
Optimization and Anti-Cancer Properties of Fluoromethylketones as Covalent Inhibitors for Ubiquitin C-Terminal Hydrolase L1

 , , , , and
, , , , and
Abstract
:
1. Introduction
2. Results and Discussion
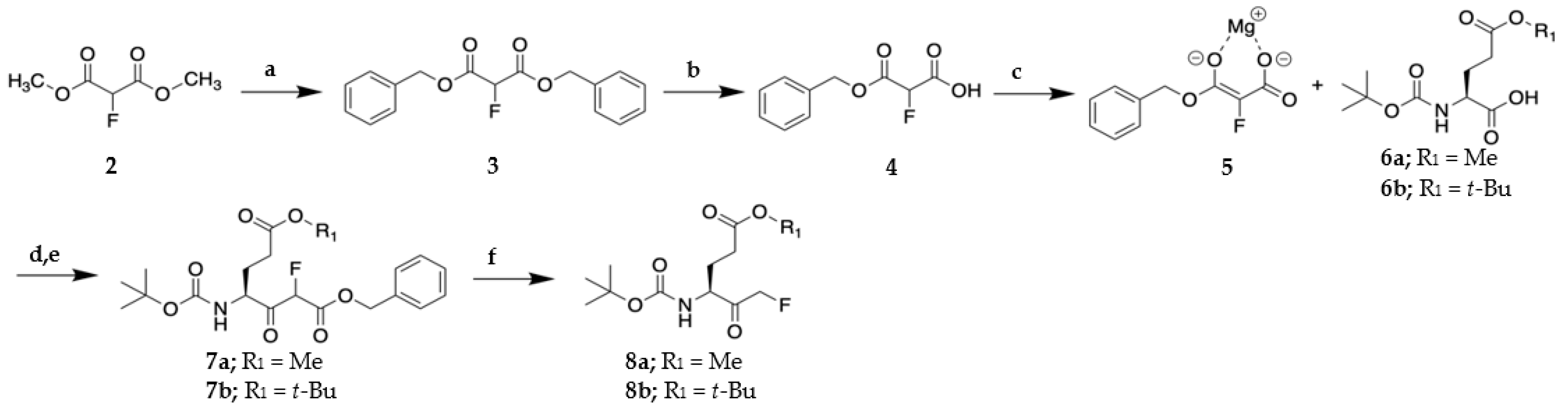
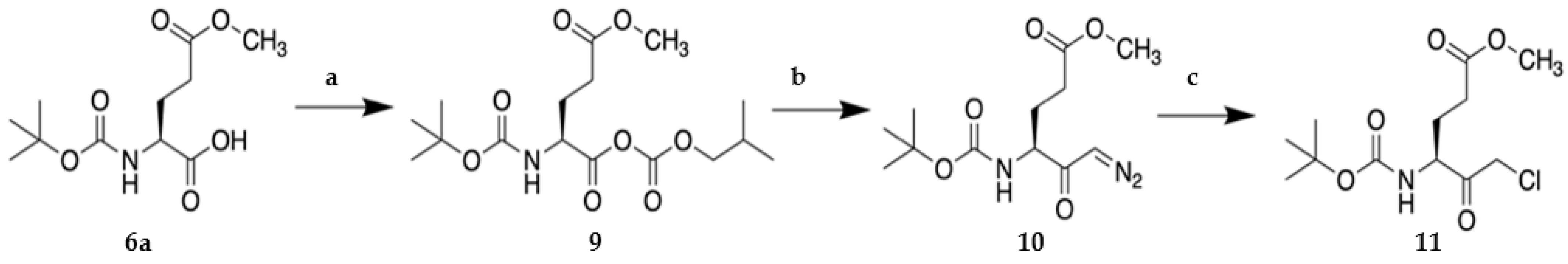
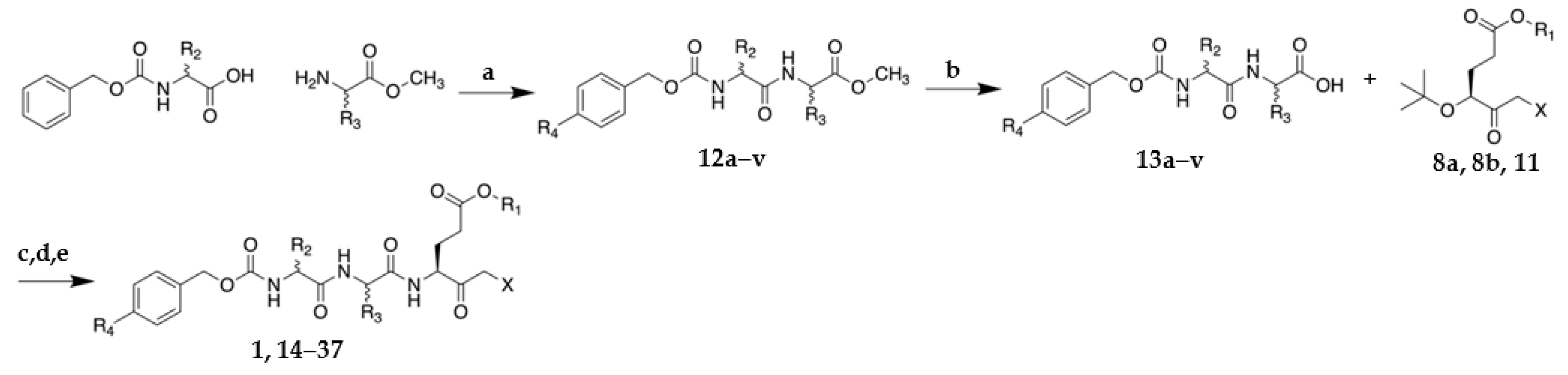
2.1. Synthesis of VAEFMK and Derivatives
2.2. Structure-Activity Relationship Studies for Tripeptide Halomethylketones
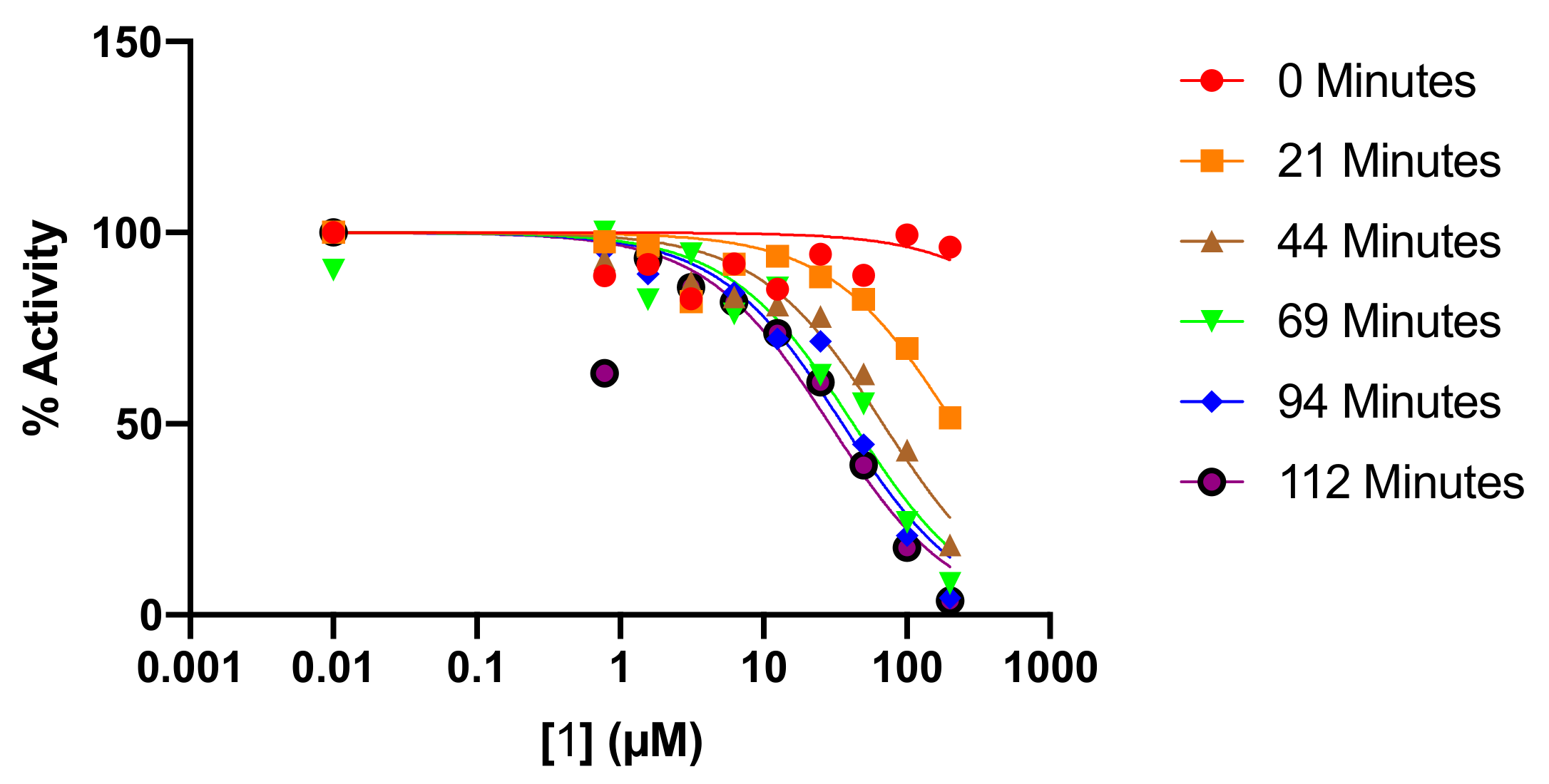
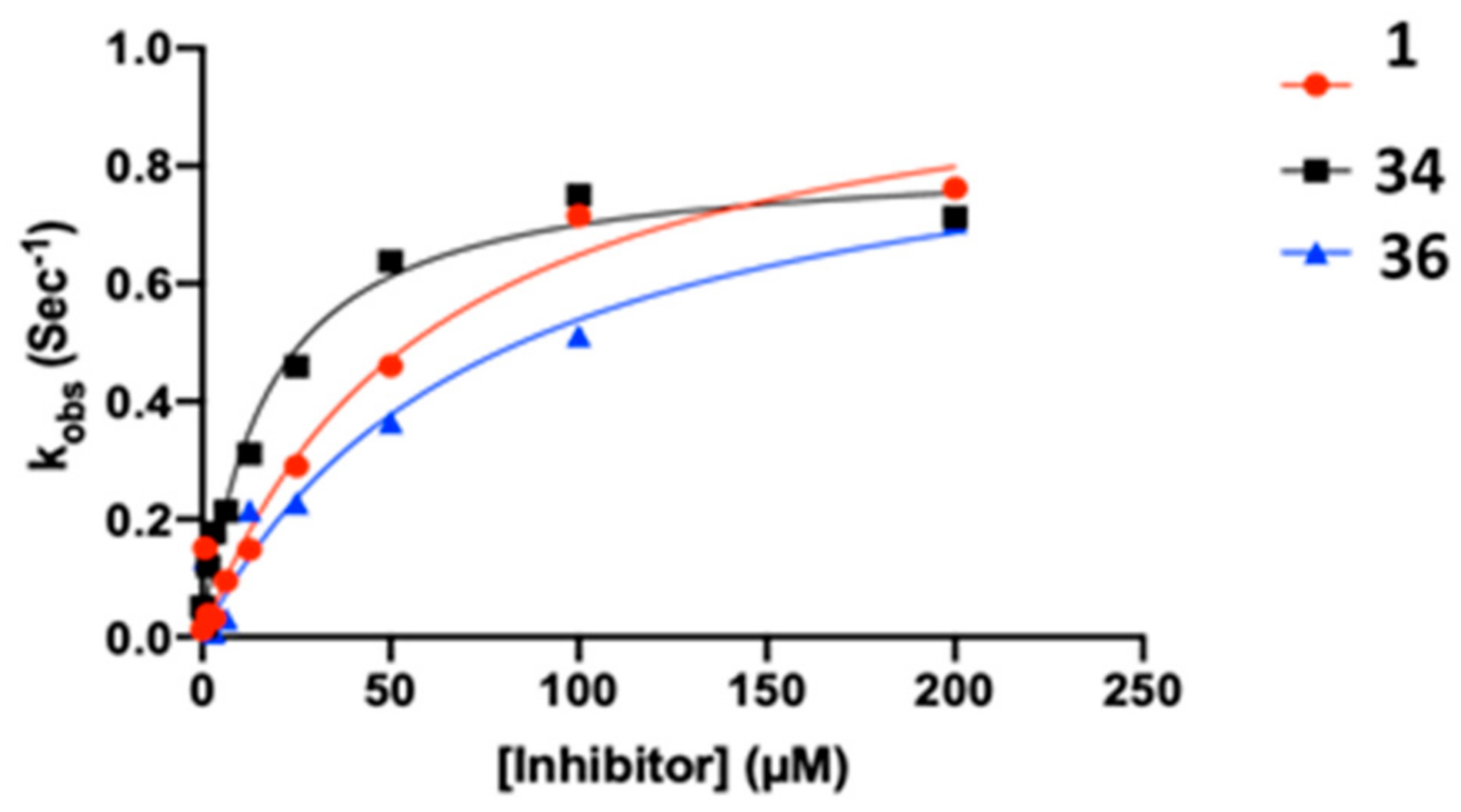
2.3. Kinetic Analysis of Fluoromethylketone Analogs
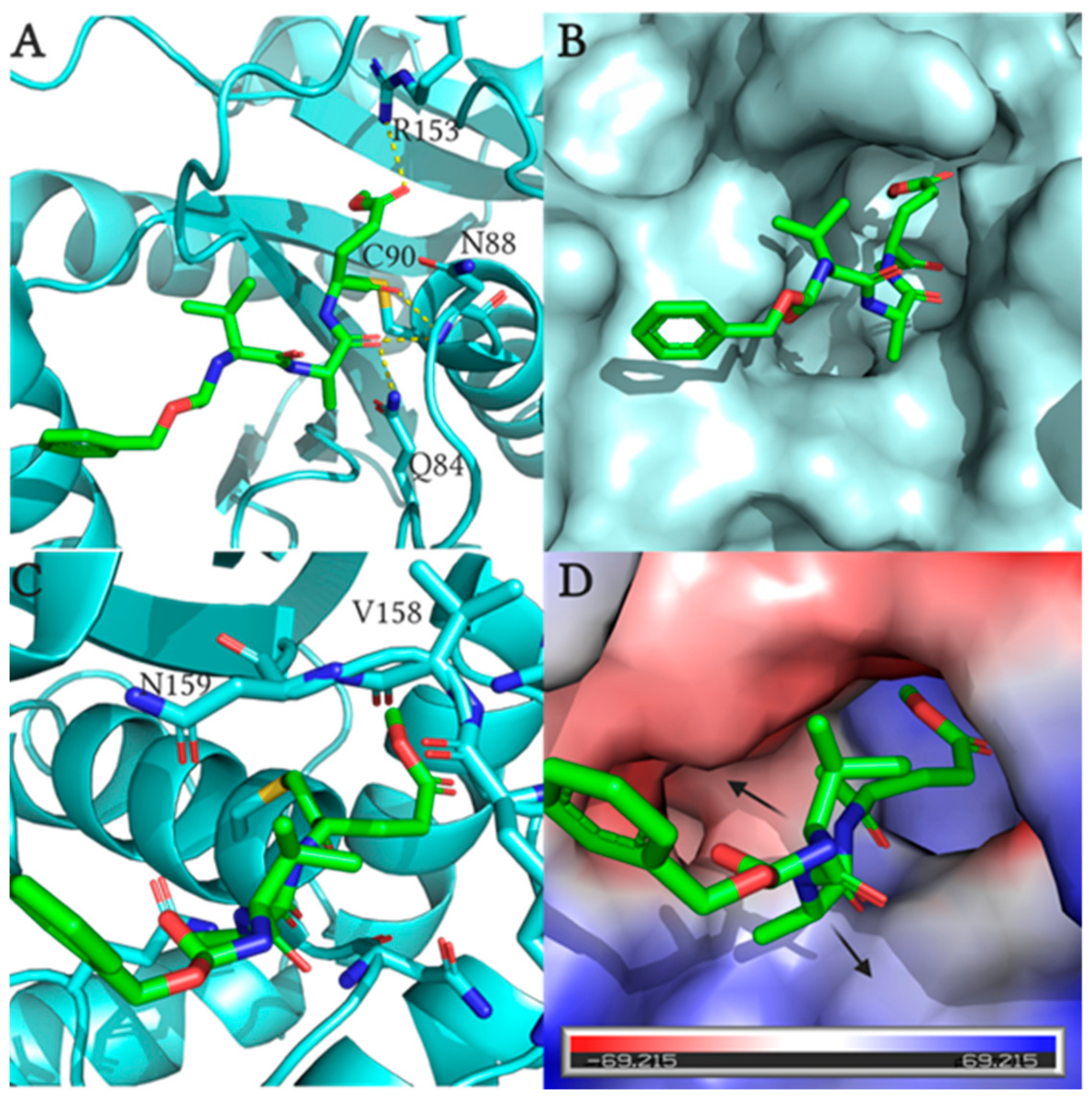
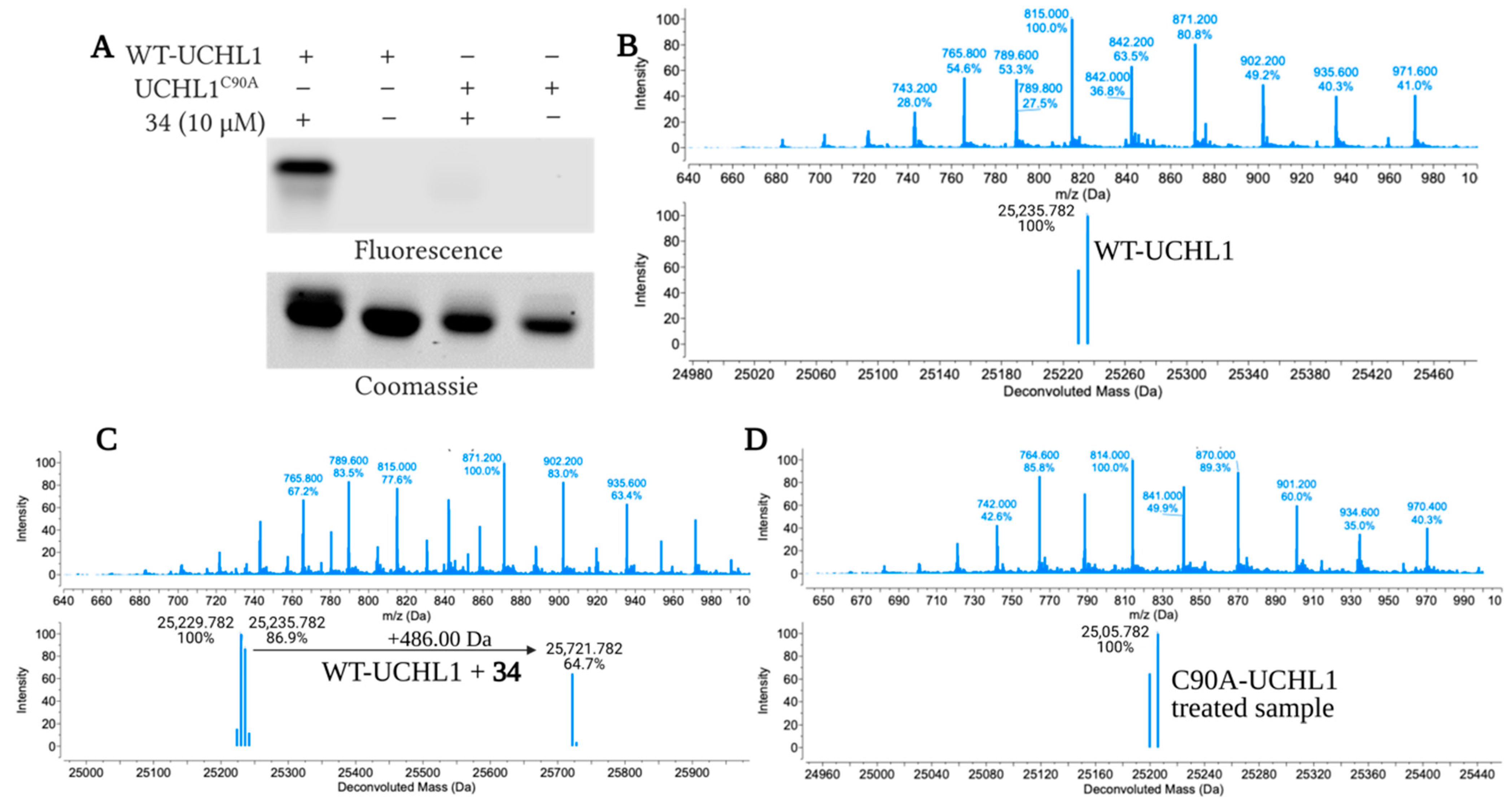
2.4. Fluoromethylketone Analogs Preclude Binding of Ub to UCHL1
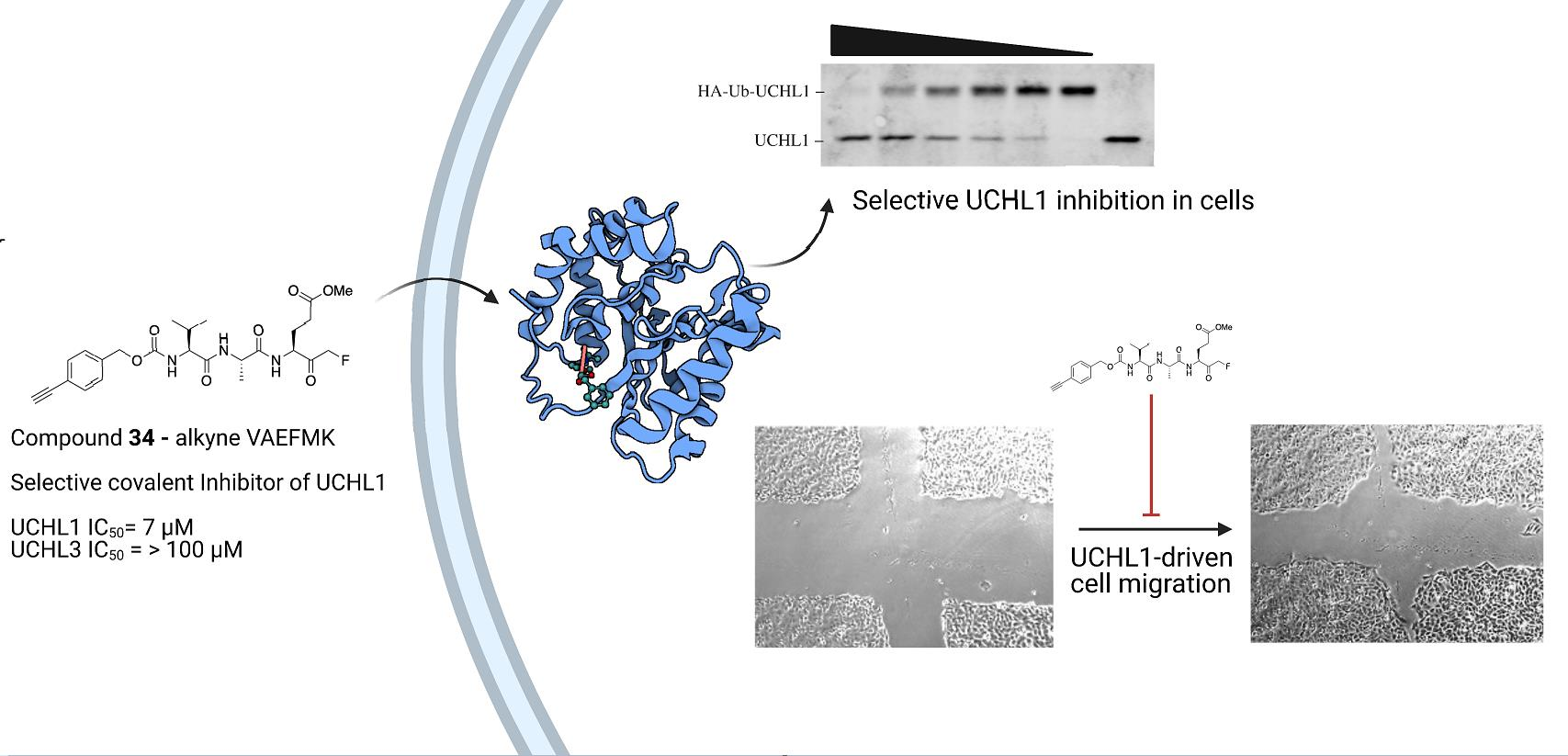
2.5. Cellular Efficacy in Cancer Cell Lines
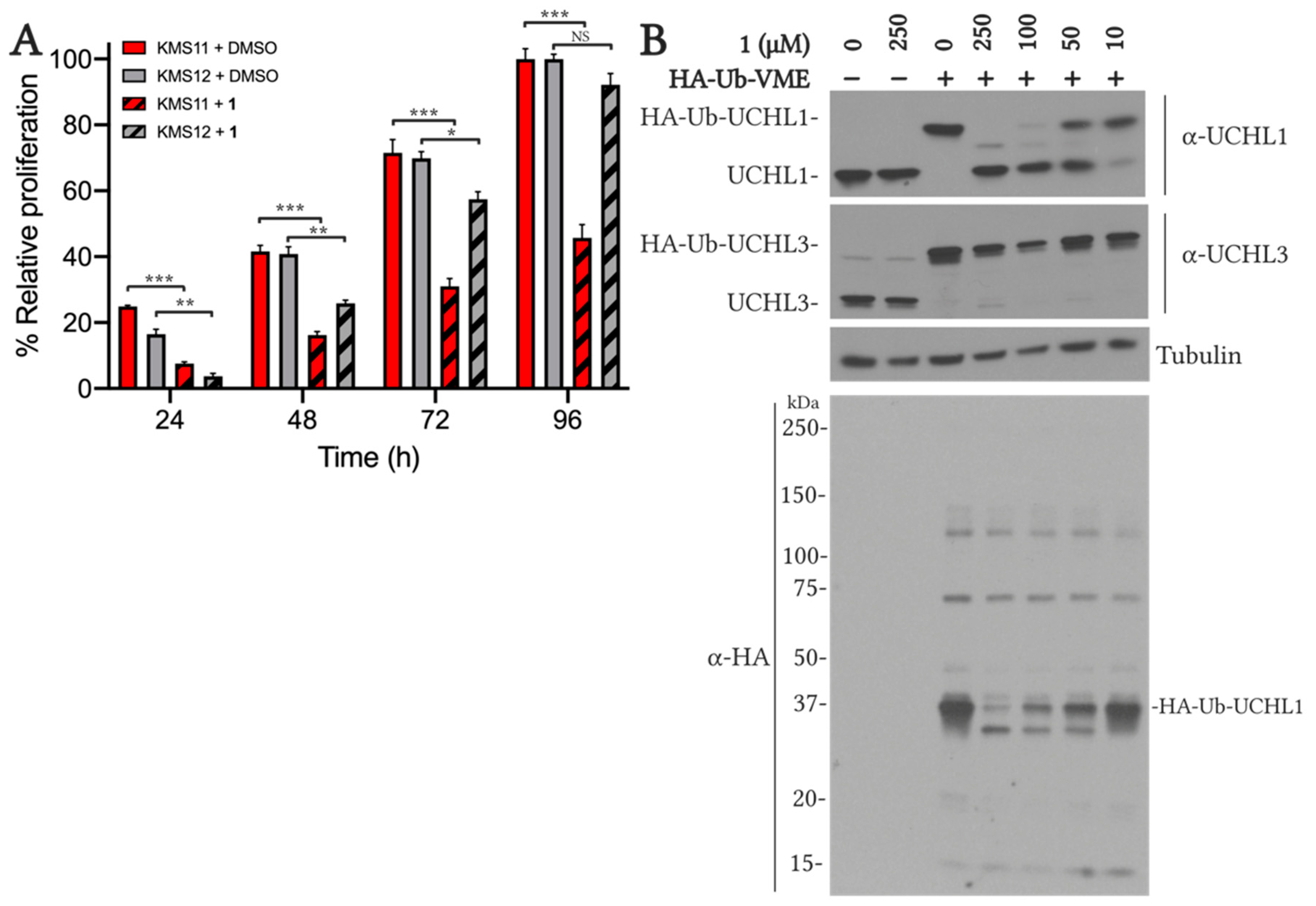
2.5.1. VAEFMK Efficacy in Myeloma Cells
2.5.2. Cell Viability and On-target Engagement of Analogs in SW1271 Small Cell Lung Cancer Cells
2.5.3. Inhibition of UCHL1 Reduces Migratory Capability of SW1271 Cells
3. Materials and Methods
3.1. Chemistry
3.1.1. General Experimental
3.1.2. General Procedure for synthesis of tripeptide halomethylketones
Dibenzyl 2-fluoromalonate (3)
3-(Benzyloxy)-2-fluoro-3-oxopropanoic acid (4)
Methyl (S)-4-((tert-butoxycarbonyl)amino)-6-fluoro-5-oxohexanoate (8a)
Methyl ((benzyloxy)carbonyl)-L-phenylalanyl-L-alaninate (12g)
((Benzyloxy)carbonyl)-L-phenylalanyl-L-alanine (13g)
Cbz-L-Phe-L-Ala-L-Glu(OMe)-fluoromethylketone (21)
3.2. Biological Evaluation
3.2.1. Fluorescence-based Ub-Rho Deubiquitinase Activity Assay
3.2.2. Time-Dependent Inhibitor Analysis
3.2.3. Ub Binding Affinity Analysis Using Biolayer Interferometry
3.2.4. Protein Expression and Purification
3.2.5. Copper-Catalyzed Click Reaction and In-Gel Fluorescence for Wild-Type and C90A UCHL1
3.2.6. Mass Spectrometry Analysis of 34 Adducts on UCHL1
3.2.7. KMS Cell Proliferation Assay
3.2.8. KMS Cell Activity-Based Probe Target Engagement Assay
3.2.9. SW1271 Cell Viability Assay
3.2.10. SW1271 Cell Activity-Based Probe Target Engagement Assay
3.2.11. Copper-Catalyzed Click Reaction and In-Gel Fluorescence of Treated SW1271 Cells
3.2.12. shRNA UCHL1 Knockdown in SW1271 Cells
3.2.13. Scratch Wound Healing Cell Migration Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Acconcia, F.; Sigismund, S.; Polo, S. Ubiquitin in trafficking: The network at work. Exp. Cell Res. 2009, 315, 1610–1618. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Durocher, D. Regulation of DNA Damage Responses by Ubiquitin and SUMO. Mol. Cell 2013, 49, 795–807. [Google Scholar] [CrossRef] [Green Version]
- Myung, J.; Kim, K.B.; Crews, C.M. The ubiquitin-proteasome pathway and proteasome inhibitors. Med. Res. Rev. 2001, 21, 245–273. [Google Scholar] [CrossRef] [PubMed]
