
7-(2-Anilinopyrimidin-4-yl)-1-benzazepin-2-ones Designed by a “Cut and Glue” Strategy Are Dual Aurora A/VEGF-R Kinase Inhibitors


 , and
, and
Abstract
:1. Introduction
2. Results
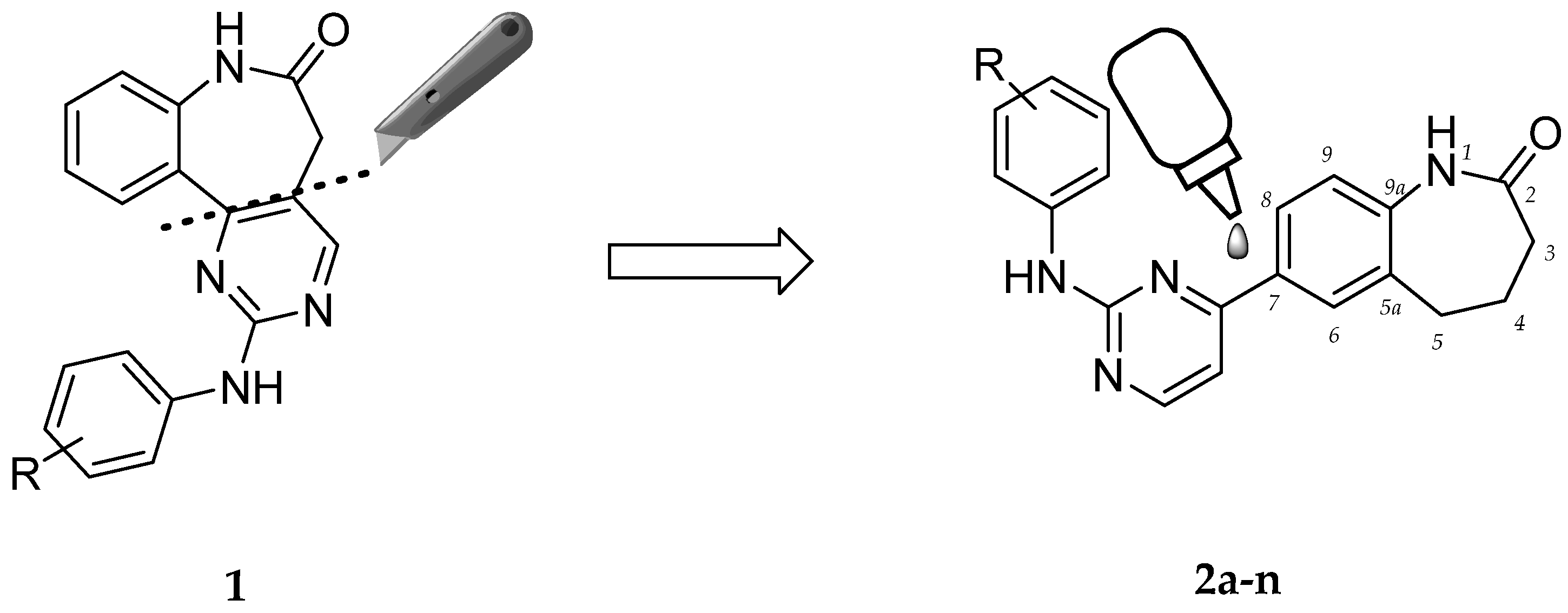
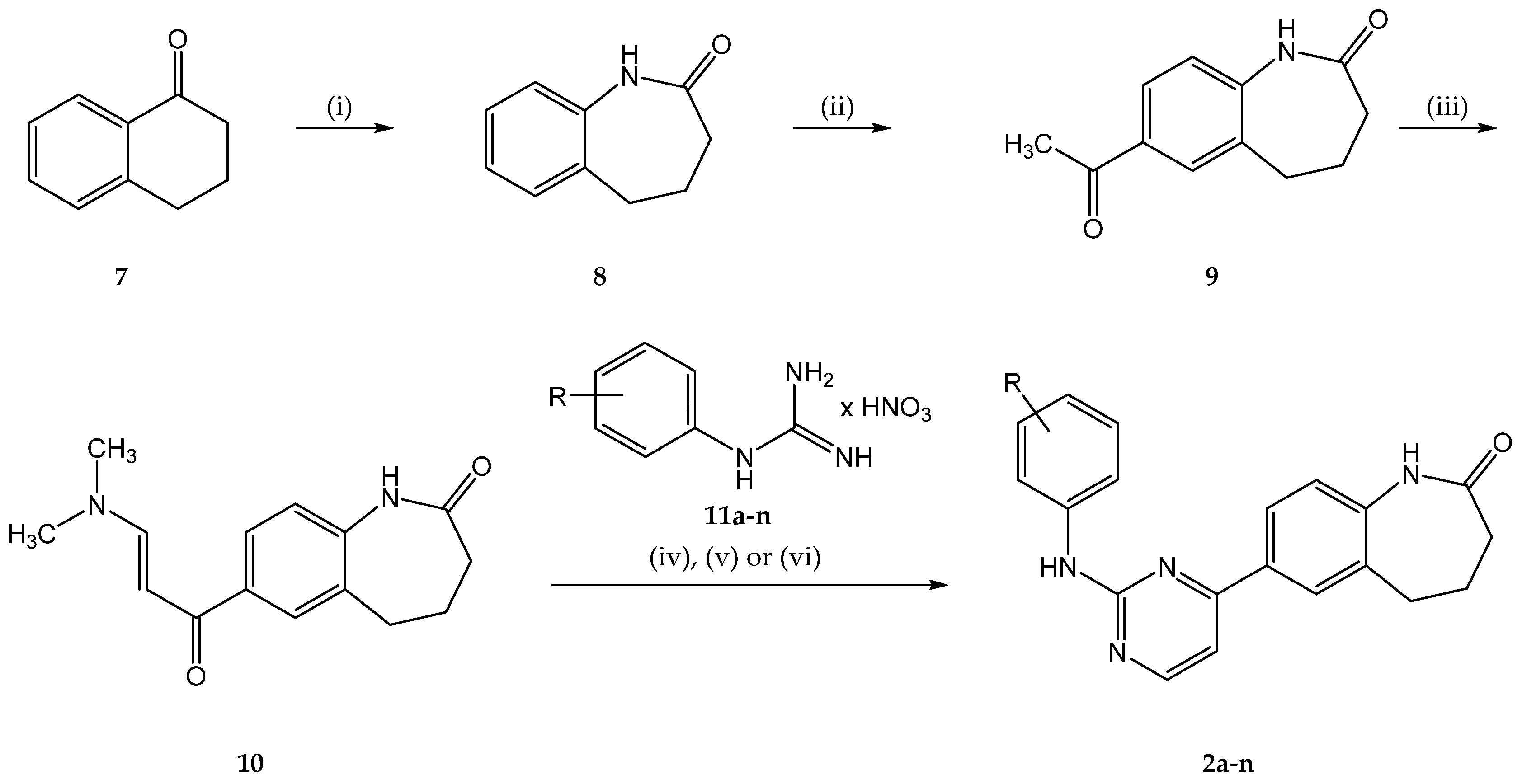
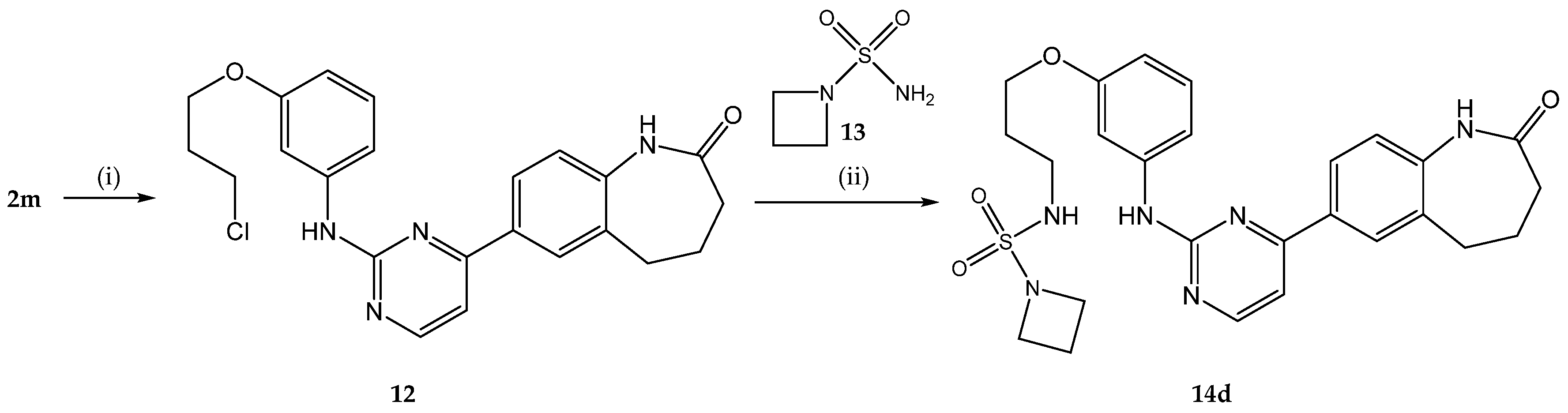
2.1. Chemistry
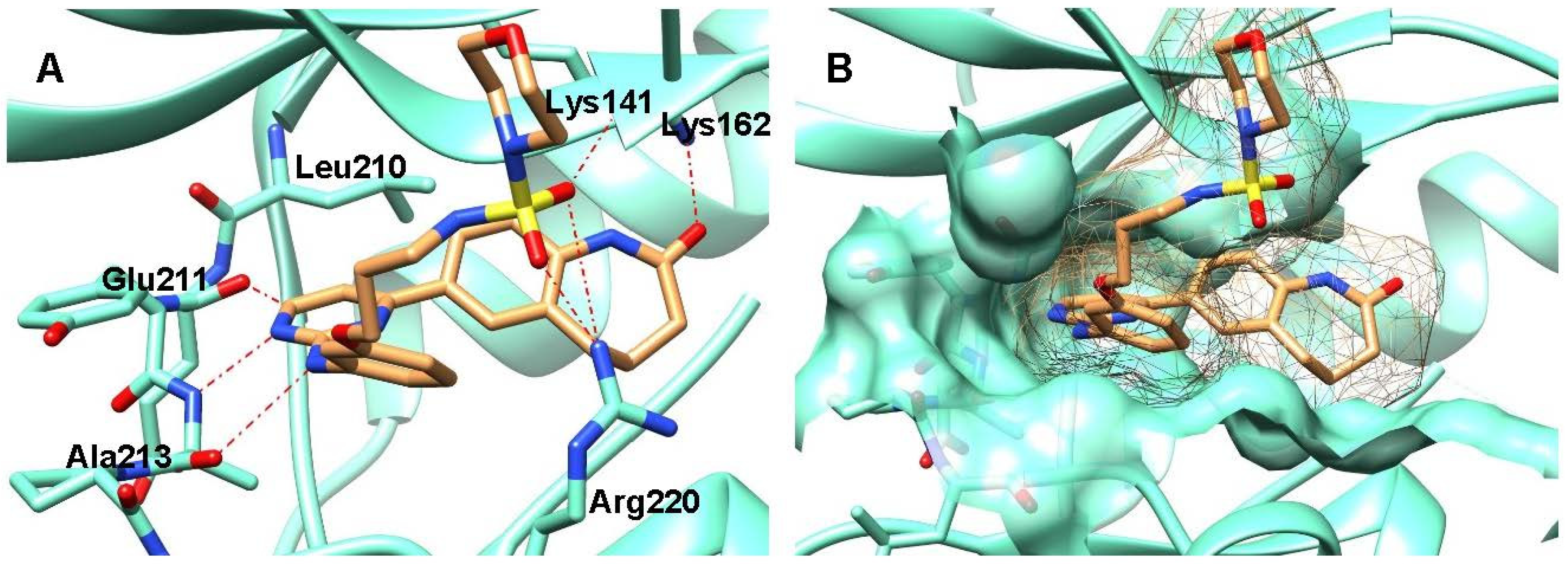
2.2. Molecular Docking
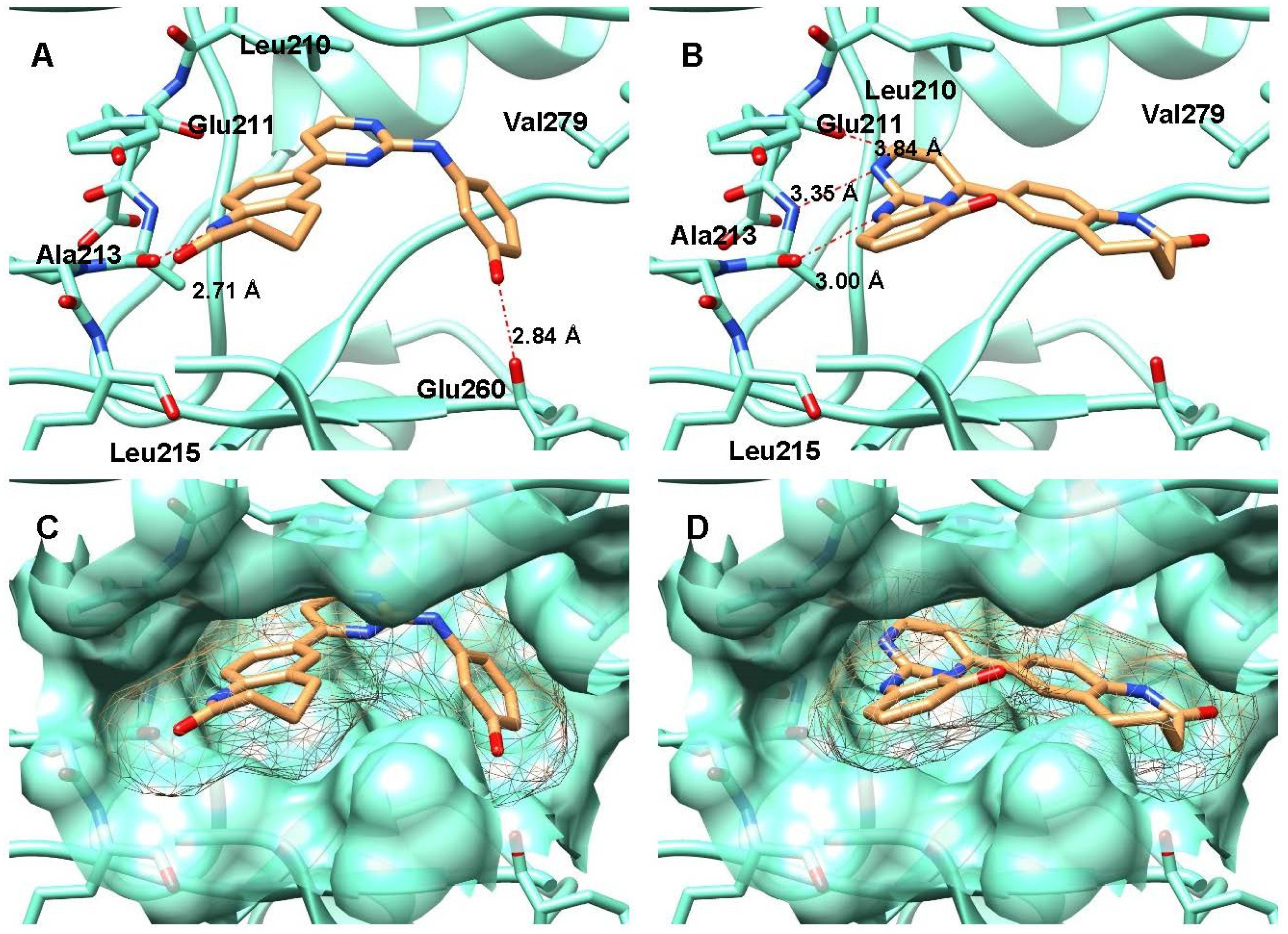
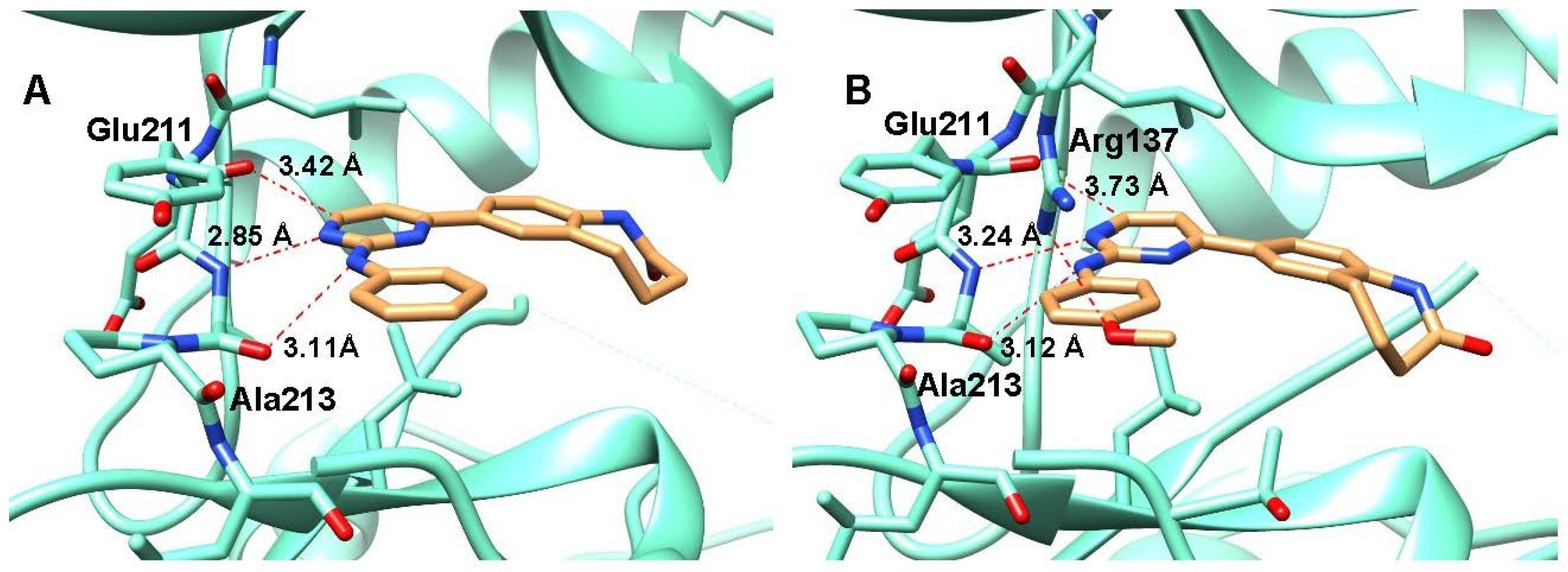
2.3. Crystal Structure Analyses
2.4. Protein Kinase Inhibition
2.5. Kinetic Solubility
2.6. Antiproliferative Activity on Cancer Cell Lines
3. Discussion
4. Materials and Methods
4.1. General Information
4.2. Syntheses
4.2.1. Syntheses of Intermediates
4.2.2. General Procedure A for the Synthesis of 7-(2-Anilinopyrimidin-4-yl)-1,3,4,5-tetrahydro-2H-1-benzazepin-2-ones 2b,d,e,f,g,h,i,k,l
4.2.3. General Procedure B for the Synthesis of 7-(2-Anilinopyrimidin-4-yl)-1,3,4,5-tetrahydro-2H-1-benzazepin-2-ones 2c,m,n
4.2.4. General Procedure C for the Synthesis of 2-Methyl-1,2,5-thiadiazolidine-1,1-dioxide Derivatives 14a,e
4.2.5. General Procedure D for the Synthesis of N-Alkylated Sulfamide Derivatives 14b,c,d,f,g
4.2.6. Detailed Chemical Synthesis Procedures and Characterization
4.3. Molecular Docking
4.4. Crystallization of Aurora A Complexes, Data Collection and Structure Determination
4.5. Protein Kinase Inhibition Assays
4.6. Kinetic Solubility
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Ferguson, F.M.; Gray, N.S. Kinase inhibitors: The road ahead. Nat. Rev. Drug Discov. 2018, 17, 353–377. [Google Scholar] [CrossRef] [PubMed]
- Lightfoot, H.L.; Goldberg, F.W.; Sedelmeier, J. Evolution of small molecule kinase drugs. ACS Med. Chem. Lett. 2019, 10, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R. A historical overview of protein kinases and their targeted small molecule inhibitors. Pharmacol. Res. 2015, 100, 1–23. [Google Scholar] [CrossRef]
- Egert-Schmidt, A.-M.; Dreher, J.; Dunkel, U.; Kohfeld, S.; Preu, L.; Weber, H.; Ehlert, J.E.; Mutschler, B.; Totzke, F.; Schächtele, C.; et al. Identification of 2-anilino-9-methoxy-5,7-dihydro-6H-pyrimido5,4-d1benzazepin-6-ones as dual PLK1/VEGF-R2 kinase inhibitor chemotypes by structure-based lead generation. J. Med. Chem. 2010, 53, 2433–2442. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Wu, H.; Wang, L.; Liu, Y.; Knapp, S.; Liu, Q.; Gray, N.S. Exploration of type II binding mode: A privileged approach for kinase inhibitor focused drug discovery? ACS Chem. Biol. 2014, 9, 1230–1241. [Google Scholar] [CrossRef]
