
Site-Selective Artificial Ribonucleases: Renaissance of Oligonucleotide Conjugates for Irreversible Cleavage of RNA Sequences

 ,
,
Abstract
:1. Introduction
1.1. From Antisense Oligonucleotides to Site-Selective Ribonucleases
1.2. Initial Stage of aRNase Development: Screening of Chemical Groups and General Structures
2. Synthetic Approaches Applied for the Generation of Site-Selective Artificial Ribonucleases
2.1. Fragment Conjugation in Solution: Application to Peptidyl-Oligonucleotide Conjugate Synthesis
2.2. Fragment Conjugation on the Solid Support
2.3. Solid-Phase Synthesis
3. In Vitro Characteristics of Artificial Ribonucleases
3.1. Chemical Moieties Used for Creating aRNases and the Corresponding Mechanism of Cleavage
3.1.1. Acridines and Azacrowns
3.1.2. Trisbenzimidazole
3.1.3. Peptide [(LeuArg)nGly]m
3.2. RNA-Recognition Domains of ss-aRNases: Structure and Modification
3.2.1. Principles of Short RNA Target Cleavage
3.2.2. Principles of Long RNA Target Cleavage
3.3. Specificity of Cleavage
3.3.1. Site-Selectivity
3.3.2. Non-Complementary Substrates
3.4. Chemical Modifications of Oligonucleotide Domain: Influence on ss-aRNase Performance
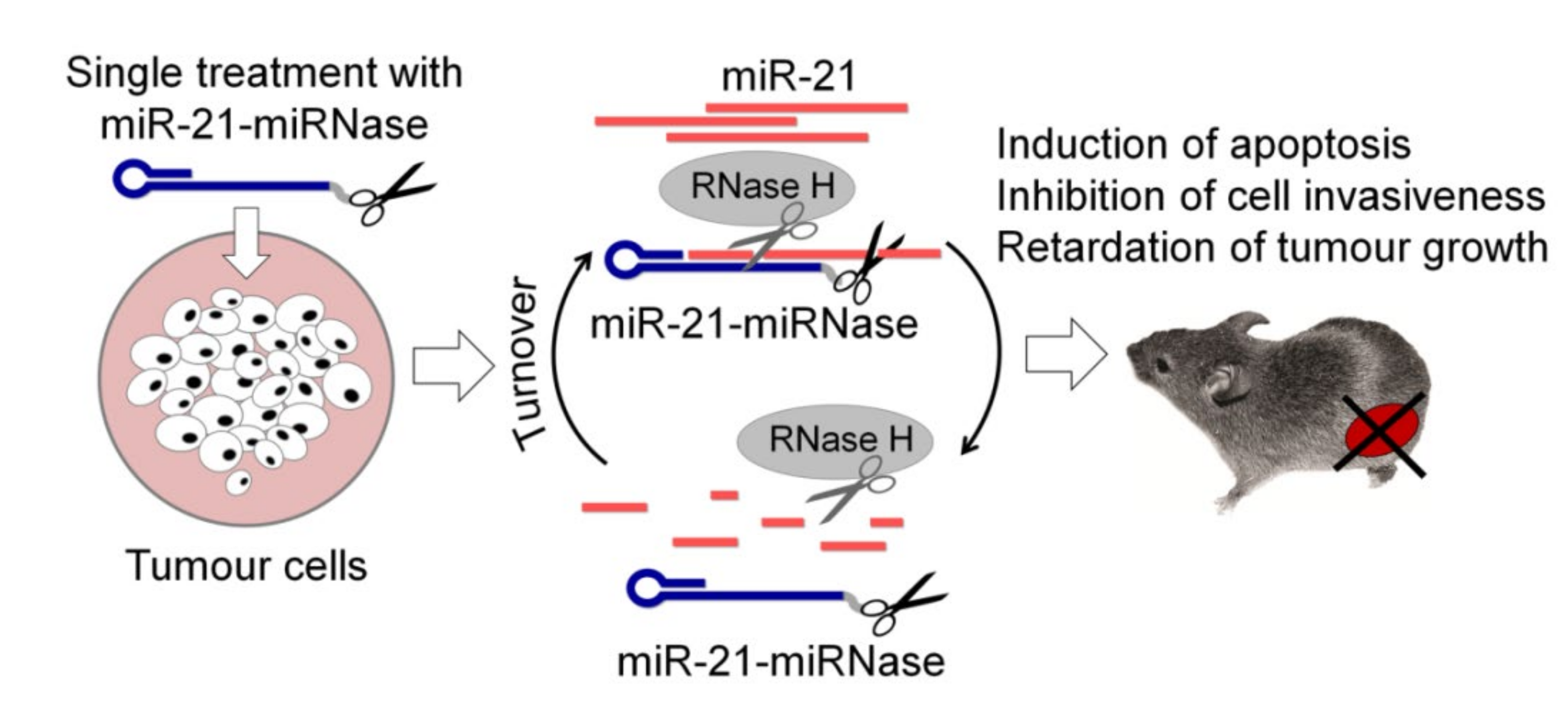
4. Therapeutic Application of Sequence-Specific aRNases in Cell Cultures and In Vivo
5. Conclusions
5.1. General Principles of ss-aRNases Design
5.2. Prospects of ss-aRNases Applications
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Belikova, A.; Zarytova, V.; Grineva, N. Synthesis of ribonucleosides and diribonucleoside phosphates containing 2-chloro-ethylamine and nitrogen mustard residues. Tetrahedron Lett. 1967, 8, 3557–3562. [Google Scholar] [CrossRef]
- Grineva, N.I.; Karpova, G.G. Complementary addressed alkylation of ribosomal RNA with alkylating derivatives of oligonucleotides. Mol. Biol. 1974, 8, 832–844. [Google Scholar] [PubMed]
- Grineva, N.I.; Karpova, G.G.; Kuznetsova, L.M.; Uimitova, T.A.; Venkstern, T.B. Complementarily addressed alkylation of yeast tRNA 1 Val with chloroethylmethylaminobenzylidene d(pC-G)-A. Proof of the modification of the third nucleotide located at the 5′-terminus of the complete binding site of the reagent. Mol. Biol. 1976, 10, 1260–1271. [Google Scholar]
- Paterson, B.M.; Roberts, B.E.; Kuff, E.L. Structural gene identification and mapping by DNA-mRNA hybrid-arrested cell-free translation. Proc. Natl. Acad. Sci. USA 1977, 74, 4370–4374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zamecnik, P.C.; Stephenson, M.L. Inhibition of Rous sarcoma virus replication and cell transformation by a specific oligodeoxynucleotide. Proc. Natl. Acad. Sci. USA 1978, 75, 280–284. [Google Scholar] [CrossRef] [Green Version]
- Stephenson, M.L.; Zamecnik, P.C. Inhibition of Rous sarcoma viral RNA translation by a specific oligodeoxyribonucleotide. Proc. Natl. Acad. Sci. USA 1978, 75, 285–288. [Google Scholar] [CrossRef] [Green Version]
- Eckstein, F. Phosphorothioate oligodeoxynucleotides: What is their origin and what is unique about them? Antisense Nucleic Acid Drug Dev. 2000, 10, 117–121. [Google Scholar] [CrossRef]
- Agrawal, S.; Jiang, Z.; Zhao, Q.; Shaw, D.; Cai, Q.; Roskey, A.; Channavajjala, L.; Saxinger, C.; Zhang, R. Mixed-backbone oligonucleotides as second generation antisense oligonucleotides: In vitro and in vivo studies. Proc. Natl. Acad. Sci. USA 1997, 94, 2620–2625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurreck, J. Antisense technologies. Improvement through novel chemical modifications. JBIC J. Biol. Inorg. Chem. 2003, 270, 1628–1644. [Google Scholar] [CrossRef] [PubMed]
- Miroshnichenko, S.K.; Patutina, O.A.; Burakova, E.A.; Chelobanov, B.P.; Fokina, A.A.; Vlassov, V.V.; Altman, S.; Zenkova, M.A.; Stetsenko, D.A. Mesyl phosphoramidate antisense oligonucleotides as an alternative to phosphorothioates with improved biochemical and biological properties. Proc. Natl. Acad. Sci. USA 2019, 116, 1229–1234. [Google Scholar] [CrossRef] [Green Version]