- Schulman, B.A.; Wade Harper, J. Ubiquitin-like protein activation by E1 enzymes: The apex for downstream signalling pathways. Nat. Rev. Mol. Cell Biol. 2009, 10, 319–331. [Google Scholar] [CrossRef] [Green Version]
- Ye, Y.; Rape, M. Building ubiquitin chains: E2 enzymes at work. Nat. Rev. Mol. Cell Biol. 2009, 10, 755–764. [Google Scholar] [CrossRef] [Green Version]
- Deshaies, R.J.; Joazeiro, C.A.P. RING Domain E3 Ubiquitin Ligases. Annu. Rev. Biochem. 2009, 78, 399–434. [Google Scholar] [CrossRef]
- Komander, D. The emerging complexity of protein ubiquitination. Biochem. Soc. Trans. 2009, 37, 937–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahighi, S.; Ikeda, F.; Kawasaki, M.; Akutsu, M.; Suzuki, N.; Kato, R.; Kensche, T.; Uejima, T.; Bloor, S.; Komander, D.; et al. Specific Recognition of Linear Ubiquitin Chains by NEMO Is Important for NF-κB Activation. Cell 2009, 136, 1098–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haakonsen, D.L.; Rape, M. Branching Out: Improved Signaling by Heterotypic Ubiquitin Chains. Trends Cell Biol. 2019, 29, 704–716. [Google Scholar] [CrossRef]
- Komander, D.; Clague, M.J.; Urbé, S. Breaking the chains: Structure and function of the deubiquitinases. Nat. Rev. Mol. Cell Biol. 2009, 10. [Google Scholar] [CrossRef]
- Abdul Rehman, S.A.; Kristariyanto, Y.A.; Choi, S.Y.; Nkosi, P.J.; Weidlich, S.; Labib, K.; Hofmann, K.; Kulathu, Y. MINDY-1 Is a Member of an Evolutionarily Conserved and Structurally Distinct New Family of Deubiquitinating Enzymes. Mol. Cell 2016, 63, 146–155. [Google Scholar] [CrossRef] [Green Version]
- Hewings, D.S.; Heideker, J.; Ma, T.P.; AhYoung, A.P.; El Oualid, F.; Amore, A.; Costakes, G.T.; Kirchhofer, D.; Brasher, B.; Pillow, T.; et al. Reactive-site-centric chemoproteomics identifies a distinct class of deubiquitinase enzymes. Nat. Commun. 2018, 9, 1162. [Google Scholar] [CrossRef]
- Jara, J.H.; Frank, D.D.; Özdinler, P.H. Could Dysregulation of UPS be a Common Underlying Mechanism for Cancer and Neurodegeneration? Lessons from UCHL1. Cell Biochem. Biophys. 2013, 67, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.; Zhang, Y.; Galardy, P. DUBs and cancer: The role of deubiquitinating enzymes as oncogenes, non-oncogenes and tumor suppressors. Cell Cycle 2009, 8, 1688–1697. [Google Scholar] [CrossRef]
- Lim, K.-H.; Baek, K.-H. Deubiquitinating enzymes as therapeutic targets in cancer. Curr. Pharm. Des. 2013, 19, 4039–4052. [Google Scholar] [CrossRef]
- Atkin, G.; Paulson, H. Ubiquitin pathways in neurodegenerative disease. Front. Mol. Neurosci. 2014, 7, 63. [Google Scholar] [CrossRef] [Green Version]
- Todi, S.V.; Paulson, H.L. Balancing act: Deubiquitinating enzymes in the nervous system. Trends Neurosci. 2011, 34, 370–382. [Google Scholar] [CrossRef] [Green Version]