- Berger, B. Dissertation; Technische Universität Braunschweig: Braunschweig, Germany, 2007. [Google Scholar]
- Karatas, M. Dissertation; Technische Universität Braunschweig: Braunschweig, Germany, 2020. [Google Scholar]
- Zhu, X.-D.; Tang, Z.-Y.; Sun, H.-C. Targeting angiogenesis for liver cancer: Past, present, and future. Genes Dis. 2020, 7, 328–335. [Google Scholar] [CrossRef]
- Wilhelm, S.M.; Carter, C.; Tang, L.; Wilkie, D.; McNabola, A.; Rong, H.; Chen, C.; Zhang, X.; Vincent, P.; McHugh, M.; et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004, 64, 7099–7109. [Google Scholar] [CrossRef] [Green Version]
- Yakes, F.M.; Chen, J.; Tan, J.; Yamaguchi, K.; Shi, Y.; Yu, P.; Qian, F.; Chu, F.; Bentzien, F.; Cancilla, B.; et al. Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth. Mol. Cancer Ther. 2011, 10, 2298–2308. [Google Scholar] [CrossRef] [Green Version]
- Vader, G.; Lens, S.M.A. The Aurora kinase family in cell division and cancer. Biochim. Biophys. Acta 2008, 1786, 60–72. [Google Scholar] [CrossRef]
- Willems, E.; Dedobbeleer, M.; Digregorio, M.; Lombard, A.; Lumapat, P.N.; Rogister, B. The functional diversity of Aurora kinases: A comprehensive review. Cell Div. 2018, 13, 7. [Google Scholar] [CrossRef] [Green Version]
- Gautschi, O.; Heighway, J.; Mack, P.C.; Purnell, P.R.; Lara, P.N.; Gandara, D.R. Aurora kinases as anticancer drug targets. Clin. Cancer Res. 2008, 14, 1639–1648. [Google Scholar] [CrossRef] [Green Version]
- Yan, M.; Wang, C.; He, B.; Yang, M.; Tong, M.; Long, Z.; Liu, B.; Peng, F.; Xu, L.; Zhang, Y.; et al. Aurora-A kinase: A potent oncogene and target for cancer therapy. Med. Res. Rev. 2016, 36, 1036–1079. [Google Scholar] [CrossRef] [PubMed]
- Lok, W.; Klein, R.Q.; Saif, M.W. Aurora kinase inhibitors as anti-cancer therapy. Anticancer Drugs 2010, 21, 339–350. [Google Scholar] [CrossRef]
- Kitzen, J.J.E.M.; de Jonge, M.J.A.; Verweij, J. Aurora kinase inhibitors. Crit. Rev. Oncol. Hematol. 2010, 73, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Katayama, H.; Sen, S. Aurora kinase inhibitors as anticancer molecules. Biochim. Biophys. Acta 2010, 1799, 829–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liewer, S.; Huddleston, A. Alisertib: A review of pharmacokinetics, efficacy and toxicity in patients with hematologic malignancies and solid tumors. Expert Opin. Investig. Drugs 2018, 27, 105–112. [Google Scholar] [CrossRef]