- Hutcherson, S.L.; Lanz, R. A randomized controlled clinical trial of intravitreous fomivirsen for treatment of newly diagnosed peripheral cytomegalovirus retinitis in patients with aids. Am. J. Ophthalmol. 2002, 133, 467–474. [Google Scholar]
- Li, N.; Li, Q.; Tian, X.-Q.; Qian, H.-Y.; Yang, Y.-J. Mipomersen is a Promising Therapy in the Management of Hypercholesterolemia: A Meta-Analysis of Randomized Controlled Trials. Am. J. Cardiovasc. Drugs 2014, 14, 367–376. [Google Scholar] [CrossRef]
- Hua, Y.; Sahashi, K.; Hung, G.; Rigo, F.; Passini, M.A.; Bennett, C.F.; Krainer, A.R. Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes Dev. 2010, 24, 1634–1644. [Google Scholar] [CrossRef] [Green Version]
- Stein, C.A. Eteplirsen Approved for Duchenne Muscular Dystrophy: The FDA Faces a Difficult Choice. Mol. Ther. 2016, 24, 1884–1885. [Google Scholar] [CrossRef] [Green Version]
- Keam, S.J. Inotersen: First Global Approval. Drugs 2018, 78, 1371–1376. [Google Scholar] [CrossRef] [PubMed]
- Elbashir, S.M.; Harborth, J.; Lendeckel, W.; Yalcin, A.; Weber, K.; Tuschl, T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nat. Cell Biol. 2001, 411, 494–498. [Google Scholar] [CrossRef] [PubMed]
- Burnett, J.C.; Rossi, J.J. RNA-Based Therapeutics: Current Progress and Future Prospects. Chem. Biol. 2012, 19, 60–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breaker, R.R.; Joyce, G.F. A DNA enzyme that cleaves RNA. Chem. Biol. 1994, 1, 223–229. [Google Scholar] [CrossRef]
- Zhou, W.; Ding, J.; Liu, J. Theranostic DNAzymes. Theranostics 2017, 7, 1010–1025. [Google Scholar] [CrossRef] [PubMed]
- Terns, M.P. CRISPR-Based Technologies: Impact of RNA-Targeting Systems. Mol. Cell 2018, 72, 404–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niittymäki, T.; Lönnberg, H. Artificial ribonucleases. Org. Biomol. Chem. 2006, 4, 15–25. [Google Scholar] [CrossRef]
- Matsumura, K.; Endo, M.; Komiyama, M. Lanthanide complex–oligo-DNA hybrid for sequence-selective hydrolysis of RNA. J. Chem. Soc. Chem. Commun. 1994, 17. [Google Scholar] [CrossRef]
- Hall, J.; Hüsken, D.; Pieles, U.; Moser, H.E.; Häner, R. Efficient sequence-specific cleavage of RNA using novel europium complexes conjugated to oligonucleotides. Chem. Biol. 1994, 1, 185–190. [Google Scholar] [CrossRef]
- Magda, D.; Miller, R.A.; Sessler, J.L.; Iverson, B.L. Site-Specific Hydrolysis of UNA by Europium(III) Texaphyrin Conjugated to a Synthetic Oligodeoxyribonucleotide. J. Am. Chem. Soc. 1994, 116, 7439–7440. [Google Scholar] [CrossRef]
- Vlassov, V.V.; Williams, N.; Raines, R.T.; Kierzek, R.; Dallas, A.; Lyo, M.; Mironova, N.L.; Kuznetsova, I.L.; Kuzuya, A.; Francois, J.C.; et al. Artificial Nucleases, 1st ed.; Zenkova, M.A., Ed.; Springer: Berlin/Heidelberg, Germany, 2004. [Google Scholar]
- Gnaccarini, C.; Peter, S.; Scheffer, U.; Vonhoff, S.; Klussmann, S.; Göbel, M.W. Site-Specific Cleavage of RNA by a Metal-Free Artificial Nuclease Attached to Antisense Oligonucleotides. J. Am. Chem. Soc. 2006, 128, 8063–8067. [Google Scholar] [CrossRef]
- Barbier, B.; Brack, A. Search for catalytic properties of simple polypeptides. Orig. Life Evol. Biosphere 1987, 17, 381–390. [Google Scholar] [CrossRef]
- Brack, A.; Barbier, B. Chemical activity of simple basic peptides. Orig. Life Evol. Biosphere 1990, 20, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Mironova, N.L.; Pyshnyi, D.V.; Ivanova, E.M.; Zenkova, M.A.; Gross, H.J.; Vlassov, V.V. Artificial ribonucleases: Oligonucleotide-peptide conjugates cleaving RNA at GpX and PyPA. Dokl. Akad. Nauk. 2002, 385, 1–5. [Google Scholar]
- Danneberg, F.; Ghidini, A.; Dogandzhiyski, P.; Kalden, E.; Strömberg, R.; Göbel, M.W. Sequence-specific RNA cleavage by PNA conjugates of the metal-free artificial ribonuclease tris(2-aminobenzimidazole). Beilstein J. Org. Chem. 2015, 11, 493–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dogandzhiyski, P.; Ghidini, A.; Danneberg, F.; Strömberg, R.; Göbel, M.W. Studies on Tris(2-aminobenzimidazole)-PNA Based Artificial Nucleases: A Comparison of Two Analytical Techniques. Bioconjug. Chem. 2015, 26, 2514–2519. [Google Scholar] [CrossRef] [PubMed]