- Kemp, M. Recent Advances in The Discovery of Deubiquitinating Enzyme Inhibitors, 1st ed.; Elsevier B.V.: Amsterdam, The Netherlands, 2016. [Google Scholar]
- Schauer, N.; Magin, R.S.; Liu, X.; Doherty, L.; Buhrlage, S. Advances in Discovering Deubiquitinating Enzyme (DUB) Inhibitors. J. Med. Chem. 2019. [Google Scholar] [CrossRef] [PubMed]
- Gong, B.; Radulovic, M.; Figueiredo-Pereira, M.E.; Cardozo, C. The Ubiquitin-Proteasome System: Potential Therapeutic Targets for Alzheimer’s Disease and Spinal Cord Injury. Front. Mol. Neurosci. 2016, 9, 4. [Google Scholar] [CrossRef] [Green Version]
- Fang, Y.; Fu, D.; Shen, X.Z. The potential role of ubiquitin c-terminal hydrolases in oncogenesis. Biochim. Biophys. Acta Rev. Cancer 2010, 1806, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Bishop, P.; Rocca, D.; Henley, J.M. Ubiquitin C-terminal hydrolase L1 (UCH-L1): Structure, distribution and roles in brain function and dysfunction. Biochem. J. 2016, 473, 2453–2462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Day, I.N.M.; Thompson, R.J. UCHL1 (PGP 9.5): Neuronal biomarker and ubiquitin system protein. Prog. Neurobiol. 2010, 90, 327–362. [Google Scholar] [CrossRef] [PubMed]
- Leroy, E.; Boyer, R.; Auburger, G.; Leube, B.; Ulm, G.; Mezey, E.; Harta, G.; Brownstein, M.J.; Jonnalagada, S.; Chernova, T.; et al. The ubiquitin pathway in Parkinson’s disease. Nature 1998, 395, 451–452. [Google Scholar] [CrossRef]
- Janda, E.; Isidoro, C.; Carresi, C.; Mollace, V. Defective autophagy in Parkinson’s disease: Role of oxidative stress. Mol. Neurobiol. 2012, 46, 639–661. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, K.; Li, H.; Kawamura, R.; Osaka, H.; Wang, Y.L.; Hara, Y.; Hirokawa, T.; Manago, Y.; Amano, T.; Noda, M.; et al. Alterations of structure and hydrolase activity of parkinsonism-associated human ubiquitin carboxyl-terminal hydrolase L1 variants. Biochem. Biophys. Res. Commun. 2003, 304, 176–183. [Google Scholar] [CrossRef]
- Setsuie, R.; Wada, K. The functions of UCH-L1 and its relation to neurodegenerative diseases. Neurochem. Int. 2007, 51, 105–111. [Google Scholar] [CrossRef]
- Hussain, S.; Bedekovics, T.; Liu, Q.; Hu, W.; Jeon, H.; Johnson, S.H.; Vasmatzis, G.; May, D.G.; Roux, K.J.; Galardy, P.J. UCH-L1 bypasses mTOR to promote protein biosynthesis and is required for MYC-driven lymphomagenesis in mice. Blood 2018, 132, 2564–2574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedekovics, T.; Hussain, S.; Feldman, A.L.; Galardy, P.J. UCH-L1 is induced in germinal center B cells and identifies patients with aggressive germinal center diffuse large B-cell lymphoma. Blood 2016, 127, 1564–1574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carolan, B.J.; Heguy, A.; Harvey, B.G.; Leopold, P.L.; Ferris, B.; Crystal, R.G. Up-regulation of expression of the ubiquitin carboxyl-terminal hydrolase L1 gene in human airway epithelium of cigarette smokers. Cancer Res. 2006, 66, 10729–10740. [Google Scholar] [CrossRef] [Green Version]
