
Facile Synthesis of Saikosaponins

1
Department of Chemistry, University of Science and Technology of China, 96 Jinzhai Road, Hefei 230026, China
2
State Key Laboratory of Bio-organic and Natural Products Chemistry, Center for Excellence in Molecular Synthesis, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China
3
School of Chemistry and Materials Science, Hangzhou Institute for Advanced Study, University of Chinese Academy of Sciences, 1 Sub-lane Xiangshan, Hangzhou 310024, China
*
Authors to whom correspondence should be addressed.
Molecules 2021, 26(7), 1941; https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26071941
Submission received: 18 February 2021
/
Revised: 3 March 2021
/
Accepted: 10 March 2021
/
Published: 30 March 2021
(This article belongs to the Special Issue Recent Advances in Glycochemistry)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Saikosaponin A (SSa) and D (SSd) are typical oleanane-type saponins featuring a unique 13,28-epoxy-ether moiety at D ring of the aglycones, which exhibit a wide range of biological and pharmacological activities. Herein, we report the first synthesis of saikosaponin A/D (1–2) and their natural congeners, including prosaikosaponin F (3), G (4), saikosaponin Y (5), prosaikogenin (6), and clinoposaponin I (7). The present synthesis features ready preparation of the aglycones of high oxidation state from oleanolic acid, regioselective glycosylation to construct the β-(1→3)-linked disaccharide fragment, and efficient gold(I)-catalyzed glycosylation to install the glycans on to the aglycones.
1. Introduction
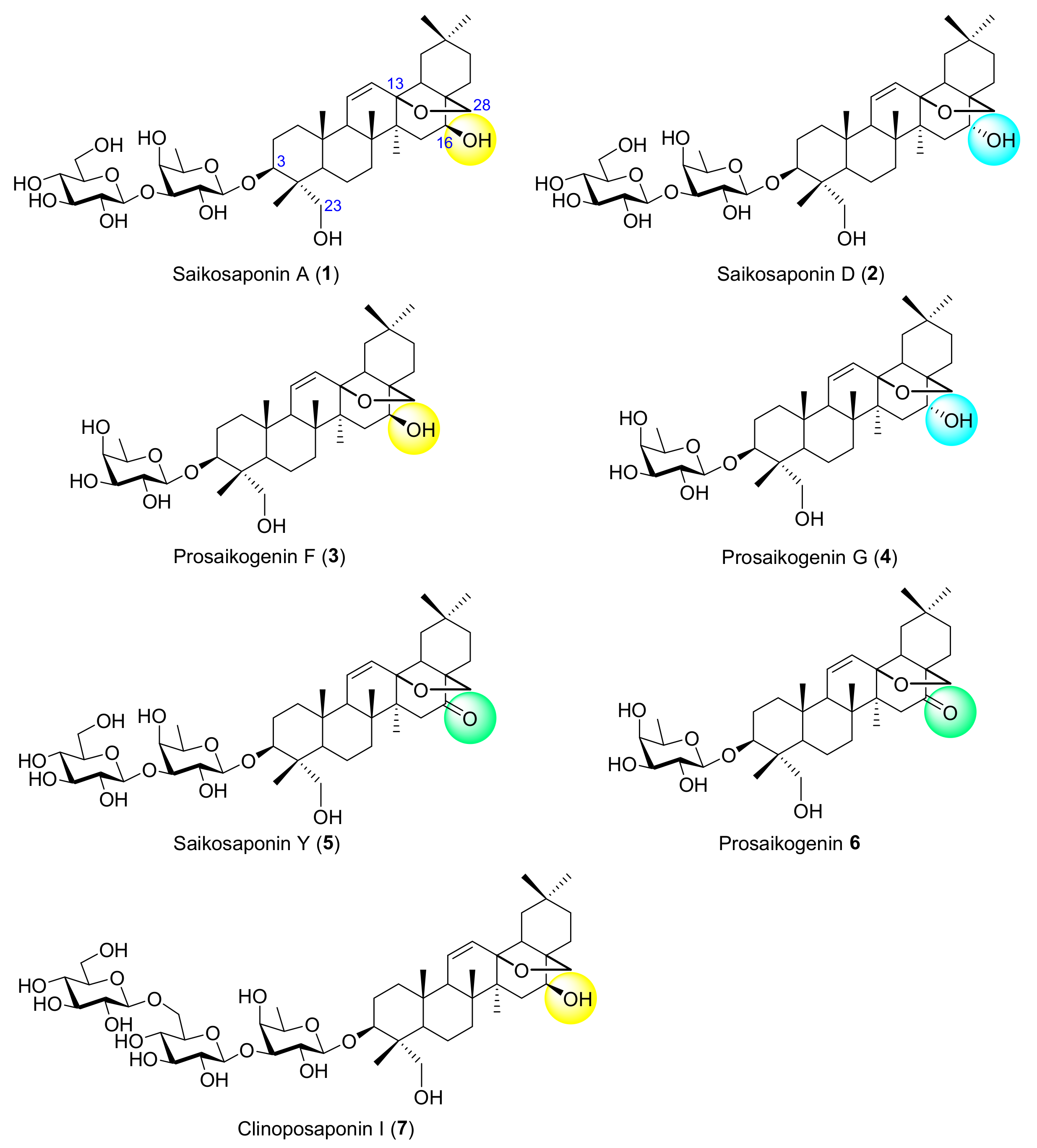
The roots of Radix Bupleuri, one of the most common traditional Chinese medicines, have been frequently used for the treatment of common cold with fever, influenza, inflammation, and infectious diseases [1,2]. Saikosaponin A (SSa, 1) and D (SSd, 2) are the major compounds in Radix Bupleuri and are used as the markers for evaluation of the quality of the medicine (Figure 1) [3]. Indeed, these two triterpene glycosides together with their congeners exhibit a variety of pharmacological activities, including anti-inflammatory, anti-tumor, neuro-regulation, immuno-regulation, hepatoprotection, and antiviral effects [1,4]. Structurally, SSa (1) and SSd (2) are a pair of the oleanane-type disaccharide stereoisomers, with the aglycones, namely saikogenin F (SGF) and saikogenin G (SGG), respectively, featuring a 13,28-epoxy-ether moiety at the D ring and an opposite chirality at 16-OH. Interestingly, SSa (1) was reported to show a strong anti-inflammatory effect, whereas SSd (2) showed a strong antitumor effect [1]. The corresponding monosaccharides, named prosaikogenin F (PSF, 3) and G (PSG, 4) were identified as metabolites of SSa and SSd [5], which showed hemolytic activity and the absorbability on red blood cells [6,7]. Saikosaponin Y (5) is likely a biogenetic precursor/product of SSa and SSd, which has a higher oxidation state at C-16; this congener displayed potent cytotoxic and antiviral activities [8,9,10]. Monosaccharide 6, named posaikogenin, was found as an artifact which might be derived from disaccharide 5. A number of saikosaponin-like compounds have also been identified from Clinopodium gracile and other plants, among which Clinoposaponin I (7) bearing a trisaccharide residue is a typical one [11,12,13]. All these seven saponins (1–7) belong to type I saikosaponins, which bear the 13,28-epoxy-ether moiety, and this epoxy-ether moiety is found crucial to their cytotoxic activities.
