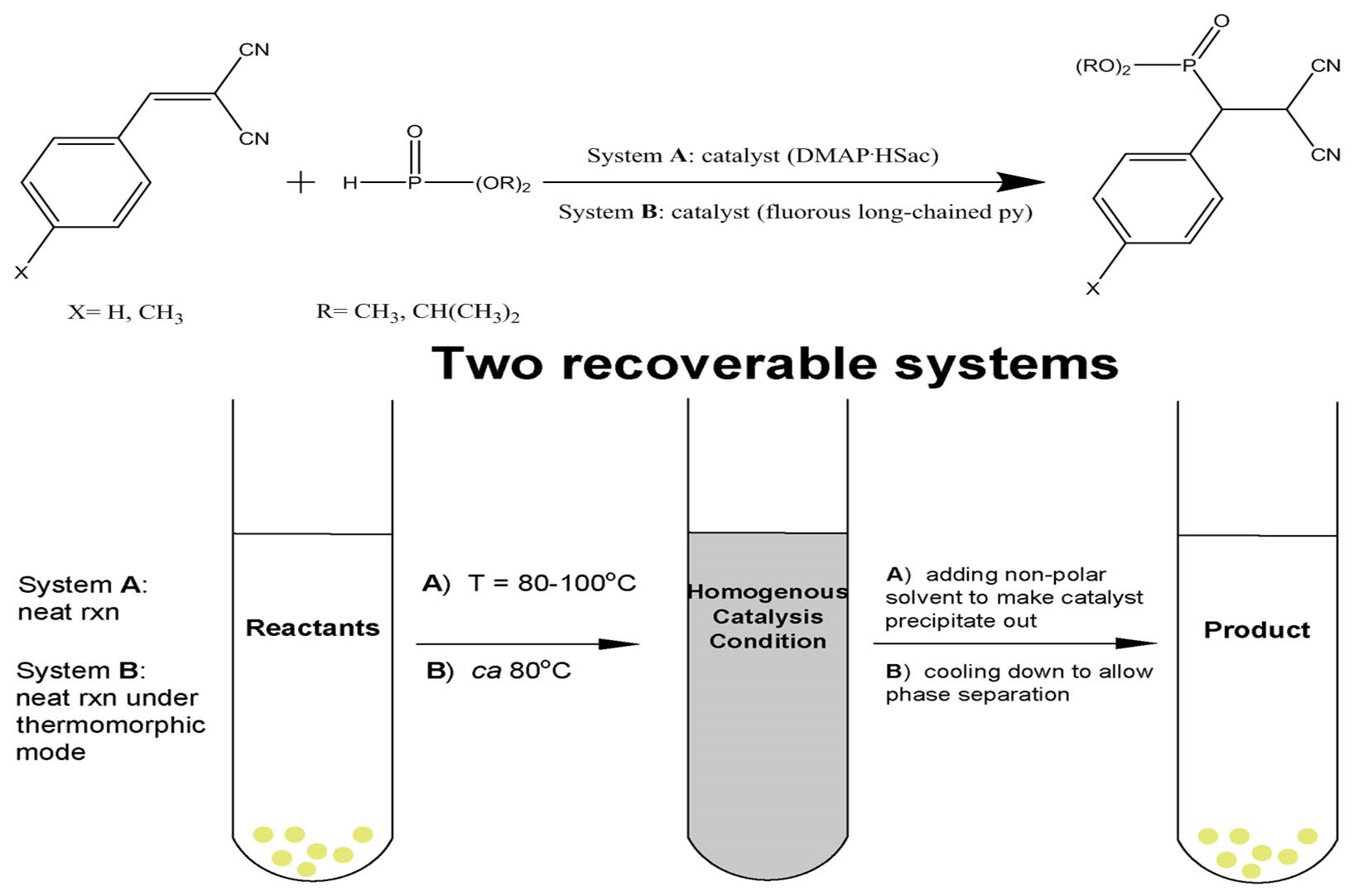
3) was selected to demonstrate the feasibility of recycling usage with
A as a catalyst using a solvent free method, at 80 °C mainly for 1 h. In this time, the reaction was successfully carried out to afford the product in quantitative conversions, as shown in
Scheme 2 and
Table 1. At the end of each cycle, the product mixtures were cooled to room temperature, and non-polar solvent (e.g., ether) was added to the reaction tube and centrifuged, and the catalyst was recovered by decantation. The recovered catalyst
A was dried under vacuum and proceeded to the next cycle. The products were quantified with GC/MS analysis by comparison to internal standard (anisole). As shown in
Table 1,
A-catalyzed phospha-Michael addition of
1a with
3 could give rise to good yield and recycling results for a total of eight times without using any solvent.