- Shimada, Y.; Kudo, Y.; Maehara, S.; Matsubayashi, J.; Otaki, Y.; Kajiwara, N.; Ohira, T.; Minna, J.D.; Ikeda, N. Ubiquitin C-terminal hydrolase-L1 has prognostic relevance and is a therapeutic target for high-grade neuroendocrine lung cancers. Cancer Sci. 2020, 111, 610–620. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Gharib, T.G.; Huang, C.-C.; Thomas, D.G.; Shedden, K.A.; Taylor, J.M.G.; Kardia, S.L.R.; Misek, D.E.; Giordano, T.J.; Iannettoni, M.D. Proteomic analysis of lung adenocarcinoma: Identification of a highly expressed set of proteins in tumors. Clin. Cancer Res. 2002, 8, 2298–2305. [Google Scholar] [PubMed]
- Sanchez-Diaz, P.C.; Chang, J.C.; Moses, E.S.; Dao, T.; Chen, Y.; Hung, J.Y. Ubiquitin carboxyl-Terminal esterase L1 (UCHL1) is associated with stem-like cancer cell functions in pediatric high-grade glioma. PLoS ONE 2017, 12, e0176879. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.; Bedekovics, T.; Chesi, M.; Bergsagel, L.P.; Galardy, P.J. UCHL1 is a biomarker of aggressive multiple myeloma required for disease progression. Oncotarget 2015, 5. [Google Scholar] [CrossRef]
- Hussain, S.; Bedekovics, T.; Ali, A.; Zaid, O.; May, D.G.; Roux, K.J.; Galardy, P.J. A cysteine near the C-terminus of UCH-L1 is dispensable for catalytic activity but is required to promote AKT phosphorylation, eIF4F assembly, and malignant B-cell survival. Cell Death Discov. 2019, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Otsuki, T.; Yata, K.; Takata-Tomokuni, A.; Hyodoh, F.; Miura, Y.; Sakaguchi, H.; Hatayama, T.; Hatada, S.; Tsujioka, T.; Sato, Y. Expression of protein gene product 9· 5 (PGP9· 5)/ubiquitin-C-terminal hydrolase 1 (UCHL-1) in human myeloma cells. Br. J. Haematol. 2004, 127, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.Y.; Yang, M.; Zhao, M.; Luo, Q.; Yang, L.; Peng, H.; Wang, J.; Huang, S.K.; Zheng, Z.X.; Yuan, X.H.; et al. The de-ubiquitinase UCHL1 promotes gastric cancer metastasis via the Akt and Erk1/2 pathways. Tumor Biol. 2015, 36, 8379–8387. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; González-Prieto, R.; Zhang, M.; Geurink, P.P.; Kooij, R.; Iyengar, P.V.; van Dinther, M.; Bos, E.; Zhang, X.; Le Dévédec, S.E.; et al. Deubiquitinase activity profiling identifies UCHL1 as a candidate oncoprotein that promotes TGFβ-induced breast cancer metastasis. Clin. Cancer Res. 2020, 26, 1460–1473. [Google Scholar] [CrossRef] [Green Version]
- Jang, M.J.; Baek, S.H.; Kim, J.H. UCH-L1 promotes cancer metastasis in prostate cancer cells through EMT induction. Cancer Lett. 2011, 302, 128–135. [Google Scholar] [CrossRef]