Notwithstanding, these naturally extracted saikosaponins are associated with systemic toxicities and side effects [14,15]. Thus, saikosaponins could induce obvious hepatoxicity and necrosis upon 15 day administration to rats [14]. Furthermore, SSd (2) alone was found to stimulate mitochondrial apoptosis in human hepatocyte cells [15]. On the other hand, these saikosaponins are difficult to purify from plants; therefore, in-depth studies have not been carried out on their structure-activity relationship (SAR) and mode of action. Chemical synthesis of saikosaponins is also a challenging task, given the scarce availability of the triterpene aglycones that bear high oxidation state at the D/E rings [16,17,18,19]. Very recently, we disclosed a site-selective C-H hydroxylation reaction at the D/E rings of pentacyclic triterpenoids, thus paving a venue for the synthesis of saikosaponins [18,20]. Herein, we report the synthesis of saikosaponin A (1), D (2), and their congeners (3–7), using the largely available oleanolic acid as a starting material.
2. Results
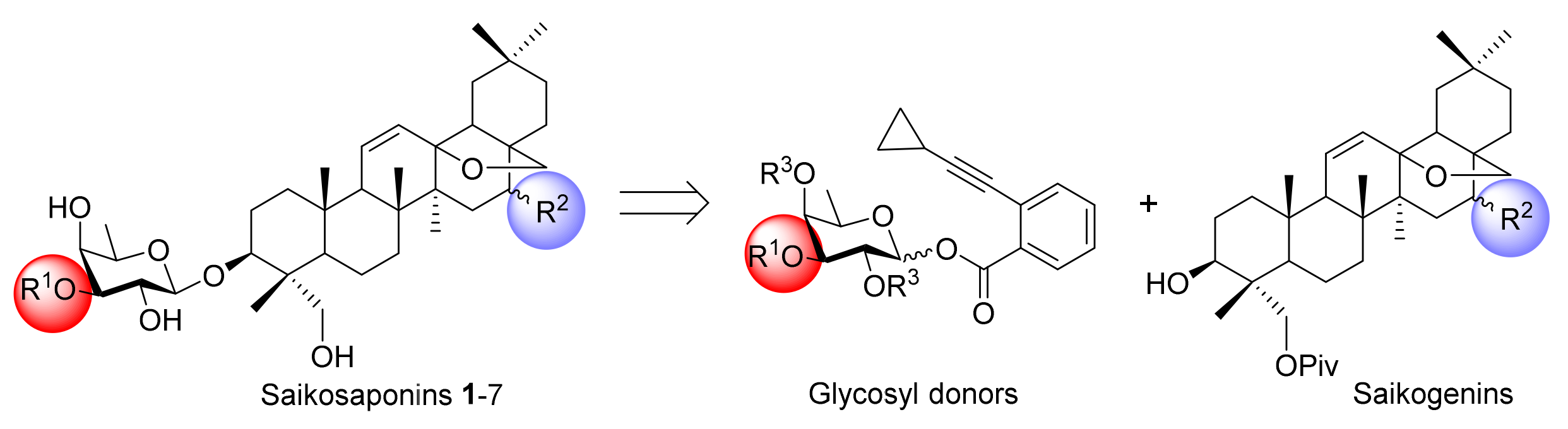
We envisioned the assembly of saikosaponins (1–7) from two pieces, i.e., a saikogenin derivative and a glycosyl donor (Scheme 1) [21]. Considering the susceptibility of the 13,28-epoxy-ether moiety in the aglycones, we planned to employ glycosyl o-alkynylbenzoates as donors that can proceed glycosylation under the mild gold(I)-catalyzed conditions.
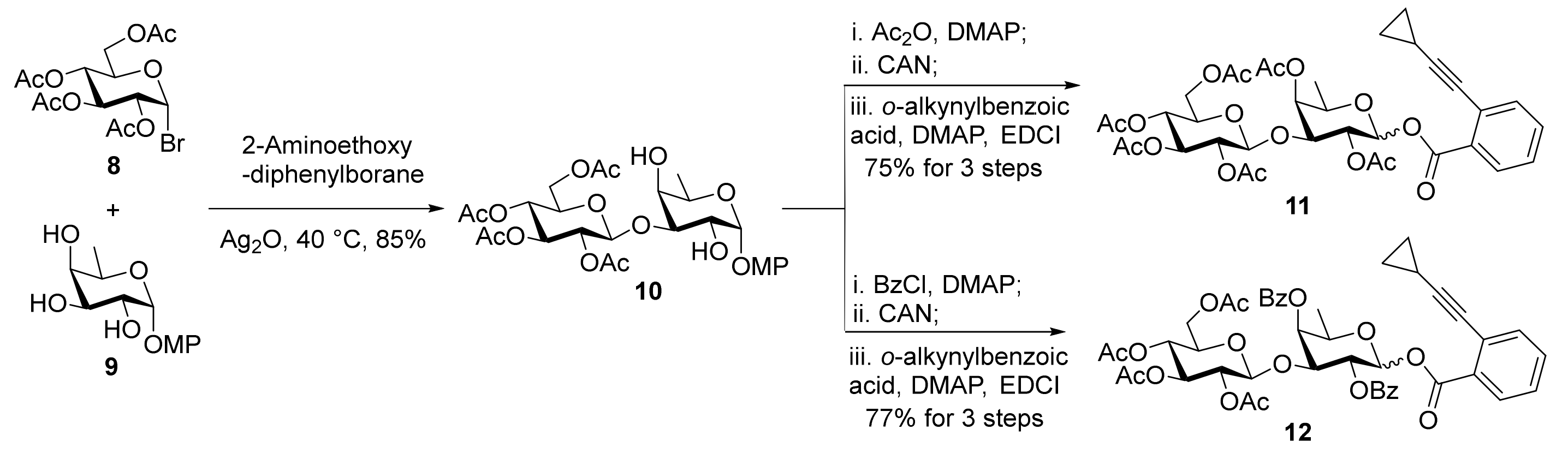
The requisite disaccharide o-alkynylbenzoates 11 and 12 were readily prepared via a regioselective glycosylation approach (Scheme 2) [22]. In view of the higher reactivity of the equatorial 3-OH versus the equatorial 2-OH and the axial 4-OH in p-methoxyphenyl β-d-fucopyranoside 9, condensation of triol 9 with glucosyl bromide 8 under the modified Taylor’s conditions led to the desired (1→3)-disaccharide 10 smoothly (85%) [23,24]. Acetylation of diol 10, oxidative cleavage of the anomeric MP group with CAN, and esterification with o-alkynylbenzoic acid furnished disaccharide o-alkynylbenzoate 11 (75% for 3 steps) [25,26]. Since the 2-O-acetyl group might result in orthoester formation or migration to the aglycone during the glycosylation reactions [27,28,29], 2,4-di-O-benzoyl o-alkynylbenzoate 12 was also prepared (77% for 3 steps).
