
Alkyl-Resorcinol Derivatives as Inhibitors of GDP-Mannose Pyrophosphorylase with Antileishmanial Activities

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. Plant Materials
3.2. Extraction and Compounds Isolation
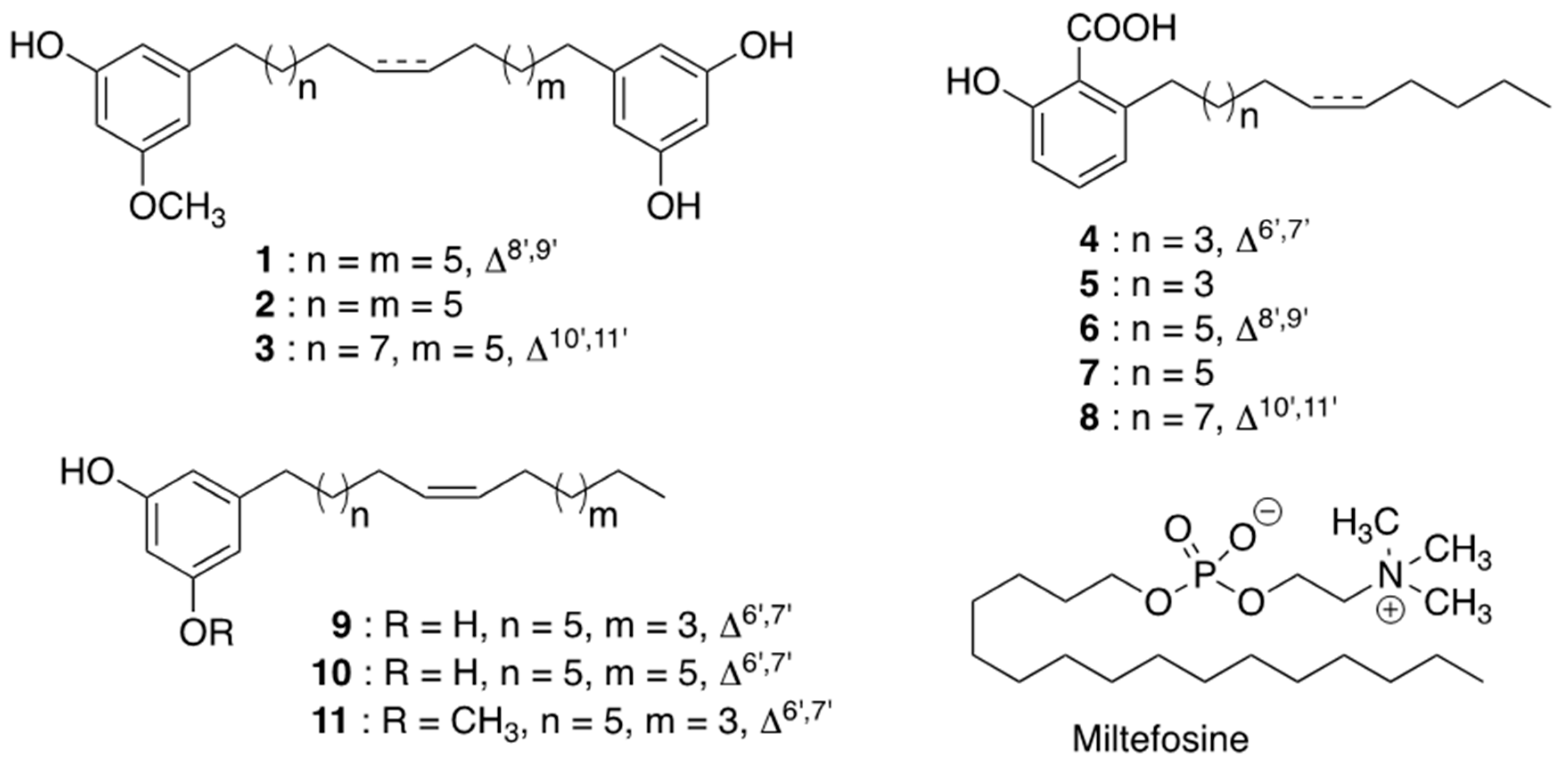
3.3. Structural Identification of Compounds (1–11, Figure 1)
3.4. GDP-MP Production and Purification
3.5. Enzyme Assays
3.6. Evaluation of Compounds on Purified Enzymes
3.7. Cell Cultures
3.8. In Vitro Evaluation of Compounds’ Cytotoxicity
3.9. In Vitro Antileishmanial Evaluation of Compounds on Axenic and Intramacrophage Amastigotes
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Roatt, B.M.; de Oliveira Cardoso, J.M.; De Brito, R.C.F.; Coura-Vital, W.; de Oliveira Aguiar-Soares, R.D.; Reis, A.B. Recent advances and new strategies on leishmaniasis treatment. Appl. Microbiol. Biotechnol. 2020, 104, 8965–8977. [Google Scholar] [CrossRef]
- World Health Organization. Leishmaniasis Fact Sheet. 2 March 2020. Available online: https://www.who.int/news-room/fact-sheets/detail/leishmaniasis (accessed on 10 March 2021).