- Ma, Y.; Zhao, M.; Zhong, J.; Shi, L.; Luo, Q.; Liu, J.; Wang, J.; Yuan, X.; Huang, C. Proteomic profiling of proteins associated with lymph node metastasis in colorectal cancer. J. Cell. Biochem. 2010, 110, 1512–1519. [Google Scholar] [CrossRef]
- Miyoshi, Y.; Nakayama, S.; Torikoshi, Y.; Tanaka, S.; Ishihara, H.; Taguchi, T.; Tamaki, Y.; Noguchi, S. High expression of ubiquitin caboxy-terminal hydrolase-L1 and -L3 mRNA predicts early recurrence in patients with invasive breast cancer. Cancer Sci. 2006, 97, 523–529. [Google Scholar] [CrossRef]
- Goto, Y.; Zeng, L.; Yeom, C.J.; Zhu, Y.; Morinibu, A.; Shinomiya, K.; Kobayashi, M.; Hirota, K.; Itasaka, S.; Yoshimura, M.; et al. UCHL1 provides diagnostic and antimetastatic strategies due to its deubiquitinating effect on HIF-1α. Nat. Commun. 2015, 6, 6153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.J.; Kim, Y.M.; Lim, S.; Nam, Y.K.; Jeong, J.; Kim, H.-J.; Lee, K.-J. Ubiquitin C-terminal hydrolase-L1 is a key regulator of tumor cell invasion and metastasis. Oncogene 2009, 28, 117–127. [Google Scholar] [CrossRef] [Green Version]
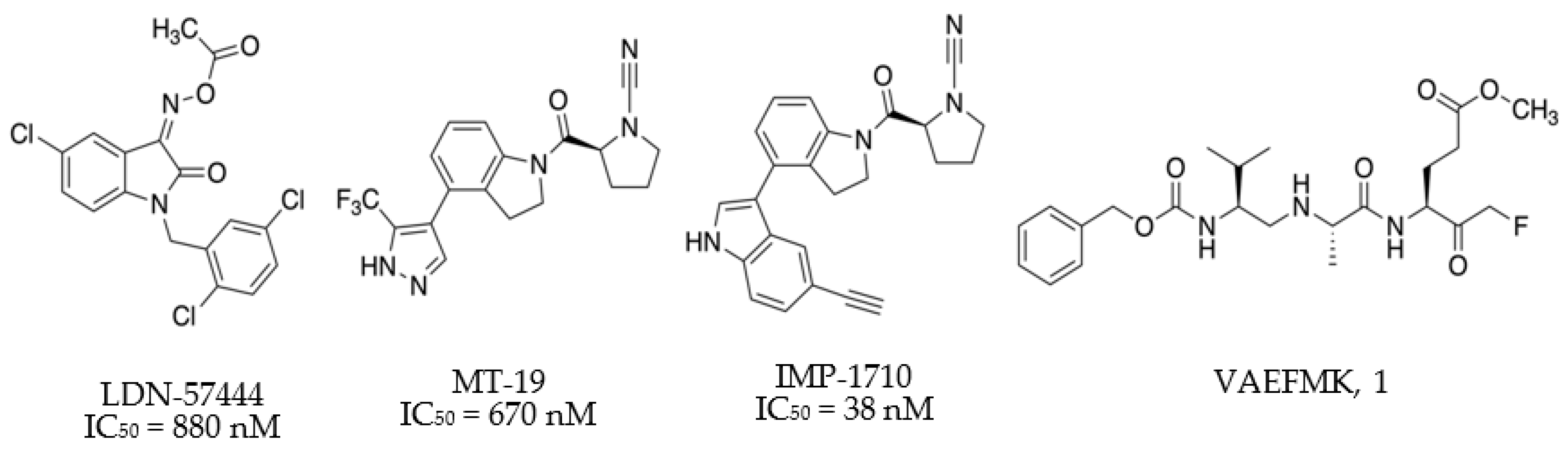
- Liu, Y.; Lashuel, H.; Choi, S.; Xing, X.; Case, A.; Ni, J.; Yeh, L.-A.; Cuny, G.D.; Stein, R.L.; Lansbury, P.T. Discovery of Inhibitors that Elucidate the Role of UCH-L1 Activity in the H1299 Lung Cancer Cell Line. Chem. Biol. 2003, 10, 837–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panyain, N.; Godinat, A.; Lanyon-Hogg, T.; Lachiondo-Ortega, S.; Will, E.J.; Soudy, C.; Mason, K.; Elkhalifa, S.; Smith, L.M.; Harrigan, J.A.; et al. Discovery of a potent and selective covalent inhibitor and activity-based probe for the deubiquitylating enzyme UCHL1, with anti-fibrotic activity. J. Am. Chem. Soc. 2020. [Google Scholar] [CrossRef]
- Krabill, A.D.A.D.; Chen, H.; Hussain, S.; Feng, C.; Abdullah, A.; Das, C.; Aryal, U.K.; Post, C.B.C.B.; Wendt, M.K.M.K.; Galardy, P.J.; et al. Ubiquitin C-Terminal Hydrolase L1: Biochemical and Cellular Characterization of a Covalent Cyanopyrrolidine-Based Inhibitor. ChemBioChem 2020, 21, 712–722. [Google Scholar] [CrossRef] [PubMed]
- Kattenhorn, L.M.; Korbel, G.A.; Kessler, B.M.; Spooner, E.; Ploegh, H.L. A Deubiquitinating Enzyme Encoded by HSV-1 Belongs to a Family of Cysteine Proteases that Is Conserved across the Family Herpesviridae. Mol. Cell 2005, 19, 547–557. [Google Scholar] [CrossRef]
- Davies, C.W.; Chaney, J.; Korbel, G.; Ringe, D.; Petsko, G.A.; Ploegh, H.; Das, C. The co-crystal structure of ubiquitin carboxy-terminal hydrolase L1 (UCHL1) with a tripeptide fluoromethyl ketone (Z-VAE(OMe)-FMK). Bioorganic Med. Chem. Lett. 2012, 22, 3900–3904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powers, J.C.; Asgian, J.L.; James, K.E. Irreversible Inhibitors of Serine, Cysteine, and threonine proteases. Chem. Rev. 2002. [Google Scholar] [CrossRef] [PubMed]