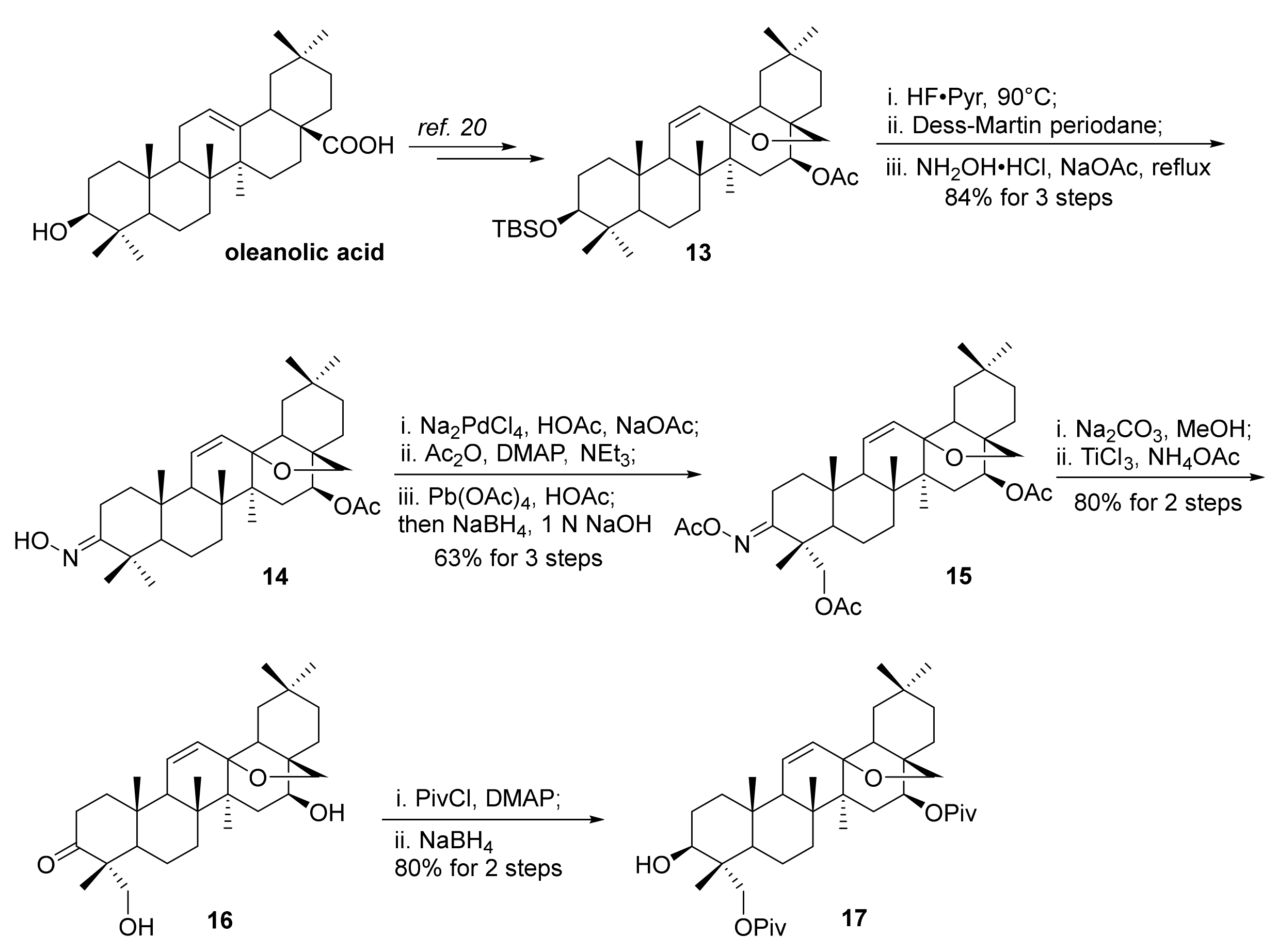
The synthesis of saikogenins commenced with known compound 13, which was readily prepared from oleanolic acid in 7 steps (Scheme 3) [20]. Desilylation with the Olah reagent, oxidation of the resultant 3-OH, and subsequent oxime formation with hydroxylamine hydrochloride afforded oxime 14 (84%). It was noted that desilylation with TBAF under reflux conditions led to unmask of the 16-O-acetyl group. Selective hydroxylation of the C-4 equatorial methyl group was then achieved following Baldwin’s method to furnish 15 (63%) [30,31,32,33,34]. that involved (1) cyclopalladation of the equatorial methyl group from oxime 14 with Na2PdCl4, (2) acetylation of the oxime hydroxyl group, and (3) oxidation with Pb(OAc)4 followed by reductive workup with NaBH4. Hydrolysis of 15 under basic conditions and then with TiCl3/NH4OAc gave 3-one-16-β-ol 16. The nascent 16-β-OH and 23-OH were firstly protected with acetyl groups; however, subsequent reduction of the 3-ketone with NaBH4 led to a mixture, due to acetyl group migration under basic conditions [35,36]. Thus, diol 16 was masked with the bulkier pivaloyl groups and then the carbonyl was reduced with NaBH4 to afford the desired aglycone derivative 17 (80%) [37].
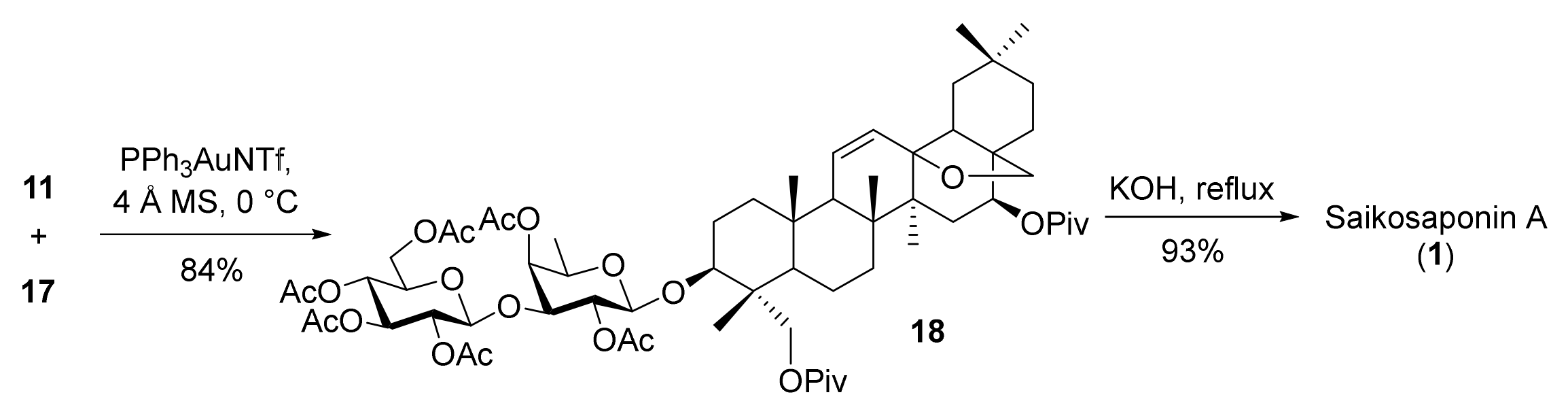
As expected, condensation of 17 with disaccharide o-alkynylbenzoate 11 proceeded smoothly under the catalysis of PPh3AuNTf2 (0.1 equiv.) [38], leading to glycoside 18 (84%) (Scheme 4). Alternatively, coupling of 17 with a relevant disaccharide trichloroacetimidate under the promotion of TMSOTf (0.1 equiv.) failed to provide glycoside 18, due to decomposition of 17 under the acidic conditions [5,39]. Finally, saponification of 18 under basic and reflux conditions furnished SSa (1) in 93% yield [40].