The phospha-Michel addition reaction of
1a with
3 showed high yield and good recycling result for eight cycles by using 5 mol% catalyst at 80 °C. By further lowering the catalytic loading to 1 mol% at 100 °C, a higher yield was still obtained within 1 h (see
Table 2). Additionally, catalyst
A, which is thermally robust and is stable even when temperature is higher than 100 °C, was effectively recycled for the increased number of cycles (16 cycles) almost without a loss in catalytic activity. The average yield for all the consecutive runs was 99%, which clearly demonstrates the practical reusability of this catalyst. Thus, it can be said that catalyst
A showed a robust catalytic activity with a TON =
ca 100 and a TOF =
ca 100 and also with excellent recovery and good thermal stability.
- b.
A-catalyzed phospha-Michael addition of diisopropyl phosphite (1a) with 2-(4-methylbenzylidene)malononitrile (4)
The catalyst
A -catalyzed phospha-Michael addition reaction of diisopropyl phosphite (
1a) with 2-(4-methylbenzylide)nemalononitrile (
4) proceeded with a good yield at 80 °C in 2.5–3.5 h. However, the introduction of the electron donating group (X = CH
3) into the benzene ring of compound
4 reduced the rate of the reaction, leading to increased reaction time (
Table 3) compared to the unsubstituted one (
3, X = H).
- c.
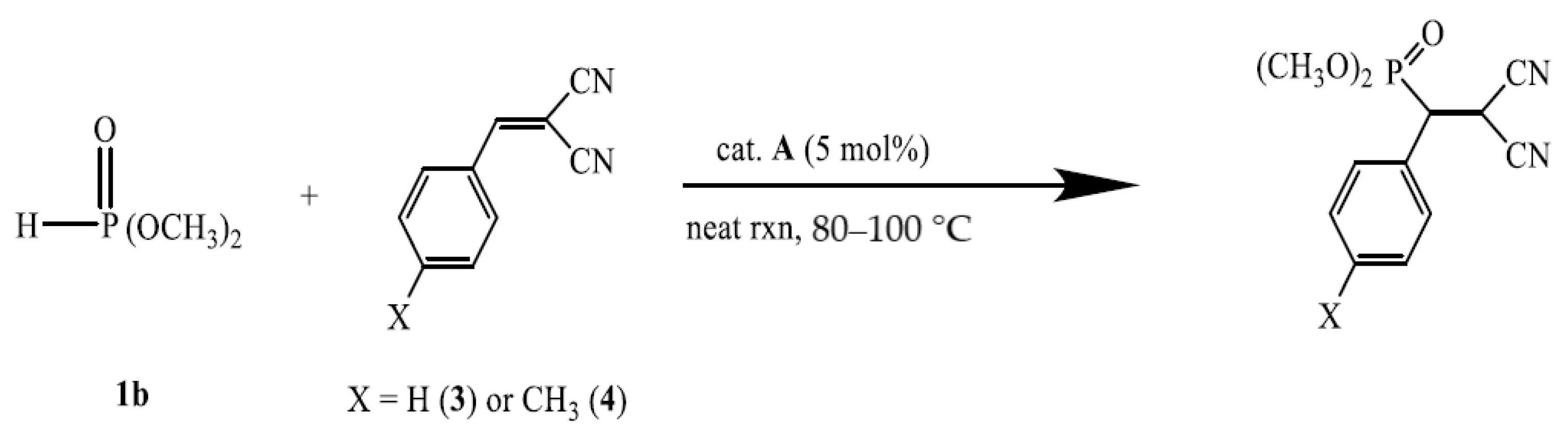
A-catalyzed phospha-Michel addition of dimethyl phosphite (1b) and 2-benzylidenemalononitrile (3)
Table 4 shows the result of the reactivity and recycling of the
A-catalyzed phospha-Michael addition reaction of dimethyl phosphite (
1b) with 2-benzylidenemalononitrile (
3). The results of
Table 1 show a slightly shorter reaction time at later stages of recycling than those of
Table 4. The change of the isopropyl group to methyl group on the phosphite substrate (see
Scheme 2 and
Scheme 3) did not show much change in the rate of the reaction. Thus, this change does not have a significant effect on the nucleophilicity of the phosphite substrate.