- Palmer, J. Process for Forming a Fluoromethyl Ketone. U.S. Patent No. 5,210,272, 11 May 1993. [Google Scholar]
- Witte, M.D.; Descals, C.V.; De Lavoir, S.V.P.; Florea, B.I.; Van Der Marel, G.A.; Overkleeft, H.S. Bodipy-VAD-Fmk, a useful tool to study yeast peptide N-glycanase activity. Org. Biomol. Chem. 2007, 5, 3690–3697. [Google Scholar] [CrossRef]
- Morris, T.S.; Frormann, S.; Shechosky, S.; Lowe, C.; Laid, M.S.; Gauss-mfiller, V.; Purcell, R.H.; Emerson, S.U.; Vederas, J.C.; Malcolm, B.A. In Vitro and Ex Vivo Inhibition of Hepatitis A Virus 3C Proteinase by a Peptidyl Monofluoromethyl Ketone. Bioorg. Med. Chem. 1997, 5, 797–807. [Google Scholar] [CrossRef]
- Misaghi, S.; Pacold, M.E.; Blom, D.; Ploegh, H.L.; Korbel, G.A. Using a small molecule inhibitor of peptide: N-glycanaseto probe its role in glycoprotein turnover. Chem. Biol. 2004, 11, 1677–1687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassiepen, U.; Eidhoff, U.; Meder, G.; Bulber, J.F.; Hein, A.; Bodendorf, U.; Lorthiois, E.; Martoglio, B. A sensitive fluorescence intensity assay for deubiquitinating proteases using ubiquitin-rhodamine110-glycine as substrate. Anal. Biochem. 2007, 371, 201–207. [Google Scholar] [CrossRef]
- Boudreaux, D.A.; Maiti, T.K.; Davies, C.W.; Das, C. Ubiquitin vinyl methyl ester binding orients the misaligned active site of the ubiquitin hydrolase UCHL1 into productive conformation. Proc. Natl. Acad. Sci. USA 2010, 107, 9117–9122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koharudin, L.M.I.; Liu, H.; Di Maio, R.; Kodali, R.B.; Graham, S.H.; Gronenborn, A.M. Cyclopentenone prostaglandin-induced unfolding and aggregation of the Parkinson disease-associated UCH-L1. Proc. Natl. Acad. Sci. USA 2010, 107, 6835–6840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Contu, V.R.; Kotake, Y.; Toyama, T.; Okuda, K.; Miyara, M.; Sakamoto, S.; Samizo, S.; Sanoh, S.; Kumagai, Y.; Ohta, S. Endogenous neurotoxic dopamine derivative covalently binds to Parkinson’s disease-associated ubiquitin C-terminal hydrolase L1 and alters its structure and function. J. Neurochem. 2014, 130, 826–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, T.; Oh, C.; Liao, L.; Zhang, X.; Lopez, K.M.; Gibbs, D. Noncanonical transnitrosylation network contributes to synapse loss in Alzheimer’ s disease. Science (80-.) 2020, 0843, 1–18. [Google Scholar]