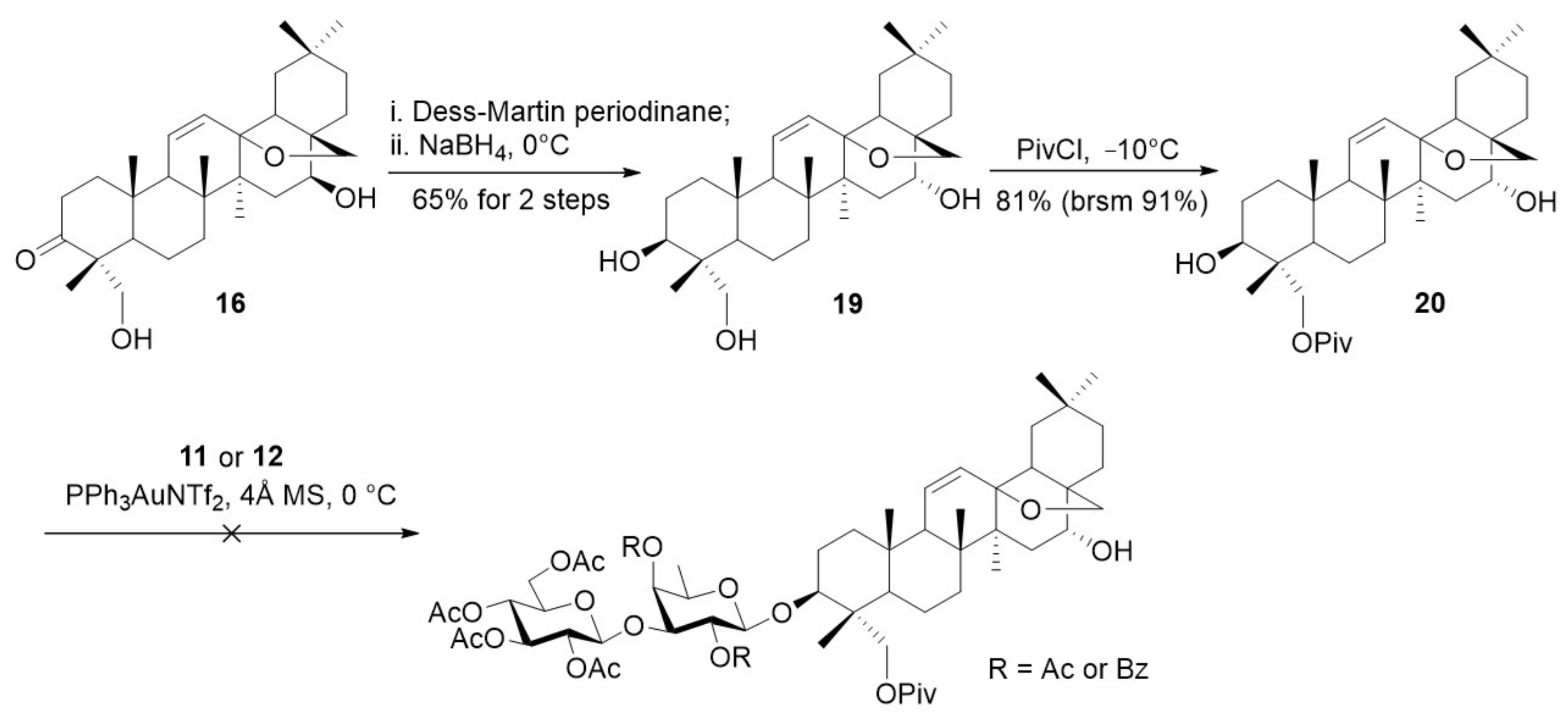
SSd (2) could be synthesized by modification of the synthetic approach to SSa (1) (Scheme 5). Thus, the configuration of 16-β-OH in 16 was inverted via Dess-Martin oxidation and NaBH4 reduction, giving SGG (19) in good yield (65%). Selective protection of the primary 23-OH with pivaloyl group provide diol 20 (81%). It was reported that regioselective acetylation of the triterpene C3-OH in the presence of C16-OH was feasible [41]. Thus, we attempted a regioselective glycosylation of 3,16-diol 20. Disappointingly, the glycosylation reactions with disaccharide donors 11 and 12 were found to futile, leading to complex mixtures.