- Tayyar, Y.; Shiels, R.; Bulmer, A.C.; Lam, A.K.; Clarke, D.; Idris, A.; McMillan, N.A. Development of an intravaginal ring for the topical delivery of Aurora kinase A inhibitor, MLN8237. PLoS ONE 2019, 14, e0225774. [Google Scholar] [CrossRef]
- Huisgen, R.; Ugi, I.; Brade, H.; Rauenbusch, E. Medium sized rings. III. Properties and reactions of the 1,2-benzolactams. Justus Liebigs Ann. Chem. 1954, 586, 30–51. [Google Scholar] [CrossRef]
- Augustine, R.L.; Pierson, W.G. Synthesis of dl-deethylibogamine. J. Org. Chem. 1969, 34, 1070–1075. [Google Scholar] [CrossRef]
- Chen, W.-Y.; Gilman, N.W. Synthesis of 7-phenylpyrimido[5,4-d][1]benzazepin-2-ones. J. Heterocycl. Chem. 1983, 20, 663–666. [Google Scholar] [CrossRef]
- Moffat, D.F.C.; Allen, R.A.; Rapecki, S.E.; Davis, P.D.; O’Connell, J.; Hutchings, M.C.; King, M.A.; Boyce, B.A.; Perry, M.J. 4-Thiophenoxy-N-(3,4,5-trialkoxyphenyl)pyrimidine-2-amines as potent and selective inhibitors of the T-cell tyrosine kinase p56lck. Curr. Med. Chem. 2004, 11, 747–753. [Google Scholar] [CrossRef]
- Dodson, C.A.; Kosmopoulou, M.; Richards, M.W.; Atrash, B.; Bavetsias, V.; Blagg, J.; Bayliss, R. Crystal structure of an Aurora-A mutant that mimics Aurora-B bound to MLN8054: Insights into selectivity and drug design. Biochem. J. 2010, 427, 19–28. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [Green Version]
- Iversen, P.W.; Eastwood, B.J.; Sittampalam, G.S.; Cox, K.L. A comparison of assay performance measures in screening assays: Signal window, Z’ factor, and assay variability ratio. J. Biomol. Screen. 2006, 11, 247–252. [Google Scholar] [CrossRef]
- Zhang, J.H.; Chung, T.D.; Oldenburg, K.R. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 1999, 4, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Hart, M.L. Brief overview of various approaches to enhance drug solubility. J. Dev. Drugs 2013, 2. [Google Scholar] [CrossRef] [Green Version]
- Meanwell, N.A. Improving drug candidates by design: A focus on physicochemical properties as a means of improving compound disposition and safety. Chem. Res. Toxicol. 2011, 24, 1420–1456. [Google Scholar] [CrossRef] [PubMed]
- Williams, H.D.; Trevaskis, N.L.; Charman, S.A.; Shanker, R.M.; Charman, W.N.; Pouton, C.W.; Porter, C.J.H. Strategies to address low drug solubility in discovery and development. Pharmacol. Rev. 2013, 65, 315–499. [Google Scholar] [CrossRef] [PubMed]
- Kerns, E.H.; Di, L.; Carter, G.T. In vitro solubility assays in drug discovery. Curr. Drug Metab. 2008, 9, 879–885. [Google Scholar] [CrossRef] [PubMed]
- Lovering, F.; Bikker, J.; Humblet, C. Escape from flatland: Increasing saturation as an approach to improving clinical success. J. Med. Chem. 2009, 52, 6752–6756. [Google Scholar] [CrossRef] [PubMed]