- Bishop, P.; Rubin, P.; Thomson, A.R.; Rocca, D.; Henley, J.M. The Ubiquitin C-Terminal Hydrolase L1 (UCH-L1) C terminus plays a key role in protein stability, but its farnesylation is not required for membrane association in primary neurons. J. Biol. Chem. 2014, 289, 36140–36149. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Calvo, M.; Peterson, E.P.; Leiting, B.; Ruel, R.; Nicholson, D.W.; Thornberry, N.A. Inhibition of human caspases by peptide-based and macromolecular inhibitors. J. Biol. Chem. 1998, 273, 32608–32613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mons, E.; Jansen, I.D.C.; Loboda, J.; Van Doodewaerd, B.R.; Hermans, J.; Verdoes, M.; Van Boeckel, C.A.A.; Van Veelen, P.A.; Turk, B.; Ovaa, H. The Alkyne Moiety as a Latent Electrophile in Irreversible Covalent Small Molecule Inhibitors of Cathepsin K. J. Am. Chem. Soc. 2019, 141, 3507–3514. [Google Scholar] [CrossRef] [Green Version]
- Kathman, S.G.; Xu, Z.; Statsyuk, A. V A Fragment-Based Method to Discover Irreversible Covalent Inhibitors of Cysteine Proteases. J. Med. Chem. 2014, 4–9. [Google Scholar] [CrossRef]
- Strelow, J.M. A perspective on the kinetics of covalent and irreversible inhibition. SLAS Discov. 2017, 22, 3–20. [Google Scholar] [CrossRef] [Green Version]
- Resnick, E.; Bradley, A.; Gan, J.; Douangamath, A.; Krojer, T.; Sethi, R.; Geurink, P.P.; Aimon, A.; Amitai, G.; Bellini, D.; et al. Rapid Covalent-Probe Discovery by Electrophile-Fragment Screening. J. Am. Chem. Soc. 2019. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, K.D.; Lee, K.; Deshpande, S.; Duerksen-hughes, P.; Boss, J.M.; Pohl, J. The neuron-specific protein PGP 9.5 is a ubiquitin carboxyl-terminal hydrolase. Science (80-.) 1989, 246, 670–673. [Google Scholar] [CrossRef] [PubMed]
- Larsen, C.N.; Krantz, B.A.; Wilkinson, K.D. Substrate specificity of deubiquitinating enzymes: Ubiquitin C-terminal hydrolases. Biochemistry 1998, 37, 3358–3368. [Google Scholar] [CrossRef]
- Nakatsu, F.; Sakuma, M.; Matsuo, Y.; Arase, H.; Yamasaki, S.; Nakamura, N.; Saito, T.; Ohno, H. A Di-leucine signal in the ubiquitin moiety Possible involvement in ubiquitination-mediated endocytosis. J. Biol. Chem. 2000, 275, 26213–26219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shih, S.C. Monoubiquitin carries a novel internalization signal that is appended to activated receptors. EMBO J. 2000, 19, 187–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hewitt, C.S.; Krabill, A.D.; Das, C.; Flaherty, D.P. Development of Ubiquitin Variants with Selectivity for Ubiquitin C-Terminal Hydrolase Deubiquitinases. Biochemistry 2020, 1. [Google Scholar] [CrossRef]
- Ekkebus, R.; Flierman, D.; Geurink, P.P.; Ovaa, H. Catching a DUB in the act: Novel ubiquitin-based active site directed probes. Curr. Opin. Chem. Biol. 2014, 23, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Magesh, V.; Lee, J.; Kim, S.; Knaus, U.G.; Lee, K. Ubiquitin C-terminal hydrolase-L1 increases cancer cell invasion by modulating hydrogen peroxide generated via NADPH oxidase. Oncotarget 2015, 6, 16287. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Cpd | X | R1 | a R2 | a R3 | R4 | IC50 (µM) b |