- d.
A-catalyzed phospha-Michael addition of dimethyl phosphite (1b) and 2-(4-methylbenzylidene)malononitrile (4)
In
Table 5, the recycling results of the
A-catalyzed phospha-Michael addition of dimethyl phosphite (
1b) with 2-(4-methylbenzylidene)malononitrile (
4) are presented. This reaction proceeded with good yield, mainly for 2 h at 80–100 °C. However, the introduction of the electron donating group (X = CH
3) into the malononitrile substrate (
4, X = CH
3) reduced the rate of the reaction, leading to an increased reaction time and temperature (
Table 5) compared to the unsubstituted substrate (
3, X = H).
- e.
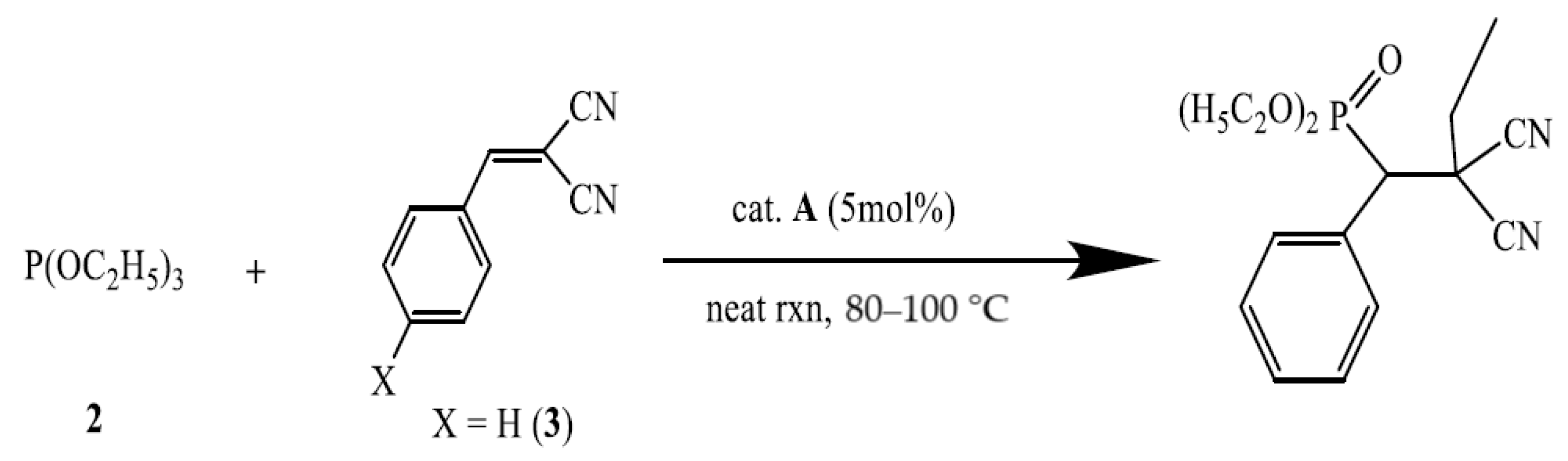
A-catalyzed phospha-Michael addition of triethyl phosphite (2) and 2-benzylidenemalononitrile (3)
As shown in
Table 6, the
A-catalyzed phospha-Michael addition of triethyl phosphite (
2) with 2-benzylidenemalononitrile (
3) demonstrated good product yield and catalytic recovery in 1 h for the first four cycles. However, it was found that the catalytic performance of the recovered cat.