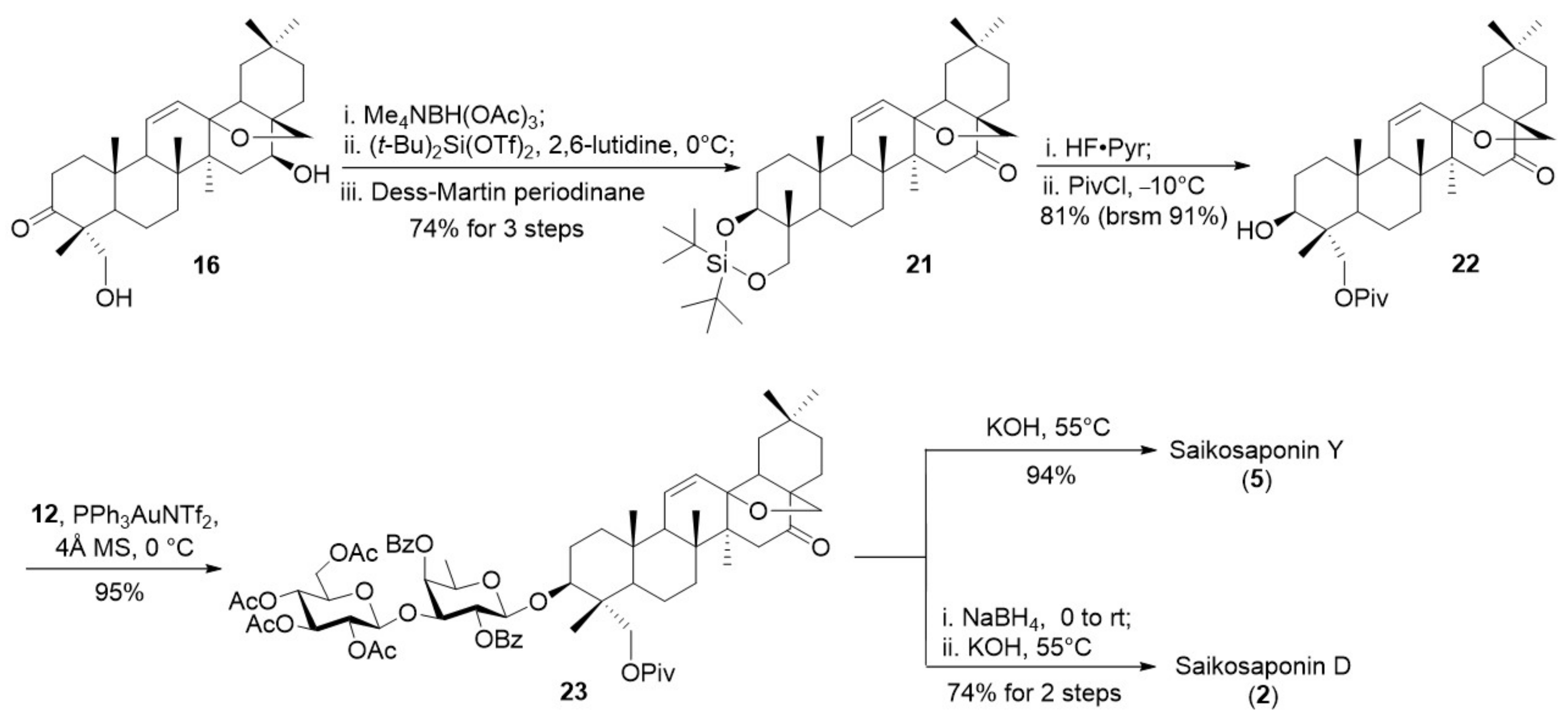
Considering the influence of the 16-α-OH on the 3-O-glycosylation, we arranged the reduction of 16-ketone at a late stage (Scheme 6). Thus, the 3-ketone in 16 was reduced with Me4NBH(OAc)3 [42,43], the resultant 3,23-diol was then protected with silylidene ketal, subsequent Dess-Martin oxidation of the remaining 16-OH led to ketone 21 (74% for 3 steps). Next, the silylidene ketal in 21 was removed and the resulting primary 23-OH was selectively protected with pivaloyl group to furnish 22 (81%). Coupling of 3-ol 22 with disaccharide o-alkynylbenzoate 11 under the optimized gold(I)-catalyzed conditions provided the desired glycoside 23 in excellent yield (95%). Ketone 23 was then subjected to reduction with NaBH4 followed by saponification with KOH at 55 °C to afford SSd (2) in 74% yield. Direct saponification of 23 furnished saikosaponin Y (5) (94%).
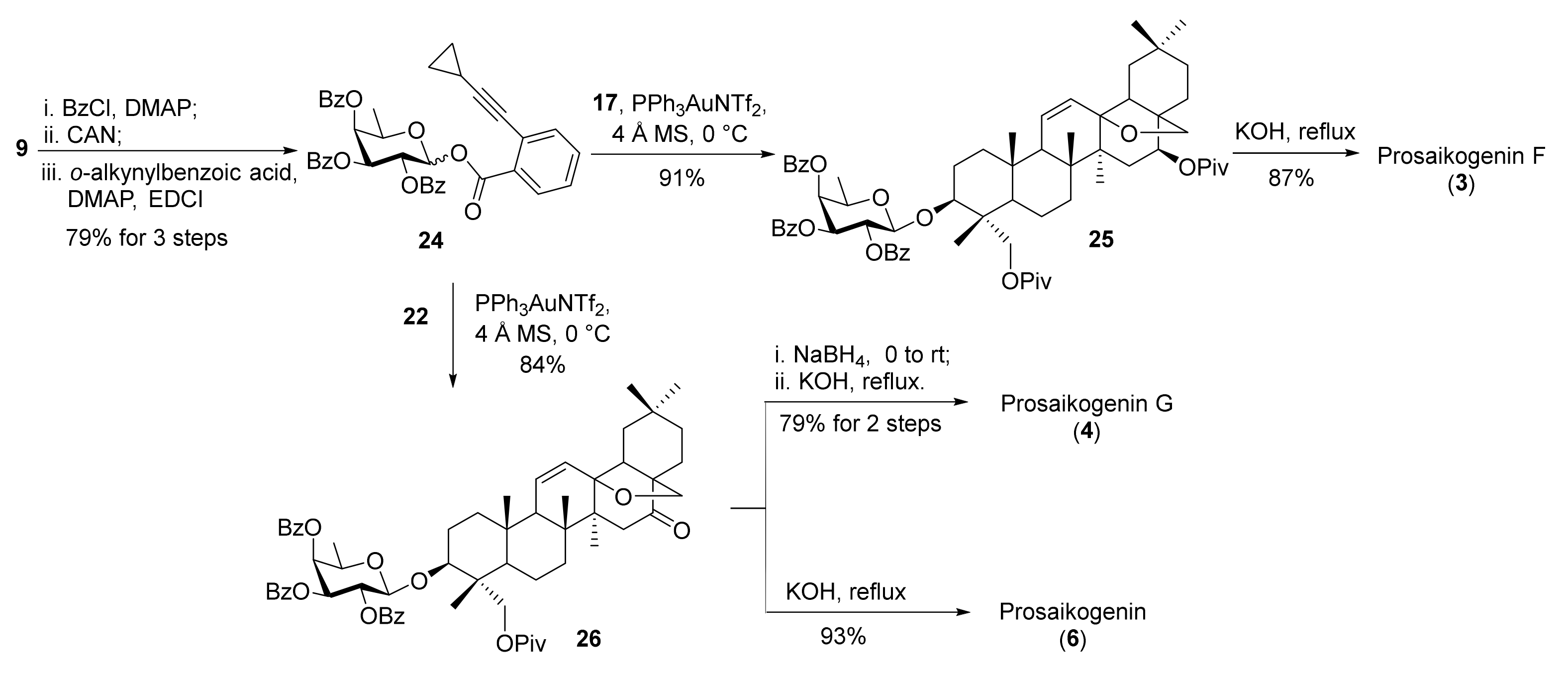
To synthesize monosaccharide saikosaponins 3, 4, and 6, fucosyl o-alkynylbenzoate 24 was prepared from p-methoxyphenyl β-d-fucopyranoside 9 via protection with benzoyl groups, oxidative cleavage of the anomeric MP group, and esterification with o-alkynylbenzoic acid (79% for 3 steps) (Scheme 7). Glycosylation of aglycone derivatives 17 and 22 with o-alkynylbenzoate 24 led to glycosides 25 (91%) and 26 (84%). Saponification of 25 and 26 afforded prosaikogenin F (3) and prosaikogenin 6 in good yields. Alternatively, reduction of the 16-ketone in 26 with NaBH4 prior to saponification furnished prosaikogenin G (4) in 79% yield.