- Zellmann, F.; Thomas, L.; Scheffer, U.; Hartmann, R.K.; Göbel, M.W. Site-Specific Cleavage of RNAs Derived from the PIM1 3′-UTR by a Metal-Free Artificial Ribonuclease. Molecules 2019, 24, 807. [Google Scholar] [CrossRef] [Green Version]
- Beloglazova, N.G.; Fabani, M.M.; Polushin, N.N.; Sil’Nikov, V.V.; Vlassov, V.V.; Bichenkova, E.V.; Zenkova, M.A. Site-Selective Artificial Ribonucleases: Oligonucleotide Conjugates Containing Multiple Imidazole Residues in the Catalytic Domain. J. Nucleic Acids 2011, 2011, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Williams, A.; Staroseletz, Y.; Zenkova, M.A.; Jeannin, L.; Aojula, H.; Bichenkova, E.V. Peptidyl–Oligonucleotide Conjugates Demonstrate Efficient Cleavage of RNA in a Sequence-Specific Manner. Bioconjug. Chem. 2015, 26, 1129–1143. [Google Scholar] [CrossRef] [PubMed]
- Patutina, O.A.; Bichenkova, E.V.; Miroshnichenko, S.K.; Mironova, N.L.; Trivoluzzi, L.T.; Burusco, K.K.; Bryce, R.A.; Vlassov, V.V.; Zenkova, M.A. miRNases: Novel peptide-oligonucleotide bioconjugates that silence miR-21 in lymphosarcoma cells. Biomater. 2017, 122, 163–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patutina, O.A.; Bazhenov, M.A.; Miroshnichenko, S.K.; Mironova, N.L.; Pyshnyi, D.V.; Vlassov, V.V.; Zenkova, M.A. Peptide-oligonucleotide conjugates exhibiting pyrimidine-X cleavage specificity efficiently silence miRNA target acting synergistically with RNase H. Sci. Rep. 2018, 8, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Murtola, M.; Wenska, M.; Strömberg, R. PNAzymes That Are Artificial RNA Restriction Enzymes. J. Am. Chem. Soc. 2010, 132, 8984–8990. [Google Scholar] [CrossRef] [PubMed]
- Ghidini, A.; Murtola, M.; Strömberg, R. Influence of conjugation and other structural changes on the activity of Cu2+ based PNAzymes. Org. Biomol. Chem. 2016, 14, 2768–2773. [Google Scholar] [CrossRef] [PubMed]
- Murtola, M.; Ghidini, A.; Virta, P.; Strömberg, R. Zinc Ion-Dependent Peptide Nucleic Acid-Based Artificial Enzyme that Cleaves RNA—Bulge Size and Sequence Dependence. Molecules 2017, 22, 1856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luige, O.; Murtola, M.; Ghidini, A.; Strömberg, R. Further Probing of Cu2+-Dependent PNAzymes Acting as Artificial RNA Restriction Enzymes. Molecules 2019, 24, 672. [Google Scholar] [CrossRef] [Green Version]
- Hall, J.; Hüsken, D.; Häner, R. Towards artificial ribonucleases: The sequence-specific cleavage of RNA in a duplex. Nucleic Acids Res. 1996, 24, 3522–3526. [Google Scholar] [CrossRef] [Green Version]
- Staroseletz, Y.; Amirloo, B.; Williams, A.; Lomzov, A.; Burusco, K.K.; Clarke, D.J.; Brown, T.; Zenkova, M.A.; Bichenkova, E.V. Strict conformational demands of RNA cleavage in bulge-loops created by peptidyl-oligonucleotide conjugates. Nucleic Acids Res. 2020, 48, 10662–10679. [Google Scholar] [CrossRef]
- Silnikov, V.N.; Vlassov, V.V. Design of site-specific RNA-cleaving reagents. Russ. Chem. Rev. 2001, 70, 491–508. [Google Scholar] [CrossRef]
- Yurchenko, L.; Silnikov, V.; Godovikova, T.; Shishkin, G.; Toulme, J.J.; Vlassov, V. Cleavage of Leishmania mini-exon sequence by oligonucleotides conjugated to a diimidazole construction. Nucleosides Nucleotides 1997, 16, 1721–1725. [Google Scholar] [CrossRef]
- Vlassov, V.; Abramova, T.; Godovikova, T.; Giege, R.; Silnikov, V. Sequence-Specific Cleavage of Yeast tRNA Phe with Oligonucleotides Conjugated to a Diimidazole Construct. Antisense Nucleic Acid Drug Dev. 1997, 7, 39–42. [Google Scholar] [CrossRef]
- Canaple, L.; Hüsken, D.; Hall, J.; Häner, R. Artificial ribonucleases: Efficient and specific in vitro cleavage of human c-raf-1 RNA. Bioconjug. Chem. 2002, 13, 945–951. [Google Scholar] [CrossRef] [PubMed]
- Staroseletz, Y.; Williams, A.; Burusco, K.K.; Alibay, I.; Vlassov, V.V.; Zenkova, M.A.; Bichenkova, E.V. ‘Dual’ peptidyl-oligonucleotide conjugates: Role of conformational flexibility in catalytic cleavage of RNA. Biomaterials 2017, 112, 44–61. [Google Scholar] [CrossRef] [PubMed]
- Patutina, O.; Chiglintseva, D.; Bichenkova, E.; Gaponova, S.; Mironova, N.; Vlassov, V.; Zenkova, M. Dual miRNases for Triple Incision of miRNA Target: Design Concept and Catalytic Performance. Molecules 2020, 25, 2459. [Google Scholar] [CrossRef] [PubMed]