- Hoelke, B.; Gieringer, S.; Arlt, M.; Saal, C. Comparison of nephelometric, UV-spectroscopic, and HPLC methods for high-throughput determination of aqueous drug solubility in microtiter plates. Anal. Chem. 2009, 81, 3165–3172. [Google Scholar] [CrossRef]
- Grever, M.R.; Schepartz, S.A.; Chabner, B.A. The National Cancer Institute: Cancer drug discovery and development program. Semin. Oncol. 1992, 19, 622–638. [Google Scholar]
- Shoemaker, R.H. The NCI60 human tumour cell line anticancer drug screen. Nat. Rev. Cancer 2006, 6, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, J.N.; Myers, T.G.; O’Connor, P.M.; Friend, S.H.; Fornace, A.J.; Kohn, K.W.; Fojo, T.; Bates, S.E.; Rubinstein, L.V.; Anderson, N.L.; et al. An information-intensive approach to the molecular pharmacology of cancer. Science 1997, 275, 343–349. [Google Scholar] [CrossRef] [Green Version]
- Monks, A.; Scudiero, D.; Skehan, P.; Shoemaker, R.; Paull, K.; Vistica, D.; Hose, C.; Langley, J.; Cronise, P.; Vaigro-Wolff, A. Feasibility of a high-flux anticancer drug screen using a diverse panel of cultured human tumor cell lines. J. Natl. Cancer Inst. 1991, 83, 757–766. [Google Scholar] [CrossRef]
- Rubinstein, L.V.; Shoemaker, R.H.; Paull, K.D.; Simon, R.M.; Tosini, S.; Skehan, P.; Scudiero, D.A.; Monks, A.; Boyd, M.R. Comparison of in vitro anticancer-drug-screening data generated with a tetrazolium assay versus a protein assay against a diverse panel of human tumor cell lines. J. Natl. Cancer Inst. 1990, 82, 1113–1118. [Google Scholar] [CrossRef] [PubMed]
- Prasad, V.V.; Gopalan, R.O. Continued use of MDA-MB-435, a melanoma cell line, as a model for human breast cancer, even in year, 2014. NPJ Breast Cancer 2015, 1, 15002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liscovitch, M.; Ravid, D. A case study in misidentification of cancer cell lines: MCF-7/AdrR cells (re-designated NCI/ADR-RES) are derived from OVCAR-8 human ovarian carcinoma cells. Cancer Lett. 2007, 245, 350–352. [Google Scholar] [CrossRef] [PubMed]
- Jani, J.P.; Arcari, J.; Bernardo, V.; Bhattacharya, S.K.; Briere, D.; Cohen, B.D.; Coleman, K.; Christensen, J.G.; Emerson, E.O.; Jakowski, A.; et al. PF-03814735, an orally bioavailable small molecule aurora kinase inhibitor for cancer therapy. Mol. Cancer Ther. 2010, 9, 883–894. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Midgley, C.A.; Scaërou, F.; Grabarek, J.B.; Griffiths, G.; Jackson, W.; Kontopidis, G.; McClue, S.J.; McInnes, C.; Meades, C.; et al. Discovery of N-phenyl-4-(thiazol-5-yl)pyrimidin-2-amine aurora kinase inhibitors. J. Med. Chem. 2010, 53, 4367–4378. [Google Scholar] [CrossRef] [Green Version]