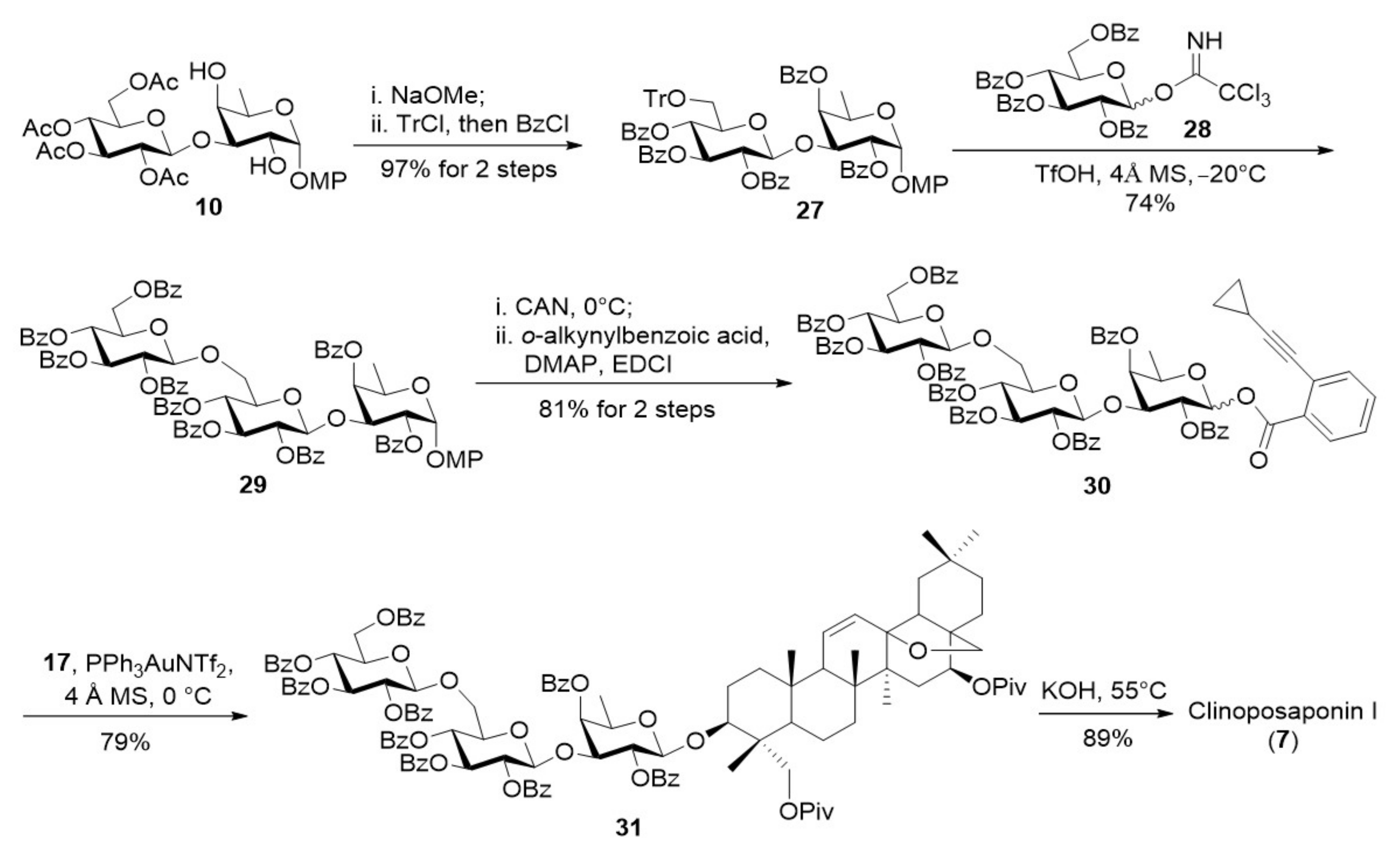
The synthesis of trisaccharide clinoposaponin I (7) was depicted in Scheme 8. Removal of the acetyl groups on disaccharide 10 followed by selective tritylation of the primary hydroxyl group and subsequent benzoylation led to disaccharide 27 (97%). Trityl ether 27 was successfully glycosylated with glucosyl trichloroacetimidate 28 under the action of TfOH (4 Å MS, CH2Cl2, −20 °C), affording trisaccharide 29 in 74% yield. Oxidative cleavage of the anomeric MP group on 29 followed by esterification provided the desired trisaccharide o-alkynylbenzoate 30 (81%). Under similar conditions as used in the previous glycosylations with mono- and disaccharide donors, coupling of aglycone 17 with trisaccharide o-alkynylbenzoate 30 led to the desired glycoside 31 in good yield (79%). Finally, saponification of 31 furnished clinoposaponin I (7) in 89% yield.
The analytical data of the synthetic saikosaponins 1–7 were in good agreement with those reported in the literatures for the natural products.
3. Experimental Section
3.1. General Information
Commercial reagents were used without further purification and made in China unless specified. Crushed 4Å molecular sieves (MS) were activated through flame-drying under high vacuum immediately prior to use. Dry CH2Cl2, DMF, and toluene were obtained by drying with activated MS. Dry pyridine and NEt3 were obtained by drying with anhydrous KOH. Anhydrous THF was obtained by refluxing with Na under argon. Thin layer chromatography (TLC) was performed on TLC silica gel 60 F254 (Merck, Darmstadt, Germany). The TLC plates were visualized with UV light and/or by staining with EtOH/H2SO4 (10%, v/v). Flash column chromatography was performed on silica gel SiliaFlash P60 (40−63 μm, Silicycle, Quebec, QC, Canada). NMR spectra were measured on a Bruker AM 400 (Switzerland), Agilent 500, or 600 MHz (U.S.) NMR spectrometer at 25 °C. 1H and 13C NMR signals were calibrated to the residual proton and carbon resonance of the solvent (CDCl3: δH = 7.26 ppm, δC = 77.16 ppm; CD3OD: δH = 3.31 ppm, δC = 49.00 ppm; DMSO-d6: δH = 2.50 ppm, δC = 39.52 ppm). High-resolution mass spectra were recorded with Shimadzu Biotech Axima Performance FTMS, maXis 4G FTMS, Thermo Scientific Q Exactive HF Orbitrap-FTMS, or Agilent-TOF/LC-MS 1260-6230 FTMS. Optical rotations were measured on an Anton Paar MCP5500 polarimeter.