- Miroshnichenko, S.K.; Amirloo, B.; Bichenkova, E.V.; Vlassov, V.V.; Zenkova, M.A.; Patutina, O.A. 2′OMe Modification of Anti-miRNA-21 Oligonucleotide–Peptide Conjugate Improves Its Hybridization Properties and Catalytic Activity. Russ. J. Bioorg. Chem. 2019, 45, 803–812. [Google Scholar] [CrossRef]
- Patutina, O.A.; Miroshnichenko, S.K.; Mironova, N.L.; Sen’Kova, A.V.; Bichenkova, E.V.; Clarke, D.J.; Vlassov, V.V.; Zenkova, M.A. Catalytic Knockdown of miR-21 by Artificial Ribonuclease: Biological Performance in Tumor Model. Front. Pharmacol. 2019, 10, 879. [Google Scholar] [CrossRef] [Green Version]
- Virta, P.; Katajisto, J.; Niittymäki, T.; Lönnberg, H. Solid-supported synthesis of oligomeric bioconjugates. Tetrahedron 2003, 59, 5137–5174. [Google Scholar] [CrossRef]
- Murtola, M.; Strömberg, R. PNA based artificial nucleases displaying catalysis with turnover in the cleavage of a leukemia related RNA model. Org. Biomol. Chem. 2008, 6, 3837–3842. [Google Scholar] [CrossRef]
- Scheffer, U.; Strick, A.; Ludwig, V.; Peter, S.; Kalden, E.; Göbel, M.W. Metal-Free Catalysts for the Hydrolysis of RNA Derived from Guanidines, 2-Aminopyridines, and 2-Aminobenzimidazoles. J. Am. Chem. Soc. 2005, 127, 2211–2217. [Google Scholar] [CrossRef]
- Venkatesan, N.; Kim, B.H. Peptide Conjugates of Oligonucleotides: Synthesis and Applications. Chem. Rev. 2006, 106, 3712–3761. [Google Scholar] [CrossRef]
- Polushin, N.N. The precursor strategy: Terminus methoxyoxalamido modifiers for single and multiple functionalization of oligodeoxyribonucleotides. Nucleic Acids Res. 2000, 28, 3125–3133. [Google Scholar] [CrossRef] [Green Version]
- Beloglazova, N.G.; Fabani, M.M.; Zenkova, M.A.; Bichenkova, E.V.; Polushin, N.N.; Sil’nikov, V.V.; Douglas, K.T.; Vlassov, V.V. Sequence-specific artificial ribonucleases. I. Bis-imidazole-containing oligonucleotide conjugates prepared using precursor-based strategy. Nucleic Acids Res. 2004, 32, 3887–3897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zarytova, V.F.; Ivanova, E.M.; Yarmolyuk, S.N.; Alekseeva, I.V. Synthesis of arginine-containing oligonucleotide-(5’→N)-peptides. Biopolym. Cell 1988, 4, 220–222. [Google Scholar] [CrossRef]
- Burns, J.A.; Butler, J.C.; Moran, J.; Whitesides, G.M. Selective reduction of disulfides by tris(2-carboxyethyl)phosphine. J. Org. Chem. 1991, 56, 2648–2650. [Google Scholar] [CrossRef]
- Pounder, R.J.; Stanford, M.J.; Brooks, P.; Richards, S.P.; Dove, A.P. Metal free thiol-maleimide “Click” reaction as a mild functionalisation strategy for degradable polymers. Chem. Commun. 2008, 5158–5160. [Google Scholar] [CrossRef] [PubMed]
- Ede, N.J.; Tregear, G.W.; Haralambidis, J. Routine Preparation of Thiol Oligonucleotides: Application to the Synthesis of Oligonucleotide-Peptide Hybrids. Bioconjug. Chem. 1994, 5, 373–378. [Google Scholar] [CrossRef]
- Murtola, M.; Ossipov, D.; Sandbrink, J.; Strömberg, R. RNA Cleavage by 2,9-Diamino-1,10-Phenanthroline PNA Conjugates. Nucleosides Nucleotides Nucleic Acids 2007, 26, 1479–1483. [Google Scholar] [CrossRef]
- Sandbrink, J.; Murtola, M.; Strömberg, R. Solid Support Post-Conjugation of Amino Acids and a Phenanthroline Derivative to a Central Position in Peptide Nucleic Acids. Nucleosides Nucleotides Nucleic Acids 2007, 26, 1485–1489. [Google Scholar] [CrossRef]
- Niittymäki, T.; Virta, P.; Ketomäki, K.; Lönnberg, H. Di(azacrown) Conjugates of 2‘-O-Methyl Oligoribonucleotides as Sequence-Selective Artificial Ribonucleases. Bioconjug. Chem. 2007, 18, 1583–1592. [Google Scholar] [CrossRef] [PubMed]
- Gaglione, M.; Milano, G.; Chambery, A.; Moggio, L.; Romanelli, A.; Messere, A. PNA-based artificial nucleases as antisense and anti-miRNA oligonucleotide agents. Mol. Biosyst. 2011, 7, 2490–2499. [Google Scholar] [CrossRef] [Green Version]
- Grandas, A.; Marchán, V.; Debéthune, L.; Pedroso, E. Stepwise solid-phase synthesis of nucleopeptides. Curr. Protoc. Nucleic Acid Chem. 2007, 31, 4.22.1–4.22.54. [Google Scholar] [CrossRef] [Green Version]