A slightly diminished after each step to the 8
th cycle. In
Scheme 4, the different kind of phosphite (
2) used in this reaction is seen to yield an alkyl substituted product.
In summary, in the
A-catalyzed phospha-Michel addition of phosphite reagents (
1a, 1b or
2) with 2-benzylidenemalononitrile-type substrates (
3 or
4), the methyl, ethyl and isopropyl substituents of the phosphite did not show a significant difference in their yields and reaction times. However, after several recycles, the triethyl substituted phosphite did not maintain its reaction speed as the previous cycles (see
Table 6). Additionally, for the phosphorus-acceptor saturated bond containing 2-benzylidenemalononitrile-type substrates (
3 or
4), the unsubstituted compound
3 showed a shorter reaction time than the CH
3 substituted compound
4. Here, the electron releasing CH
3 group was found to retard the speed of the reaction. Hosseini-Sarvari and Etemad reported a nanosized zinc oxide catalyzed similar reaction by using [P(O)(OEt)
2] phosphite substrate showing a similar trend of shorter reaction time for the unsubstituted malononitrile substrate (2.5 h) than the methyl substituent (5 h) [
23,
25].
For the base
A-catalyzed reactions of diorganophosphite compounds (
1a or
1b) with 2-benzylidenemalononitrile-type substrates (
3 or
4), the reactions followed the cited reaction mechanism [
39,
40], demonestrating the base assisted P-H bond cleavage and adding the H and P-containing moiety into the two sides of the double bond (see
Figure S1 in SM). However, the
A-catalyzed similar reaction of tris(organo)phosphite (
2) with 2-benzylidenemalononitrile (
3) followed a different mechanism through the O-C bond cleavage of one -OC
2H
5 group to add the phosphonate group [P(O)(O C
2H
5)