3.2. Synthesis of Compounds 1–7, 10–12, 14–27, and 29–31
3.2.1. Synthesis of Compound 10
2,3,4,6-Tetra-O-acetyl-α-D-glucopyranosyl bromide 8 (115 mg, 0.3 mmol), compound 9 (50 mg, 0.2 mmol), silver(I) oxide (65 mg, 0.3 mmol), and 2-aminoethyl diphenylborinate (9 mg, 0.04 mmol) were added to an oven-dried round bottom flask under an argon atmosphere. Dry acetonitrile (1 mL) was added and the resulting mixture was stirred at room temperature. After 60 h, the reaction was quenched with a few drops of methanol, and the resulting mixture was diluted with CH2Cl2 and filtered through a plug of Celite. The crude product was purified by silica gel chromatography (petroleum ether/EtOAc = 2/1) to give compound 10 (76 mg, 85%) as a white foam. Rf = 0.2 (silica, PE/EtOAc = 1:1); [α = 82.8 (c = 1.0, CHCl3); 1H NMR (500 MHz,CDCl3): δ 7.08–6.93 (m, 2H), 6.87–6.75 (m, 2H), 5.41 (d, J = 4.1 Hz, 1H), 5.26 (m, 1H), 5.10–5.02 (m, 2H), 4.86 (d, J = 7.9 Hz, 1H), 4.24–4.20(m, 1H), 4.19–4.04 (m, 3H), 3.97 (m, 1H), 3.90 (s, 1H), 3.77 (d, J = 3.6 Hz, 1H), 3.76 (s, 3H), 2.08 (s, 3H), 2.07 (s,3H), 2.03 (s, 3H), 2.02 (s, 3H), 1.27 (d, J = 6.4 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ 170.58, 170.18, 169.88, 169.41, 118.13, 114.64, 101.59, 98.42, 81.23, 72.30, 71.90, 71.40, 70.99, 68.46, 67.53, 66.19, 61.96, 55.62, 20.74, 20.69, 20.60, 20.57, 16.18. ESI-HRMS (m/z) calcd for C27H36NaO15 [M + Na]+ 623.1946, found 623.1951.
3.2.2. Synthesis of Compound 11
To a solution of compound 10 (63 mg, 0.1 mmol) in pyridine (1 mL), Ac2O (0.1 mL, 1.05 mmol) and DMAP (15 mg, 0.03 mmol) were added. The mixture was stirred at room temperature for 8 h before it was quenched with NaHCO3 and extracted with EtOAc. The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated under vacuum. The residue was purified by flash column chromatography on silica gel (petroleum ether/EtOAc = 1/1) to give the corresponding acetylated product (73 mg, 96%) as a white solid.
To a stirred solution of the above product (50 mg, 0.07 mmol) in CH3CN/H2O (0.8 mL/0.2 mL), ceric ammonium nitrate (CAN) (89 mg, 0.16 mmol) was added. The mixture was stirred at room temperature for 2 h before it was quenched with Na2S2O3 and extracted with EtOAc. The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated under vacuum. The residue was purified by flash column chromatography (petroleum ether/EtOAc, 1:1) to give the corresponding lactol product (33 mg, 80%) as an orange foam. The α/β anomers were difficult to separate.
The lactol (40 mg, 0.07 mmol), ortho-(cyclopropylethynyl) benzoic acid (18 mg, 0.09 mmol), EDCI (24 mg, 0.09mmol), and DMAP (12 mg, 0.09 mmol) were dissolved in CH2Cl2 (2 mL). The mixture was stirred at room temperature for 4 h and concentrated under vacuum. The residue was purified by flash chromatography (petroleum ether/EtOAc = 3:1) to give compound 11 (50 mg, 98%; α/β = 1:2.5) as a white foam.
11α: Rf = 0.3 (silica, PE/EtOAc = 1:1); [α = −3.3 (c = 1.0, CHCl3); 1H NMR (500 MHz, CDCl3): δ 7.94 (d, 1H), 7.55–7.44 (m, 2H), 7.42–7.30 (m, 1H), 6.56 (d, J = 3.6 Hz, 1H), 5.42 (d, J = 3.5 Hz, 1H), 5.37 (dd, J = 10.4, 3.6 Hz, 1H), 5.16–5.03 (m, 2H), 4.91 (dd, J = 9.4, 7.9 Hz, 1H), 4.69 (d, J = 7.9 Hz, 1H), 4.52 (q, J = 6.5 Hz, 1H), 4.37 (dd, J = 10.4, 3.5 Hz, 1H), 4.23 (m, 1H), 4.15 (m, 1H), 3.68 (m, 1H), 2.16 (s, 3H), 2.10 (s, 3H), 2.06 (s, 3H), 2.00 (s, 3H), 1.98 (s, 3H), 1.95 (s, 3H), 1.18 (d, J = 6.5 Hz, 3H), 1.0–0.8 (m, 4H). 13C NMR (125 MHz, CDCl3): δ 172.30, 170.92, 170.57, 170.45, 169.99, 169.45, 169.10, 164.64, 135.12, 132.37, 131.02, 127.50, 124.77, 75.18, 72.65, 72.54, 71.97, 71.40, 69.29, 68.63, 68.25, 61.53, 20.87, 20.77, 20.73, 20.72, 20.52, 16.28, 9.18, 9.16.
11β: Rf = 0.25 (silica, PE/EtOAc = 1:1); [α = −80.5 (c = 1.0, CHCl3); 1H NMR (500 MHz, CDCl3): δ 7.93 (d, J = 7.9 Hz, 1H), 7.46 (d, J = 7.6 Hz, 1H), 7.41 (t, J = 7.5 Hz, 1H), 7.28 (m, 1H), 5.82 (d, J = 8.3 Hz, 1H), 5.41 (t, J = 8.7 Hz, 1H), 5.30 (s, 1H), 5.10 (m, 2H), 4.91 (t, J = 8.4 Hz, 1H), 4.62 (d, J = 7.8 Hz, 1H), 4.26 (m, 1H), 4.15 (m, 1H), 3.94 (m, 2H), 3.69–3.58 (m, 1H), 2.15 (s, 3H), 2.10 (s, 3H), 1.99 (s, 9H), 1.97 (s, 3H), 1.51 (m, 1H), 1.20 (d, J = 5.3 Hz, 3H), 0.89 (m, 4H). 13C NMR (125 MHz, CDCl3): δ 170.84, 170.43, 170.41, 169.37, 169.32, 169.16, 163.64, 134.53, 132.58, 131.04, 129.35, 127.23, 125.79, 100.76, 100.45, 92.44, 74.46, 72.60, 71.95, 71.62, 71.19, 70.90, 70.00, 68.25, 61.43, 20.88, 20.76, 20.68, 20.48, 16.20, 9.02. ESI-HRMS (m/z) calcd for C36H42NaO17 [M + Na]+ 769.2314, found 7769.2311.
3.2.3. Synthesis of Compound 12
To a stirred solution of compound 10 (830 mg, 1.4 mmol) in pyridine (5 mL), benzoyl chloride (0.48 mL, 4.14 mmol) and DMAP (50 mg, 0.4 mmol) were added. The mixture was stirred at room temperature for 8 h before it was quenched with NaHCO3 and extracted with EtOAc. The combined organic phases were washed with brine, dried over anhydrous Na2SO4, and filtered. The filtrate was concentrated under vacuum. The residue was purified by flash column chromatography on silica gel (petroleum ether/EtOAc = 3/1) to give the corresponding benzoylated product (1.07 g, 96%) as a white solid.