- Emilsson, G.M.; Nakamura, S.; Roth, A.; Breaker, R.R. Ribozyme speed limits. RNA 2003, 9, 907–918. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Breaker, R.R. Kinetics of RNA degradation by specific base catalysis of transesterification involving the 2γ-hydroxyl group. J. Am. Chem. Soc. 1999, 121, 5364–5372. [Google Scholar] [CrossRef]
- Lönnberg, H. Cleavage of RNA phosphodiester bonds by small molecular entities: A mechanistic insight. Org. Biomol. Chem. 2011, 9, 1687–1703. [Google Scholar] [CrossRef]
- Kuzuya, A.; Mizoguchi, R.; Morisawa, F.; Komiyama, M. Novel approach for SNP genotyping based on site-selective RNA scission. Nucleic Acids Symp. Ser. 2002, 2, 129–130. [Google Scholar] [CrossRef] [Green Version]
- Kuzuya, A.; Machida, K.; Shi, Y.; Tanaka, K.; Komiyama, M. Site-Selective RNA Activation by Acridine-Modified Oligodeoxynucleotides in Metal-Ion Catalyzed Hydrolysis: A Comprehensive Study. ACS Omega 2017, 2, 5370–5377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sunami, T.; Kondo, J.; Hirao, I.; Watanabe, K.; Miura, K.-I.; Takénaka, A. Structure of d(GCGAAAGC) (hexagonal form): A base-intercalated duplex as a stable structure. Acta Crystallogr. Sect. D Biol. Crystallogr. 2003, 60, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Zellmann, F.; Göbel, M.W. A Trisbenzimidazole Phosphoramidite Building Block Enables High-Yielding Syntheses of RNA-Cleaving Oligonucleotide Conjugates. Molecules 2020, 25, 1842. [Google Scholar] [CrossRef] [PubMed]
- Kuzuya, A.; Mizoguchi, R.; Morisawa, F.; Komiyama, M. Site-selective RNA scission at two sites for precise genotyping of SNPs by mass spectrometry. Chem. Commun. 2003, 3, 770–771. [Google Scholar] [CrossRef] [PubMed]
- Whitney, A.; Gavory, G.; Balasubramanian, S. Site-specific cleavage of human telomerase RNA using PNA-neocuproine.Zn(II) derivatives. Chem. Commun. 2003, 36–37. [Google Scholar] [CrossRef]
- Mironova, N.L.; Pyshnyi, D.V.; Ivanova, E.M.; Zenkova, M.A.; Gross, H.J.; Vlassov, V.V. Covalently attached oligodeoxyribonucleotides induce RNase activity of a short peptide and modulate its base specificity. Nucleic Acids Res. 2004, 32, 1928–1936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenberg, H.F. RNase A ribonucleases and host defense: An evolving story. J. Leukoc. Biol. 2008, 83, 1079–1087. [Google Scholar] [CrossRef] [Green Version]
- Kovaľ, T.; Stergaard, L.H.; Lehmbeck, J.; Nørgaard, A.; Lipovová, P.; Dušková, J.; Skálová, T.; Trundová, M.; Kolenko, P.; Fejfarová, K.; et al. Structural and catalytic properties of S1 Nuclease from aspergillus oryzae responsible for substrate recognition, cleavage, non-Specificity, and inhibition. PLoS ONE 2016, 11, e0168832. [Google Scholar] [CrossRef] [PubMed]
- Pace, C.N.; Heinemann, U.; Hahn, U.; Saenger, W. Ribonuclease T1: Structure, Function, and Stability. Angew. Chem. Int. Ed. 1991, 30, 343–360. [Google Scholar] [CrossRef]
- Calnan, B.J.; Tidor, B.; Biancalana, S.; Hudson, D.; Frankel, A.D. Arginine-mediated RNA recognition: The arginine fork. Science 1991, 252, 1167–1171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beloglazova, N.; Sil’Nikov, V.; Zenkova, M.; Vlassov, V. Cleavage of yeast tRNAPhe with complementary oligonucleotide conjugated to a small ribonuclease mimic. FEBS Lett. 2000, 481, 277–280. [Google Scholar] [CrossRef] [Green Version]
- Baker, B.F.; Lot, S.S.; Kringel, J.; Cheng-Flournoy, S.; Villiet, P.; Sasmor, H.M.; Siwkowski, A.M.; Chappell, L.L.; Morrow, R. Oligonucleotide-europium complex conjugate designed to cleave the 5′ cap structure of the ICAM-1 transcript potentiates antisense activity in cells. Nucleic Acids Res. 1999, 27, 1547–1551. [Google Scholar] [CrossRef] [Green Version]
- Ushijima, K.; Shirakawa, M.; Kagoshima, K.; Park, W.-S.; Miyano-Kurosaki, N.; Takaku, H. Anti-HIV-1 activity of an antisense phosphorothioate oligonucleotide bearing imidazole and primary amine groups. Bioorganic Med. Chem. 2001, 9, 2165–2169. [Google Scholar] [CrossRef]