- Batista, M.F.; Najera, C.A.; Meneghelli, I.; Bahia, D. The parasitic intracellular lifestyle of Trypanosomatids: Parasitophorous vacuole development and survival. Front. Cell. Dev. Biol. 2020, 8, 396. [Google Scholar] [CrossRef]
- Descoteaux, A.; Turco, S.J. Glycoconjugates in Leishmania infectivity. Biochim. Biophys. Acta 1999, 1455, 341–352. [Google Scholar] [CrossRef] [Green Version]
- Pomel, S.; Loiseau, P.M. GDP-mannose: A key-point for target identification and drug design in kinetoplastids. In Trypanosomatid Diseases: Molecular Routes to Drug Discoveries; Jäger, T., Koch, O., Flohe, L., Eds.; Wiley-VCH Verlag GmbH and Co. KGaA: Weinheim, Germany, 2013; Volume 4, pp. 315–334. [Google Scholar]
- Pomel, S.; Mao, W.; Ha-Duong, T.; Cavé, C.; Loiseau, P.M. GDP-Mannose Pyrophosphorylase: A biologically validated target for drug development against leishmaniasis. Front. Cell. Infect. Dis. 2019, 9, 186. [Google Scholar] [CrossRef]
- Garami, A.; Ilg, T. Disruption of mannose activation in Leishmania mexicana: GDP-mannose pyrophosphorylase is required for virulence, but not for viability. EMBO J. 2001, 20, 3657–3666. [Google Scholar] [CrossRef] [Green Version]
- Stewart, J.; Curtis, J.; Spurck, T.P.; Ilg, T.; Garami, A.; Baldwin, T.; Courret, N.; McFadden, G.I.; Davis, A.; Handman, E. Characterisation of a Leishmania mexicana knockout lacking guanosine diphosphate-mannose pyrophosphorylase. Int. J. Parasitol. 2005, 35, 861–873. [Google Scholar] [CrossRef]
- Pomel, S.; Rodrigo, J.; Hendra, F.; Cavé, C.; Loiseau, P.M. In silico analysis of a therapeutic target in Leishmania infantum: The guanosine-diphospho-D-mannose pyrophosphorylase. Parasite 2012, 19, 63–70. [Google Scholar] [CrossRef] [Green Version]
- Daligaux, P.; Bernadat, G.; Tran, L.; Cavé, C.; Loiseau, P.M.; Pomel, S.; Ha-Duong, T. Comparative study of structural models of Leishmania donovani and human GDP-mannose pyrophosphorylases. Eur. J. Med. Chem. 2016, 107, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Lackovic, K.; Parisot, J.P.; Sleebs, N.; Baell, J.B.; Debien, L.; Watson, K.G.; Curtis, J.M.; Handman, E.; Street, I.P.; Kedzierski, L. Inhibitors of Leishmania GDP-mannose pyrophosphorylase identified by high-throughput screening of small-molecule chemical library. Antimicrob. Agents Chemother. 2010, 54, 1712–1719. [Google Scholar] [CrossRef] [Green Version]
- Mao, W.; Daligaux, P.; Lazar, N.; Ha-Duong, T.; Cavé, C.; van Tilbeurgh, H.; Loiseau, P.M.; Pomel, S. Biochemical analysis of leishmanial and human GDP-mannose pyrophosphorylases and selection of inhibitors as new leads. Sci. Rep. 2017, 7, 751. [Google Scholar] [CrossRef] [Green Version]
- Zahid, M.S.H.; Johnson, M.M.; Tokarski, R.J., 2nd; Satoskar, A.R.; Fuchs, J.R.; Bachelder, E.M.; Ainslie, K.M. Evaluation of synergy between host and pathogen-directed therapies against intracellular Leishmania donovani. Int. J. Parasitol. Drugs Drug Resist. 2019, 10, 125–132. [Google Scholar] [CrossRef] [PubMed]