- Huck, J.J.; Zhang, M.; McDonald, A.; Bowman, D.; Hoar, K.M.; Stringer, B.; Ecsedy, J.; Manfredi, M.G.; Hyer, M.L. MLN8054, an inhibitor of Aurora A kinase, induces senescence in human tumor cells both in vitro and in vivo. Mol. Cancer Res. 2010, 8, 373–384. [Google Scholar] [CrossRef] [Green Version]
- Sells, T.B.; Chau, R.; Ecsedy, J.A.; Gershman, R.E.; Hoar, K.; Huck, J.; Janowick, D.A.; Kadambi, V.J.; LeRoy, P.J.; Stirling, M.; et al. MLN8054 and Alisertib (MLN8237): Discovery of selective oral Aurora A inhibitors. ACS Med. Chem. Lett. 2015, 6, 630–634. [Google Scholar] [CrossRef]
- OBrien, Z.; Moghaddam, M.F. A systematic analysis of physicochemical and ADME properties of all small molecule kinase inhibitors approved by US FDA from January 2001 to October 2015. Curr. Med. Chem. 2017, 24, 3159–3184. [Google Scholar] [CrossRef] [Green Version]
- Herbrink, M.; Schellens, J.H.M.; Beijnen, J.H.; Nuijen, B. Inherent formulation issues of kinase inhibitors. J. Control. Release 2016, 239, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Monks, A.; Scudiero, D.A.; Johnson, G.S.; Paull, K.D.; Sausville, E.A. The NCI anti-cancer drug screen: A smart screen to identify effectors of novel targets. Anticancer Drug Des. 1997, 12, 533–541. [Google Scholar]
- Bates, S.E.; Fojo, A.T.; Weinstein, J.N.; Myers, T.G.; Alvarez, M.; Pauli, K.D.; Chabner, B.A. Molecular targets in the National Cancer Institute drug screen. J. Cancer Res. Clin. Oncol. 1995, 121, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Kunick, C.; Bleeker, C.; Prühs, C.; Totzke, F.; Schächtele, C.; Kubbutat, M.H.G.; Link, A. Matrix compare analysis discriminates subtle structural differences in a family of novel antiproliferative agents, diaryl-3-hydroxy-2,3,3a,10a-tetrahydrobenzobcycylopentaeazepine-4,10(1H,5H)-diones. Bioorg. Med. Chem. Lett. 2006, 16, 2148–2153. [Google Scholar] [CrossRef] [PubMed]
- Paull, K.D.; Shoemaker, R.H.; Hodes, L.; Monks, A.; Scudiero, D.A.; Rubinstein, L.; Plowman, J.; Boyd, M.R. Display and analysis of patterns of differential activity of drugs against human tumor cell lines: Development of mean graph and COMPARE algorithm. J. Natl. Cancer Inst. 1989, 81, 1088–1092. [Google Scholar] [CrossRef]
- Prühs, C.; Kunick, C. Darpones and water-soluble aminobutoxylated darpone derivatives are distinguished by matrix COMPARE analysis. Bioorg. Med. Chem. Lett. 2007, 17, 1850–1854. [Google Scholar] [CrossRef]