- Dong, C.G.; Wu, W.K.; Feng, S.Y.; Wang, X.J.; Shao, J.F.; Qiao, J. Co-inhibition of microRNA-10b and microRNA-21 exerts synergistic inhibition on the proliferation and invasion of human glioma cells. Int. J. Oncol. 2012, 41, 1005–1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagenaar, T.R.; Zabludoff, S.; Ahn, S.M.; Allerson, C.; Arlt, H.; Baffa, R.; Cao, H.; Davis, S.; Garcia-Echeverria, C.; Gaur, R.; et al. Anti-miR-21 suppresses Hepatocellular carcinoma growth via broad transcriptional network deregulation. Mol. Cancer Res. 2015, 13, 1009–1021. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.-R.; Qi, H.-J.; Deng, D.-F.; Luo, Y.-Y.; Yang, S.-L. MicroRNA-21 promotes cell proliferation, migration, and resistance to apoptosis through PTEN/PI3K/AKT signaling pathway in esophageal cancer. Tumor Biol. 2016, 37, 12061–12070. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.; Yeom, C.; Choi, Y.-S.; Kim, S.; Lee, E.; Park, M.J.; Kang, S.W.; Kim, S.B.; Chang, S. Simultaneous inhibition of multiple oncogenic miRNAs by a multi-potent microRNA sponge. Oncotarget 2015, 6, 20370–20387. [Google Scholar] [CrossRef]
- Miroshnichenko, S.; Patutina, O. Enhanced Inhibition of Tumorigenesis Using Combinations of miRNA-Targeted Therapeutics. Front. Pharmacol. 2019, 10, 488. [Google Scholar] [CrossRef] [PubMed]
- Šípová, H.; Špringer, T.; Rejman, D.; Šimák, O.; Petrová, M.; Novák, P.; Rosenbergová, Š.; Páv, O.; Liboska, R.; Barvík, I.; et al. 5′-O-Methylphosphonate nucleic acids—new modified DNAs that increase the Escherichia coli RNase H cleavage rate of hybrid duplexes. Nucleic Acids Res. 2014, 42, 5378–5389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kojima, N.; Shrestha, A.R.; Akisawa, T.; Piao, H.; Kizawa, H.; Ohmiya, Y.; Kurita, R. Development of gapmer antisense oligonucleotide with deoxyribonucleic guanidine (DNG) modifications. Nucleosides, Nucleotides Nucleic Acids 2019, 39, 258–269. [Google Scholar] [CrossRef]
- Danielsen, M.B.; Lou, C.; Lisowiec-Wachnicka, J.; Pasternak, A.; Jørgensen, P.T.; Wengel, J. Gapmer Antisense Oligonucleotides Containing 2′,3′-Dideoxy-2′-fluoro-3′-C-hydroxymethyl-β-d-lyxofuranosyl Nucleotides Display Site-Specific RNase H Cleavage and Induce Gene Silencing. Chem. A Eur. J. 2020, 26, 1368–1379. [Google Scholar] [CrossRef] [PubMed]
- Lützelberger, M.; Kjems, J. Strategies to Identify Potential Therapeutic Target Sites in RNA. Handb. Exp. Pharmacol. 2006, 173, 243–259. [Google Scholar]
- Kierzek, R.; Turner, D.H.; Kierzek, E. Microarrays for identifying binding sites and probing structure of RNAs. Nucleic Acids Res. 2015, 43, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Patutina, O.A.; Miroshnichenko, S.K.; Lomzov, A.A.; Mironova, N.L.; Zenkova, M.A. Search for oligonucleotides selectively binding oncogenic miR-21. Russ. J. Bioorganic Chem. 2017, 43, 29–37. [Google Scholar] [CrossRef]
- Kristensen, L.S.; Hansen, T.B.; Venø, M.T.; Kjems, J. Circular RNAs in cancer: Opportunities and challenges in the field. Oncogene 2018, 37, 555–565. [Google Scholar] [CrossRef] [Green Version]
- Verduci, L.; Strano, S.; Yarden, Y.; Blandino, G. The circRNA–microRNA code: Emerging implications for cancer diagnosis and treatment. Mol. Oncol. 2019, 13, 669–680. [Google Scholar] [CrossRef] [Green Version]
- Bhan, A.; Soleimani, M.; Mandal, S.S. Long Noncoding RNA and Cancer: A New Paradigm. Cancer Res. 2017, 77, 3965–3981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patutina, O.; Mironova, N.; Vlassov, V.; Zenkova, M. New Approaches for Cancer Treatment: Antitumor Drugs Based on gene-Targeted Nucleic Acids. Acta Nat. 2009, 1, 44–60. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y. The novel regulatory role of lncRNA-miRNA-mRNA axis in cardiovascular diseases. J. Cell. Mol. Med. 2018, 22, 5768–5775. [Google Scholar] [CrossRef] [PubMed]
- Rupaimoole, R.; Slack, F.J. MicroRNA therapeutics: Towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 2017, 16, 203–221. [Google Scholar] [CrossRef]