2] and the ethyl groups into the two sides of the double bond (
Scheme 4).
2.1.3. Kinetic Study of A-Catalyzed Phospha-Michael Addition Reaction of Diisopropyl Phosphite (1a) with Benzylidenemalononitrile (3)
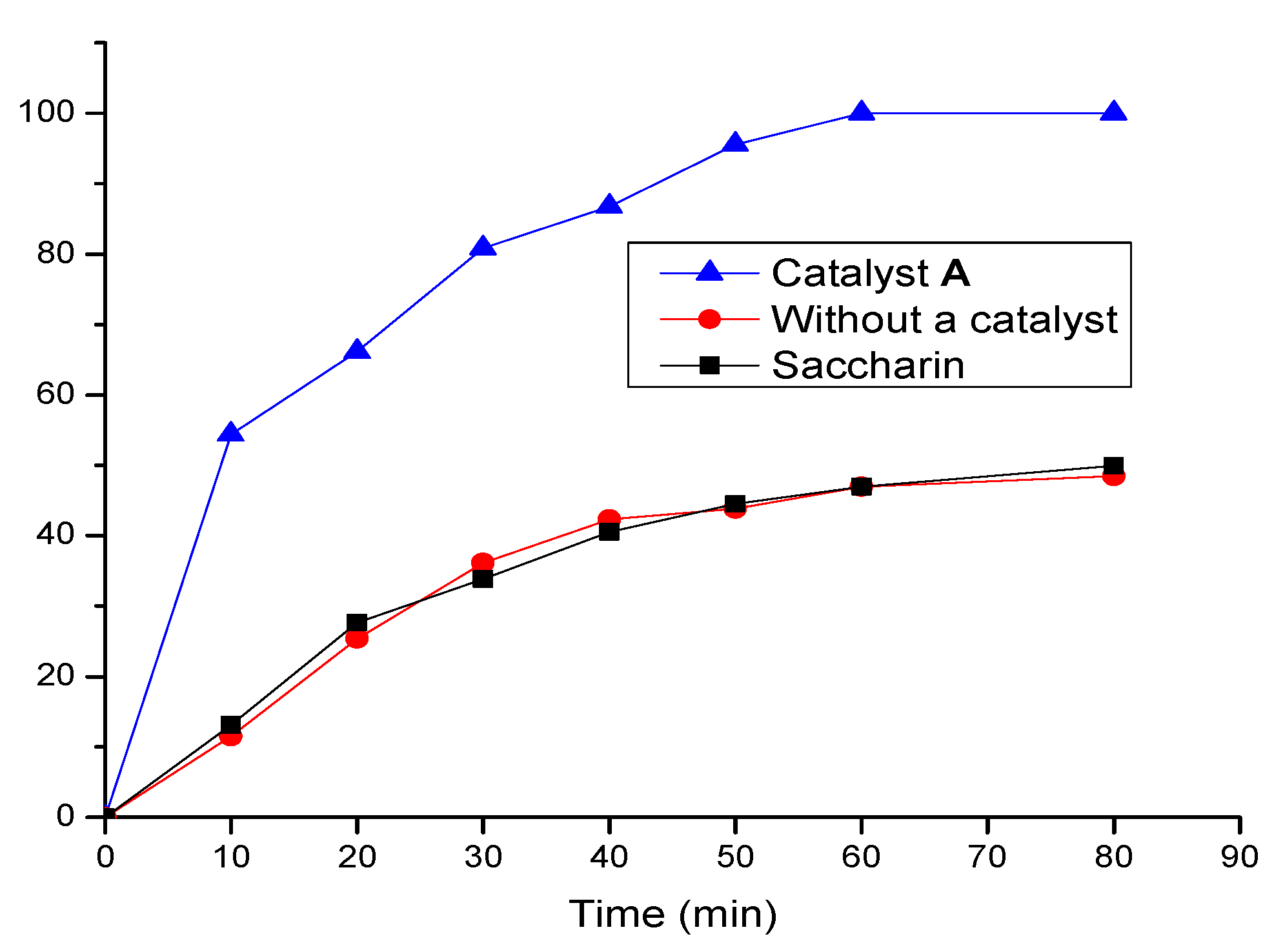
The phospha-Michael addition of diisopropyl phosphite (
1a) with benzylidenemalononitrile (
3) was also studied kinetically at 80 °C (see
Figure 1). The reaction showed a drastic increase in product concentration within the initial 10 min to 54 %, and the reaction reached 100 % yield after 1 h. The integrated rate law derived from the concentration of reactant
3 vs. time showed the ln[reactant
3] = −kt + ln[reactant
3]
o plot with a rate constant k = 0.057 and R
2 = 0.97, where [reactant
3]
o = 3 M and ln[reactant
3]
o = 1.074, as shown in
Figure S2 (in supplementary material). This kinetically monitored reaction showed the turnover frequency (TOF) to be 19.8 h
−1 at 1 h. However, for the same reaction, only about 50% product yield was found after 90 min without using catalyst
A or by using saccharine as a catalyst (see in
Figure 1). Likewise, Sobani and her coworkers reported the reaction of diethyl phosphite with 2-benzylidenemalononitrile (
3) in the absence of the catalyst to give a yield of 60% in 24 h. The reactions without a catalyst led to the formation of the desired product in low yields after a long reaction time [
25]. Furthermore, Sarvari and coworkers reported that the reaction of diethyl phosphite with the electron withdrawing group substituted 2-[(4-chlorophenyl) methylene]malononitrile in the absence of the catalyst gave no product after 24 h [
23]. Overall, these control experiments showed the same trend, which indicated that without a catalyst, the phospha-Michael addition reactions were either very slow or showed no reaction.