- De Muylder, G.; Vanhollebeke, B.; Caljon, G.; Wolfe, A.R.; McKerrow, J.; Dujardin, J.C. Naloxonazine, an amastigote-specific compound, affects Leishmania parasites through modulation of host-encoded functions. PLoS Negl. Trop. Dis. 2016, 10, e0005234. [Google Scholar] [CrossRef]
- Gupta, N.; Noël, R.; Goudet, A.; Hinsinger, K.; Michau, A.; Pons, V.; Abdelkafi, H.; Secher, T.; Shima, A.; Shtanko, O.; et al. Inhibitors of retrograde trafficking active against ricin and Shiga toxins also protect cells from several viruses, Leishmania and Chlamydiales. Chem. Biol. Interact. 2017, 267, 96–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozubek, A.; Zarnowski, R.; Stasiuk, M.; Gubernator, J. Natural amphiphilic phenols as bioactive compounds. Cell. Mol. Biol. Lett. 2001, 6, 351–355. [Google Scholar]
- Stasiuk, M.; Kosubek, A. Biological activity of phenolic lipids. Cell. Mol. Life Sci. 2010, 67, 841–860. [Google Scholar] [CrossRef] [PubMed]
- Siwko, M.E.; de Vries, A.H.; Mark, A.E.; Kosubek, A.; Marrink, S.J. Disturb or stabilize? A molecular dynamics study of the effects of resorcinolic lipids on phospholipid bilayers. Biophys. J. 2009, 96, 3140–3153. [Google Scholar] [CrossRef] [Green Version]
- Gubernator, J.; Stasiuk, M.; Kosubek, A. Dual effect of alkylresorcinols, natural amphiphilic compounds, upon liposomal permeability. Biochim. Biophys. Acta 1999, 1418, 253–260. [Google Scholar] [CrossRef] [Green Version]
- Jimenez-Romero, C.; Torres-Mendoza, D.; Urena-Gonzalez, L.D.; Ortega-Barria, E.; McPhail, K.L.; Gerwick, W.H.; Cubilla-Rios, L. Hydroxyalkenylresorcinols from Stylogyne turbacensis. J. Nat. Prod. 2007, 70, 1249–1252. [Google Scholar] [CrossRef]
- Jin, W.; Zjawiony, J.K. 5-alkylresorcinols from Merulius incarnatus. J. Nat. Prod. 2006, 69, 704–706. [Google Scholar] [CrossRef] [PubMed]
- Gény, C.; Rivière, G.; Bignon, J.; Birlirakis, N.; Guittet, E.; Awang, K.; Litaudon, M.; Roussi, F.; Dumontet, V. Anacardic acids from Knema hookeriana as modulators of Bcl-xL/Bak and Mcl-1/Bid interactions. J. Nat. Prod. 2016, 79, 838–844. [Google Scholar] [CrossRef]
- Chaturvedula, V.S.; Schilling, J.K.; Miller, J.S.; Andriantsiferana, R.; Rasamison, V.E.; Kingston, D.G.I. New cytotoxic bis 5-alkylresorcinol derivatives from the leaves of Oncostemon bojerianum from the Madagascar rainforest. J. Nat. Prod. 2002, 65, 1627–1632. [Google Scholar] [CrossRef]
- Chuang, T.H.; Wu, P.L. Cytotoxic 5-alkylresorcinol metabolites from the leaves of Grevillea robusta. J. Nat. Prod. 2007, 70, 319–323. [Google Scholar] [CrossRef]
- Suzuki, Y.; Esumi, Y.; Yamagushi, I. Structure of 5-alkylresorcinol-related analogue in rye. Phytochemistry 1999, 52, 281–289. [Google Scholar] [CrossRef]
- Kozubek, A.; Tyman, J.H.P. Resorcinolic lipids, the natural non-isoprenoid phenolic amphiphiles and their biological activity. Chem. Rev. 1999, 99, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Madrigal, R.V.; Spencer, G.F.; Plattner, R.D.; Smith, C.R., Jr. Alkyl- and alkenylresorcinols in Rapanea laetevirens seed lipids. Lipids 1977, 12, 402–406. [Google Scholar] [CrossRef]