- Paull, K.D.; Lin, C.M.; Malspeis, L.; Hamel, E. Identification of novel antimitotic agents acting at the tubulin level by computer-assisted evaluation of differential cytotoxicity data. Cancer Res. 1992, 52, 3892–3900. [Google Scholar]
- Andreani, A.; Burnelli, S.; Granaiola, M.; Leoni, A.; Locatelli, A.; Morigi, R.; Rambaldi, M.; Varoli, L.; Landi, L.; Prata, C.; et al. Antitumor activity of bis-indole derivatives. J. Med. Chem. 2008, 51, 4563–4570. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferguson, F.M.; Doctor, Z.M.; Chaikuad, A.; Sim, T.; Kim, N.D.; Knapp, S.; Gray, N.S. Characterization of a highly selective inhibitor of the Aurora kinases. Bioorg. Med. Chem. Lett. 2017, 27, 4405–4408. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Evans, P.R.; Murshudov, G.N. How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr. 2013, 69, 1204–1214. [Google Scholar] [CrossRef]
- McCoy, A.J. Acknowledging errors: Advanced molecular replacement with Phaser. Methods Mol. Biol. 2017, 1607, 421–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emsley, P. Tools for ligand validation in Coot. Acta Crystallogr. D Struct. Biol. 2017, 73, 203–210. [Google Scholar] [CrossRef] [Green Version]
- Skubák, P.; Murshudov, G.N.; Pannu, N.S. Direct incorporation of experimental phase information in model refinement. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2196–2201. [Google Scholar] [CrossRef] [Green Version]
- Williams, C.J.; Headd, J.J.; Moriarty, N.W.; Prisant, M.G.; Videau, L.L.; Deis, L.N.; Verma, V.; Keedy, D.A.; Hintze, B.J.; Chen, V.B.; et al. MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 2018, 27, 293–315. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDB Accession Code | 2a 7AYI | 2c 7AYH |
|---|---|---|
| Data collection | Data collection | |
| beamline | Swiss light source X06SA | Swiss light source X06SA |
| Resolution a (Å) | 44.43–2.86 (3.01–2.86) | 44.28–2.80 (2.95–2.80) |
| space group | P 6122 | P 6122 |
| cell dimensions | ||
| a (Å) | 82.6 | 83.4 |
| b (Å) | 82.6 | 83.4 |
| c (Å) | 170.1 | 168.2 |
| α (deg) | 90 | 90 |
| β (deg) | 90 | 90 |
| γ (deg) | 120 | 120 |
| no. unique reflections a | 8526 (1186) | 9066 (1279) |
| Completeness a (%) | 100.0 (100.0) | 99.9 (100.0) |
| I/σI a | 25.7 (2.1) | 19.3 (2.1) |
| Rmerge a | 0.038 (0.975) | 0.046 (0.968) |
| CC(1/2) | 1.000 (0.797) | 0.999 (0.818) |
| Redundancy a | 8.4 (8.6) | 8.5 (9.0) |
| no. atoms in refinement (P/L) b | 2014/25 | 2004/27 |