|---|---|---|---|---|---|---|
| 1 | F | Me | Val | Ala | H | 24 |
| 14 | Cl | Me | Val | Ala | H | >200 |
| 15 | F | Me | Val | Gly | H | 95 |
| 16 | F | H | Val | Gly | H | >200 |
| 17 | F | Me | D-Val | Ala | H | >200 |
| 18 | F | Me | Gly | Ala | H | >200 |
| 19 | F | Me | Ala | Ala | H | 76 |
| 20 | F | Me | Leu | Ala | H | 23 |
| 21 | F | Me | Phe | Ala | H | 13 |
| 22 | F | Me | Ser | Ala | H | 185 |
| 23 | F | Me | Thr | Ala | H | 100 |
| 24 | F | Me | Asn | Ala | H | 119 |
| 25 | F | Me | Asp | Ala | H | >200 |
| 26 | F | Me | Val | D-Ala | H | >200 |
| 27 | F | Me | Val | Leu | H | >200 |
| 28 | F | Me | Val | Phe | H | >200 |
| 29 | F | Me | Val | Ser | H | 100 |
| 30 | F | Me | Val | Thr | H | >200 |
| 31 | F | Me | Val | Asn | H | >200 |
| 32 | F | Me | Val | Asp | H | >200 |
| 33 | F | Me | Val | Glu | H | >200 |
| 34 | F | Me | Val | Ala | -CCH | 7.7 |
| 35 | F | Me | Val | Gly | -CCH | 62 |
| 36 | F | Me | Phe | Ala | -CCH | 11 |
| 37 | Cl | Me | Val | Ala | -CCH | >200 |
| Protein and Treatment | Calculated Deconvoluted Masses Observed (Da) a | Adduct(s) Observed (Da) |
|---|---|---|
| UCHL1 | 25,235.782 | none |
| WT-UCHL1 + 34 (1:1) | 25,235.782 and 25,721.782 | +486.00 |
| WT-UCHL1 + 34 (1:2) | 25,235.782 and 25,721.782 | +486.00 |
| UCHL1 C90A | 25,205.782 | none |
| UCHL1 C90A + 34 (1:1) | 25,205.782 | none |
| UCHL1 C90A + 34 (1:2) | 25,205.782 | none |
| Cpd | kinact (s−1) | KI (µM) | kinact/KI (M−1s−1) | R2 |
|---|---|---|---|---|
| 1 | 0.0173 | 60.04 | 288.1 | 0.9623 |
| 34 | 0.0136 | 16.79 | 810.0 | 0.9779 |
| 36 | 0.0157 | 75.32 | 208.4 | 0.9478 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krabill, A.D.; Chen, H.; Hussain, S.; Hewitt, C.S.; Imhoff, R.D.; Muli, C.S.; Das, C.; Galardy, P.J.; Wendt, M.K.; Flaherty, D.P. Optimization and Anti-Cancer Properties of Fluoromethylketones as Covalent Inhibitors for Ubiquitin C-Terminal Hydrolase L1. Molecules 2021, 26, 1227. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26051227
Krabill AD, Chen H, Hussain S, Hewitt CS, Imhoff RD, Muli CS, Das C, Galardy PJ, Wendt MK, Flaherty DP. Optimization and Anti-Cancer Properties of Fluoromethylketones as Covalent Inhibitors for Ubiquitin C-Terminal Hydrolase L1. Molecules. 2021; 26(5):1227. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26051227
Chicago/Turabian StyleKrabill, Aaron D., Hao Chen, Sajjad Hussain, Chad S. Hewitt, Ryan D. Imhoff, Christine S. Muli, Chittaranjan Das, Paul J. Galardy, Michael K. Wendt, and Daniel P. Flaherty. 2021. "Optimization and Anti-Cancer Properties of Fluoromethylketones as Covalent Inhibitors for Ubiquitin C-Terminal Hydrolase L1" Molecules 26, no. 5: 1227. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26051227