 ,
, 










{kind=link}
{kind=link}
{kind=link}
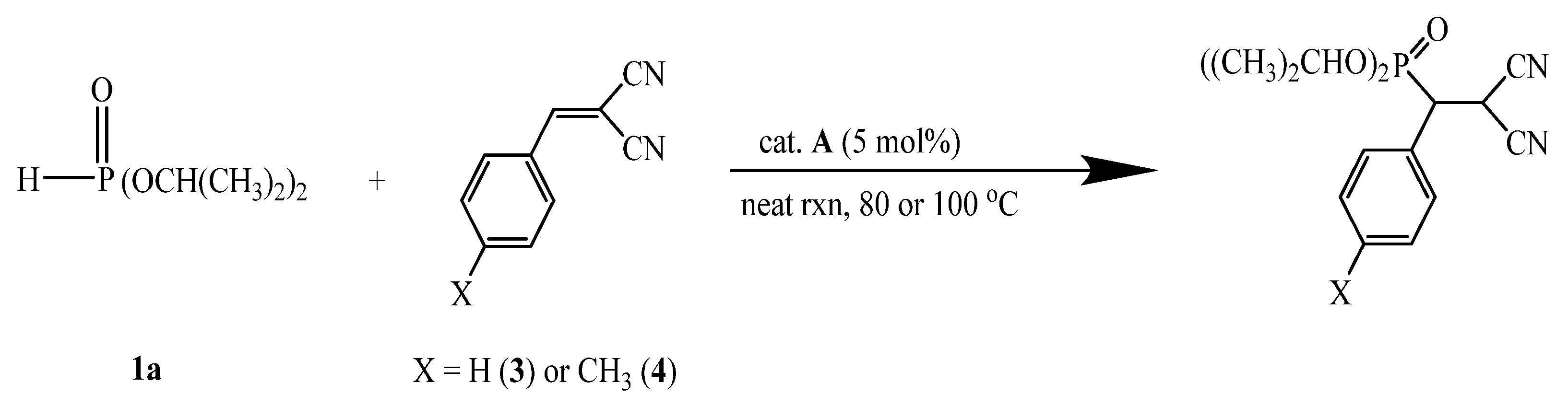{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
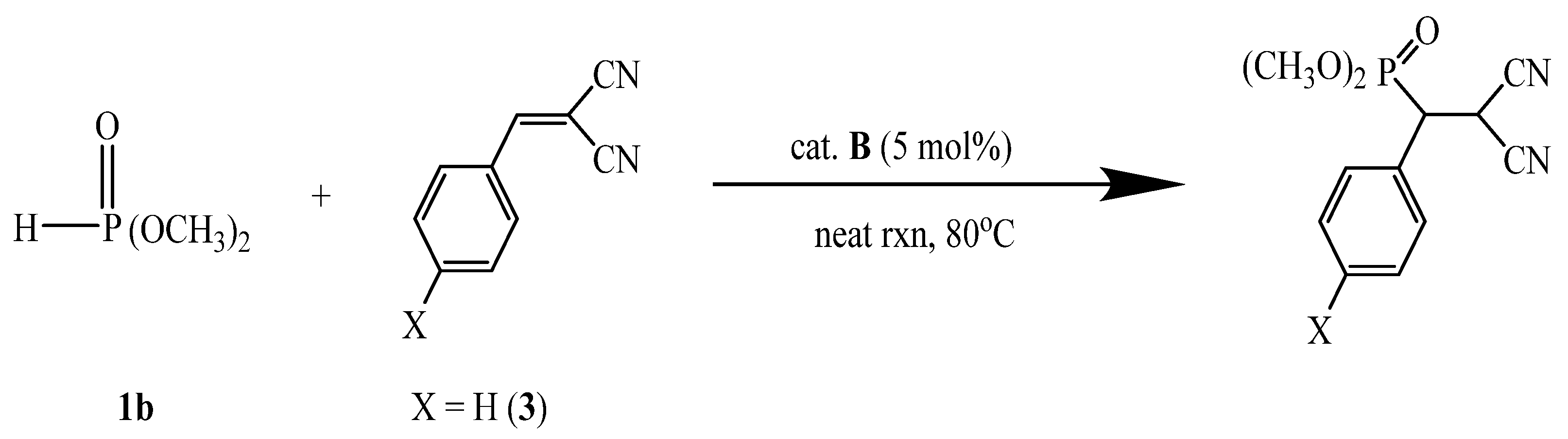{kind=link}