| Rfact (%) | 25.2 | 27.3 |
| Rfree (%) | 29.7 | 31.3 |
| Bf (P/L/O) b (Å2) | 121/115 | 133/112 |
| rmsd bond c (Å) | 0.011 | 0.011 |
| rmsd deviation angle c (deg) | 0.96 | 1.14 |
| Ramachandran favored | 94.35 | 95.12 |
| Ramachandran outlier | 0 | 0 |

| Protein Kinase | IC50 (µM) | Protein Kinase | IC50 (µM) |
|---|---|---|---|
| AKT1 | >100 | IGF1-R | >100 |
| ARK5 | >100 | INS-R | >100 |
| Aurora-A | 2.4 | MET | >100 |
| Aurora-B | 26 | PAK4 | >100 |
| CDK2/CycA | 60 | PDGFR-beta | 44 |
| CDK4/CycD1 | 19 | PDK1 | >100 |
| CK2-alpha1 | >100 | PLK1 | >100 |
| EGF-R | >100 | SAK | >100 |
| EPHB4 | >100 | SRC | 74 |
| ERBB2 | >100 | TIE2 | 19 |
| FAK | >100 | VEGF-R2 | 2.8 |
| FLT3 | 16 | VEGF-R3 | 5.9 |

| Compound | R | Aurora A | Aurora B | VEGF-R2 | VEGF-R3 |
|---|---|---|---|---|---|
| 2a | H | 2.4 µM | 26 µM | 2.8 µM | 5.9 µM |
| 2b | 4-CH3 | 2.4 µM | 16 µM | 1.8 µM | 4.3 µM |
| 2c | 4-OCH3 | 1.8 µM | 18 µM | 1.3 µM | 2.5 µM |
| 2d | 4-OC2H5 | 1.5 µM | 20 µM | 2.0 µM | 3.0 µM |
| 2e | 4-OH | 0.99 µM | 3.5 µM | 1.1 µM | 1.5 µM |
| 2f | 4-Cl | 9.4 µM | 40 µM | 11 µM | 19 µM |
| 2g | 4-Br | 3.5 µM | 18 µM | 5.4 µM | 4.1 µM |
| 2h | 4-I | 3.5 µM | 52 µM | 17 µM | 6.2 µM |
| 2i | 4-NO2 | >100 µM | >100 µM | 11 µM | 35 µM |
| 2j | 2-OH | >100 µM | >100 µM | 8.4 µM | 19 µM |
| 2k | 2-Cl | 74% | 87% | 74% | 72% |
| 2l | 2-Br | 73% | 116% | 78% | 64% |
| 2m | 3-OH | 1.7 µM | 6.7 µM | 0.78 µM | 0.54 µM |
| 2n | 3-OH, 4-OCH3 | 1.4 µM | 4.5 µM | 0.63 µM | 0.55 µM |

| Compound | R1 | R2 | Aurora-A IC50 (µM) | Kinetic Solubility (µM) |
|---|---|---|---|---|
| 2a | H | H | 2.4 | 50.3 (48.5–52.0) |
| 2m | H | -OH | 1.7 | 112 (109–114) |
| 2n | -OCH3 | -OH | 1.4 | 92.6 (92.4–92.8) |
| 14a | H |  | 1.8 | 25.3 c |
| 14b | H |  | 3.0 | 19.6 (18.8–20.4) |
| 14c | H |  | 4.5 | 25.3 (23.8–26.8) |
| 14d | H |  | 1.5 | 25.2 (20.2–30.2) |
| 14e | OCH3 |  | 1.1 | 21.4 (20.7–22.1) |
| 14f | OCH3 |  | 2.5 | 25.0 (24.4–25.7) |
| 14g | OCH3 |  | 0.82 | 34.2 (31.0–36.9) |
| Cell Line | Cancer | 2e | 2n | Cell Line | Cancer | 2e | 2n |
|---|---|---|---|---|---|---|---|
| CCRF-CEM | Leukemia | −5.60 | −5.41 | MDA-MB-435 b | Melanoma | −5.44 | −5.28 |
| HL-60(TB) | Leukemia | −5.86 | −5.50 | SK-MEL-2 | Melanoma | −5.68 | −5.04 |
| K-562 | Leukemia | −5.43 | −5.37 | SK-MEL-28 | Melanoma | −5.24 | −5.04 |
| RPMl-8226 | Leukemia | −5.49 | −5.28 | SK-MEL-5 | Melanoma | −5.51 | −5.00 |
| SR | Leukemia | −5.88 | −5.50 | UACC-257 | Melanoma | −5.38 | −4.83 |
| A549/ATCC | NSCL | −5.35 | −5.10 | UACC-62 | Melanoma | −5.04 | −5.03 |