- Marner, F.J.; Horper, W. Phenols and quinones from seeds of different Iris species. Helvet. Chim. Acta 1992, 75, 1557–1562. [Google Scholar] [CrossRef]
- Davis, A.J.; Perugini, M.A.; Smith, B.J.; Stewart, J.D.; Ilg, T.; Hodder, A.N.; Handman, E. Properties of GDP-mannose pyrophosphorylase, a critical enzyme and drug target in Leishmania mexicana. J. Biol. Chem. 2004, 279, 12462–12468. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.H.; Chung, T.D.Y.; Oldenburg, K.R. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 1999, 4, 67–73. [Google Scholar] [CrossRef]
- Saar, Y.; Ransford, A.; Waldman, E.; Mazareb, S.; Amin-Spector, S.; Plumblee, J.; Turco, S.J.; Zilberstein, D. Characterization of developmentally-regulated activities in axenic amastigotes of Leishmania donovani. Mol. Biochem. Parasitol. 1998, 95, 9–20. [Google Scholar] [CrossRef]


{kind=link}
| Name | LiGDP-MP IC50 (µM) ± SD | hGDP-MP IC50 (µM) ± SD | L. infantum Axenic Amastigotes IC50 (µM) ± SD | L. infantum Intramacrophage Amastigotes IC50 (µM) ± SD | Cytotoxicity on RAW 264.7 CC50 (µM) ± SD | Selectivity Index |
|---|---|---|---|---|---|---|
| 1 Oncostemonol D | 3.5 ± 11.4 | 1.3 ± 0.2 | 78.9 ± 3.1 | >50 | 34.4 ± 1.5 | <0.7 |
| 2 Oncostemonol I | 5.9 ± 3.5 | 13.7 ± 14.2 | ND | ND | ND | ND |
| 3 Oncostemonol J | 2.3 ± 8.1 | 1.9 ± 0.4 | 49.5 ± 4.2 | >25 | 31.8 ± 1.6 | <1.2 |
| 4 Khookerianic acid B | 120.0 ± 27.5 | 79.9 ± 5.9 | >100 | >100 | 83.1 ± 7.0 | <0.8 |
| 5 Anagigantic acid | 50.6 ± 4.8 | 38.3 ± 4.1 | >100 | >100 | >100 | / |
| 6 | 100.2 ± 16.6 | 122.2 ± 26.1 | >100 | 42.7 ± 1.3 | >100 | >2.3 |
| 7 | 16.9 ± 5.0 | 29.0 ± 2.5 | >100 | >100 | >100 | / |
| 8 | 9.8 ± 3.2 | 13.9 ± 0.6 | >100 | >100 | >100 | / |
| 9 | 11.0 ± 4.7 | 31.1 ± 10.7 | 53.3 ± 4.6 | >25 | 26.1 ± 6.4 | <1 |
| 10 | 14.1 ± 3.8 | 21.9 ± 6.3 | 25.9 ± 5.9 | >25 | 17.9 ± 1.3 | <0.7 |
| 11 | >300 | ND | 13.3 ± 4.4 | 9.5 ± 2.1 | 32.6 ± 7.4 | 3.4 |
| Miltefosine | >250 | ND | 1.0 ± 0.3 | 6.7 ± 1.7 | 54.2 ± 5.8 | 8.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Levaique, H.; Pamlard, O.; Apel, C.; Bignon, J.; Arriola, M.; Kuhner, R.; Awang, K.; Loiseau, P.M.; Litaudon, M.; Pomel, S. Alkyl-Resorcinol Derivatives as Inhibitors of GDP-Mannose Pyrophosphorylase with Antileishmanial Activities. Molecules 2021, 26, 1551. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26061551
Levaique H, Pamlard O, Apel C, Bignon J, Arriola M, Kuhner R, Awang K, Loiseau PM, Litaudon M, Pomel S. Alkyl-Resorcinol Derivatives as Inhibitors of GDP-Mannose Pyrophosphorylase with Antileishmanial Activities. Molecules. 2021; 26(6):1551. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26061551
Chicago/Turabian StyleLevaique, Hélène, Olivier Pamlard, Cécile Apel, Jérôme Bignon, Margaux Arriola, Robin Kuhner, Khalijah Awang, Philippe M. Loiseau, Marc Litaudon, and Sébastien Pomel. 2021. "Alkyl-Resorcinol Derivatives as Inhibitors of GDP-Mannose Pyrophosphorylase with Antileishmanial Activities" Molecules 26, no. 6: 1551. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26061551