- Cheng, Y.; Wang, Q.; Jiang, W.; Bian, Y.; Zhou, Y.; Gou, A.; Zhang, W.; Fu, K.; Shi, W. Emerging roles of piRNAs in cancer: Challenges and prospects. Aging 2019, 11, 9932–9946. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Dou, M.; Song, X.; Dong, Y.; Liu, S.; Liu, H.; Tao, J.; Li, W.; Yin, X.; Xu, W. The emerging role of the piRNA/piwi complex in cancer. Mol. Cancer 2019, 18, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Himeno, H.; Kurita, D.; Muto, A. TmRNA-mediated trans-translation as the major ribosome rescue system in a bacterial cell. Front. Genet. 2014, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brito, L.; Wilton, J.; Ferrándiz, M.J.; Gómez-Sanz, A.; de la Campa, A.G.; Amblar, M. Absence of tmRNA Has a against fluoroquinolones in streptococcus pneumoniae. Front. Microbiol. 2017, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukushima, A.; Fukuda, N.; Lai, Y.; Ueno, T.; Moriyama, M.; Taguchi, F.; Iguchi, A.; Shimizu, K.; Kuroda, K. Development of a Chimeric DNA-RNA Hammerhead Ribozyme Targeting SARS Virus. Intervirology 2009, 52, 92–99. [Google Scholar] [CrossRef]
- Wang, Z.; Ren, L.; Zhao, X.; Hung, T.; Meng, A.; Wang, J.; Chen, Y.-G. Inhibition of Severe Acute Respiratory Syndrome Virus Replication by Small Interfering RNAs in Mammalian Cells. J. Virol. 2004, 78, 7523–7527. [Google Scholar] [CrossRef] [Green Version]
- Piyush, R.; Rajarshi, K.; Chatterjee, A.; Khan, R.; Ray, S. Nucleic acid-based therapy for coronavirus disease 2019. Heliyon 2020, 6, e05007. [Google Scholar] [CrossRef]
- Baldassarre, A.; Paolini, A.; Bruno, S.P.; Felli, C.; Tozzi, A.E.; Masotti, A. Potential use of noncoding RNAs and innovative therapeutic strategies to target the 5′UTR of SARS-CoV-2. Epigenomics 2020, 12, 1349–1361. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Catalytic Group | Linker Type | Oligonucleotide Type, Length, nts | Target Length, nts | Nucleotide Base Specificity | Cleavage Conditions, RNA: Conjugate μM | τ½ Efficiency | Ref. |
|---|---|---|---|---|---|---|---|
| Tris(2-amino- benzimidazole) | Aminohexyl | Linear DNA, 15 | Synthetic RNA, 29 | C-G, G-A, A-U | 0.15:1.5 | 16.5 h | [26] |
| Disulfide bridge | Linear DNA, 15, 17, 20 | Synthetic RNA, 29 | G-A, C-G, G-C, U-C, C-U, G-A, A-U | 0.15:1.5 | 12.4 h/ 90% in 56 h | ||
| Aminohexyl | Linear Lys-PNA, 10, 15 | Synthetic RNA, 29 | A-U, U-C, C-U, C-G, A-G, G-A, A-A | 0.15:0.75 | 11.2 h/ 90% in 60 h | [30] | |
| Aminohexyl | Lys-PNA, bulge inducing (4 nts) 11 (7-cleaver-4) | Synthetic RNA, 15 | U-A, A-A, A-G | 4:4 4:0.8 | 9 h | [31] | |
| Aminohexyl | Linear DNA, 15 | Synthetic RNA, 22 | C-A, A-A, A-U | 0.15:3 | 14–15 h | [32] | |
| Aminohexyl | Linear DNA-LNA mixmers, 5’-end, 15 | Synthetic RNA, 22 | C-A, A-A, A-U | 0.15:0.75 | 3.5 h | [32] | |
| Aminohexyl | Linear DNA- LNA mixmers, 15 | Synthetic RNA 1, 155/412/430 * | C-A, A-A, A-U | 0.25:1 | 2.5–3 h | [32] | |
| Aminohexyl | Linear DNA, 15 | Synthetic RNA 1, 22 | C-A, A-A, A-U | 0.15:3 | 14–15 h | [71] | |
| Aminohexyl | Linear DNA- LNA mixmers 5′-end, 15 | Synthetic RNA 1, 22 | C-A, A-A, A-U | 0.15:0.75 | 3.5 h | [71] | |
| Imidazole (×24) | 41 ** | Linear DNA, 17 | tRNAPhe, 76 | C-A | 1:10 | 1 h | [33] |
| Imidazole (×4) | 41/79 ** | Linear DNA, 17 | tRNAPhe, 76 | C-A | 1:10 | 1 h | [33] |
| Zn(II)-2,9-dimethyl- phenanthroline | Diaminopropionic acid (Dap) | Lys-PNA, bulge inducing (4 nts) 11 (7-cleaver-4) | Synthetic RNA 2, 15 | A-A | 4:4, 4:1 | 11 h | [52] |
| Dap | Lys-PNA, bulge inducing (3 nts) 12 (8-cleaver-4) | Synthetic RNA 2, 15 | A-A | 4:4 | 21 h | [52] | |
| Dap and additional Gly | Lys-PNA, bulge inducing (4 nts) 11 (7-cleaver-4) | Synthetic RNA 2, 15 | A-A | 4:4 | 12 h | [52] | |
| Dap and additional Gly | Lys-PNA, bulge inducing (3 nts) 12 (8-cleaver-4) | Synthetic RNA 2, 15 | A-A | 4:4 | 15 h | [52] | |
| Cu(II)-2,9-dimethyl- phenanthroline | Dap | PNA, bulge inducing (4 nts) 11 (7-cleaver-4) | Synthetic RNA 2, 15 | A-A, G-A | 4:4 400:4 | 0.5 h | [37] |
| Dap and oligoether | Lys-PNA, bulge inducing (4 nts) 11 (7-cleaver-4) | Synthetic RNA 2, 15 | A-A | 4:4 | 1.5 h | [38] | |
| Dap and additional Gly | Lys-PNA, bulge inducing (4 nts) 11 (7-cleaver-4) | Synthetic RNA 2, 15 | A-A | 4:4 | 3 h | [38] | |