| EKVX | NSCL | −4.46 | −4.36 | IGROV1 | Ovarian | −5.60 | −5.52 |
| HOP-62 | NSCL | −4.96 | −5.09 | NCI/ADR-RES b | Ovarian | >−4.00 | −4.85 |
| HOP-92 | NSCL | −5.18 | −6.67 | OVCAR-3 | Ovarian | −5.23 | −5.00 |
| NCI-H226 | NSCL | −4.53 | −5.04 | OVCAR-4 | Ovarian | −5.25 | −5.12 |
| NCI-H23 | NSCL | >−4.00 | −4.93 | OVCAR-5 | Ovarian | −5.19 | −5.06 |
| NCI-H322M | NSCL | −5.39 | −4.51 | OVCAR-8 | Ovarian | −5.39 | −5.01 |
| NCI-H460 | NSCL | −5.50 | −5.33 | SK-OV-3 | Ovarian | −5.52 | −5.28 |
| NCI-H522 | NSCL | −5.35 | −5.00 | 786-0 | Renal | −5.36 | −5.34 |
| COLO 205 | Colon | n.a. | −4.58 | A498 | Renal | −5.32 | −4.43 |
| HCC-2998 | Colon | −5.06 | −4.64 | ACHN | Renal | −5.59 | −5.38 |
| HCT-116 | Colon | −5.44 | −5.42 | CAKl-1 | Renal | −5.38 | −5.25 |
| HCT-15 | Colon | −5.49 | −5.34 | RXF 393 | Renal | −5.73 | −5.57 |
| HT29 | Colon | −5.29 | −4.88 | SN12C | Renal | −5.21 | −5.24 |
| KM12 | Colon | −5.59 | −5.32 | TK-10 | Renal | −5.42 | −4.63 |
| SW-620 | Colon | −5.32 | −5.35 | U0-31 | Renal | −5.76 | −5.52 |
| SF-268 | CNS | −5.36 | −5.13 | PC-3 | Prostate | −5.31 | −4.90 |
| SF-539 | CNS | −5.38 | −5.35 | DU-145 | Prostate | −5.47 | −5.30 |
| SNB-75 | CNS | −5.47 | −5.66 | MCF7 | Breast | −5.37 | −5.29 |
| U251 | CNS | −5.22 | −4.99 | MDA-MB-231/ATCC | Breast | >−4.00 | >−4.00 |
| LOX IMVI | Melanoma | −5.47 | −5.26 | HS 578T | Breast | −5.31 | −5.67 |
| MALME-3M | Melanoma | −5.66 | >−4.00 | BT-549 | Breast | −5.29 | −5.25 |
| M14 | Melanoma | −5.39 | −5.36 | T-47D | Breast | −5.81 | −5.39 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karatas, M.; Chaikuad, A.; Berger, B.; Kubbutat, M.H.G.; Totzke, F.; Knapp, S.; Kunick, C. 7-(2-Anilinopyrimidin-4-yl)-1-benzazepin-2-ones Designed by a “Cut and Glue” Strategy Are Dual Aurora A/VEGF-R Kinase Inhibitors. Molecules 2021, 26, 1611. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26061611
Karatas M, Chaikuad A, Berger B, Kubbutat MHG, Totzke F, Knapp S, Kunick C. 7-(2-Anilinopyrimidin-4-yl)-1-benzazepin-2-ones Designed by a “Cut and Glue” Strategy Are Dual Aurora A/VEGF-R Kinase Inhibitors. Molecules. 2021; 26(6):1611. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26061611
Chicago/Turabian StyleKaratas, Mehmet, Apirat Chaikuad, Bianca Berger, Michael H. G. Kubbutat, Frank Totzke, Stefan Knapp, and Conrad Kunick. 2021. "7-(2-Anilinopyrimidin-4-yl)-1-benzazepin-2-ones Designed by a “Cut and Glue” Strategy Are Dual Aurora A/VEGF-R Kinase Inhibitors" Molecules 26, no. 6: 1611. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26061611