| Zn(II)-2,9-dimethyl- phenanthroline | Dap | Lys-PNA, bulge inducing (4 nts) 11 (7-cleaver-4) | Synthetic RNA, 15 | A-A, G-A | 4:4 | 7–8 h | [39] |
| Dap | Lys-PNA, bulge inducing (3 nts) 12 (8-cleaver-4) | Synthetic RNA, 15 | U-A, A-A | 4:4 | 7–8 h | [39] | |
| Cu(II)-2,9-dimethyl- phenanthroline | Dap | Lys-PNA, bulge inducing (4 nts) 11 (7-cleaver-4) | Synthetic RNA 2, 15 | A-A | 4:4 | 0.5 h | [40] |
| Dap | Lys-PNA, bulge inducing (3 nts) 12 (8-cleaver-4) | Synthetic RNA 2, 15 | U-A, A-A | 4:4 | 14–24 h | [40] | |
| Acridine+free Lu(III)/Zn(II) | Aminohexyl | Linear DNA, 18 | Synthetic RNA, 36 | C-U, U-G, | 5:10 | 5.5–115 h | [70] |
| Di(Azacrown) 3-(3-hydroxypropyl) -1,5,9-triaza- cyclododecane- Zn(II) | – | Linear 2’-OMe RNA,15 | Synthetic chimera 2’-O-Me- RNA, 19-21 | C-A | 18:18 | 90% in 120 h | [63] |
| Diethylenetriamine (DETA) | Polyethylene glycol (PEG) | Linear PNA, 14 | Synthetic RNA, 26 | G-G | 2:2, 20:2 | 90% in 24 h | [64] |
| [His(Gly)2]-Cu(II) | PEG | Linear PNA, 14 | Synthetic RNA, 26 | G-A | 2:2, 20:2 | 47.5% in 24 h | [64] |
| [(ArgLeu)4]Gly- CONH2 | Phosphor amidate | Linear DNA, 17 | tRNAPhe, 76 | C-A, U-A | 1:20 | 0.5 h | [34] |
| Phosphor amidate | Linear DNA, 17 | tRNAPhe, 76 | C-A, U-A | 1:20 | 0.75 h | [34] | |
| [(ArgLeu)2Gly]2- COOH | Phosphor amidate | Linear DNA, 17 | tRNAPhe, 76 | C-A, U-A | 1:20 | 0.9 h | [34] |
| [(ArgLeu)4]Gly | Aminohexyl and thiol-maleimide | Dual DNA, 11 + 12 | tRNAPhe, 76 | C-A, U-A | 1:20 | N.d. | [47] |
| [(ArgLeu)2Gly]2 | Aminohexyl and thiol-maleimide | Dual DNA, 11 + 12 | tRNAPhe, 76 | C-A, G-X | 1:20 | 1 h | [47] |
| Aminohexyl | Bulge-inducing DNA 11-cleaver-15 | tRNAPhe, 76 | C-A, U-A, G-X | 1:20 | 8 h | [42] | |
| Aminohexyl (C-termini) | Hairpin DNA, 14 * (9 bp stem) | miR-21, 22 | G-X | 1:20 | 17 ± 0.4 h/ 98% in 72 h | [35] | |
| Aminohexyl (C-termini) | Hairpin DNA, 16 * (6 bp stem) | miR-21, 22 | G-X | 1:20 | 83% in 72 h | ||
| Aminohexyl (C-termini) | Hairpin DNA, 16 * (9 bp stem) | miR-21, 22 | G-X | 1:20 | 57% in 72 h | ||
| Aminohexyl (C-termini) | Linear DNA, 16 | miR-21, 22 | G-X | 1:20 | 15.1 ± 0.2 h/ 93 % in 72 h | ||
| [(ArgLeu)2Gly]2 | Aminohexyl (C-termini) | Hairpin DNA, 14 * (6 bp stem) | miR-21, 22 | G-X | 1:20 | 16.2 ± 0.2 h/ 99% in 72 h | [35] |
| 50:5 | 83% in 72 h | [50] | |||||
| 25:5 | 86% in 72 h | ||||||
| 10:5 | 87% in 72 h | ||||||
| Gly(ArgLeu)4 | (N-termini) 5′pTCAA3′ + DEG or TrEG | Hairpin DNA, 12 * | miR-21, 22 | pyr-X | 1:100 | 50% in 72 h | [36] |
| miR-17, 23 | pyr-X | 1:20 | 9% in 24 h | ||||
| [(ArgLeu)2Gly]2 | Aminohexyl (C-termini) | Hairpin 2′OMe + DNA, 14 * (6 bp stem) | miR-21, 22 | G-X | 1:20 | 4.9 ± 0.1 h/ 100% in 24 h | [49] |
| G-X, pyr-A | 10:5 | 77% in 72 h | |||||
| Aminohexyl (C-termini); thiohexyl (N-termini) | Dual DNA with 2′-aminoadenines, 10 + 8 | miR-17, 23 | pyr-A | 1:20 | 32% in 48 h | [48] | |
| miR-21, 22 | 30% in 48 h | ||||||
| miR-155, 23 | 57% in 48 h | ||||||
| miR-18a, 22 | 23% in 48 h |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Staroseletz, Y.; Gaponova, S.; Patutina, O.; Bichenkova, E.; Amirloo, B.; Heyman, T.; Chiglintseva, D.; Zenkova, M. Site-Selective Artificial Ribonucleases: Renaissance of Oligonucleotide Conjugates for Irreversible Cleavage of RNA Sequences. Molecules 2021, 26, 1732. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26061732
Staroseletz Y, Gaponova S, Patutina O, Bichenkova E, Amirloo B, Heyman T, Chiglintseva D, Zenkova M. Site-Selective Artificial Ribonucleases: Renaissance of Oligonucleotide Conjugates for Irreversible Cleavage of RNA Sequences. Molecules. 2021; 26(6):1732. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26061732
Chicago/Turabian StyleStaroseletz, Yaroslav, Svetlana Gaponova, Olga Patutina, Elena Bichenkova, Bahareh Amirloo, Thomas Heyman, Daria Chiglintseva, and Marina Zenkova. 2021. "Site-Selective Artificial Ribonucleases: Renaissance of Oligonucleotide Conjugates for Irreversible Cleavage of RNA Sequences" Molecules 26, no. 6: 1732. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26061732