To a stirred solution of the above product (800 mg, 0.99 mmol) in CH3CN/H2O (12 mL/3 mL), ceric ammonium nitrate (1.35 g, 2.5 mmol) was added. The mixture was stirred at room temperature for 2 h before it was quenched with Na2S2O3 and extracted with EtOAc. The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated under vacuum. The residue was purified by flash column chromatography (petroleum ether/EtOAc = 1:1) to give the corresponding lactol product (560 mg, 81%) as an orange foam. The α/β anomers were difficult to separate.
The lactol (400 mg, 0.07 mmol), ortho-(cyclopropylethynyl) benzoic acid (167 mg, 0.89 mmol), EDCI (172 mg, 0.89 mmol), and DMAP (110 mg, 0.89 mmol) were dissolved in CH2Cl2 (5 mL). The mixture was stirred at room temperature for 4 h and then concentrated under vacuum. The residue was purified by flash chromatography (petroleum ether/EtOAc = 3:1) to give compound 12 (552 mg, 99%; α/β = 1:2.3) as a white foam.
12α: Rf = 0.2 (silica, PE/EtOAc = 3:1); [α = 107.9 (c = 1.0, CHCl3); 1H NMR (500 MHz, CDCl3): δ 8.11 (d, J = 7.5 Hz, 2H), 7.94 (d, J = 7.6 Hz, 2H), 7.90 (d, J = 7.8 Hz, 1H), 7.66–7.44 (m, 6H), 7.37 (m, 3H), 6.79 (d, J = 3.5 Hz, 1H), 5.84 (m, 1H), 5.75 (d, J = 2.5 Hz, 1H), 5.05–4.92 (m, 2H), 4.78 (m, 2H), 4.72–4.57 (m, 2H), 4.14 (m, 2H), 3.65 (m, 1H), 2.01 (s, 3H), 1.96 (s, 3H), 1.85 (s, 3H), 1.44 (m, 1H), 1.36 (s, 3H), 1.31–1.24 (m, 3H), 0.87 (m, 4H). 13C NMR (125 MHz, CDCl3): δ 170.90, 170.32, 169.32, 168.83, 166.02, 165.43, 164.59, 135.00, 133.62, 133.31, 132.32, 131.06, 130.95, 130.14, 129.83, 129.79, 129.29, 128.68, 128.60, 127.44, 124.76, 100.92, 99.57, 91.39, 75.07, 74.29, 73.08, 72.75, 71.85, 71.22, 69.36, 68.88, 68.15, 61.60, 20.80, 20.69, 20.60, 19.75, 16.66, 9.24, 9.19.
12β: Rf = 0.15 (silica, PE/EtOAc = 3:1); [α = 47.7 (c = 1.0, CHCl3); 1H NMR (500 MHz, CDCl3): δ 8.13 (d, J = 7.5 Hz, 2H), 7.97 (d, J = 7.6 Hz, 2H), 7.93 (d, J = 7.9 Hz, 1H), 7.57 (dt, J = 22.9, 7.4 Hz, 2H), 7.49 (t, J = 7.6 Hz, 2H), 7.41 (q, J = 7.7 Hz, 3H), 7.36 (m, 1H), 7.22 (d, J = 7.4 Hz, 1H), 6.10 (d, J = 8.4 Hz, 1H), 5.93–5.83 (m, 1H), 5.64 (d, J = 3.2 Hz, 1H), 5.03–4.93 (m, 2H), 4.79 (m, 1H), 4.67 (d, J = 7.8 Hz, 1H), 4.22 (m, 1H), 4.13 (m, 1H), 3.63 (d, J = 8.5 Hz, 1H), 2.02 (s, 3H), 1.96 (s, 3H), 1.86 (s, 3H), 1.50 (m, 1H), 1.44 (s, 3H), 1.32 (d, J = 6.3 Hz, 3H), 0.85 (m, 4H). 13C NMR (125 MHz, CDCl3): δ 170.91, 170.35, 169.32, 168.93, 166.14, 165.10, 163.84, 134.44, 133.63, 133.27, 132.48, 131.09, 130.23, 129.81, 129.48, 129.32, 128.71, 128.53, 127.20, 125.76, 110.13, 101.15, 100.42, 92.71, 78.23, 74.46, 72.78, 72.53, 71.86, 71.46, 71.12, 70.33, 68.08, 61.47, 31.75, 29.77, 20.82, 20.67, 20.60, 19.79, 16.65. ESI-HRMS (m/z) calcd for C46H46NaO17 [M + Na]+ 893.2627, found 893.2632.
3.2.4. Synthesis of Compound 14
Compound 13 (100 mg, 0.17 mmol) was dissolved in pyridine (5 mL), to which was added HF·pyr (0.2 mL) at room temperature. After stirring at 90 °C for 24 h, the mixture was quenched with saturated NaHCO3 and extracted with EtOAc. The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated under vacuum. The residue was purified by flash column chromatography (petroleum ether/EtOAc = 2:1) to give the 3-ol product (76 mg, 88%) as a white foam. Rf = 0.2 (silica, PE/EtOAc = 5:1).
The above product (3.6 g, 7.2 mmol) was dissolved in CH2Cl2 (40 mL), to which Dess-Martin periodinane (DMP) (3.7 g, 8.6 mmol) was added. After being stirred for 2 h at room temperature, the reaction mixture was quenched with a saturated aqueous Na2S2O3 solution, and the aqueous phase was extracted with CH2Cl2. The combined organic extracts were dried over anhydrous Na2SO4 and concentrated in vacuo.
The crude product was dissolved in MeOH/CH2Cl2 (80 mL/40 mL), to which NH2OH·HCl (692 mg, 10.8 mmol) and NaOAc (1.76 g, 21.6 mmol) were added at room temperature. After stirring two hours at reflux, the reaction mixture was diluted by addition of brine, then extracted with CH2Cl2. The extract was dried over anhydrous Na2SO4, followed by filtration and concentration in vacuo. The residue was purified by silica gel flash column chromatography (petroleum ether/EtOAc = 6/1 to 3/1) to give compound 14 (3.55 g, 96%) as a white foam. Rf = 0.5 (silica, PE/EtOAc = 2:1); [α = 76.7 (c = 1.0, CHCl3); 1H NMR (500 MHz, CDCl3): δ 9.30–9.23 (m, 1H), 5.88–5.86 (m, 1H), 5.43–5.38 (m, 2H), 3.97 (d, J = 7.4 Hz, 1H), 3.18 (d, J = 7.6, 1H), 3.11–3.07 (m, 1H), 2.26–2.19 (m, 1H), 2.04 (s, 3H), 1.14 (s, 3H), 1.10 (s, 3H), 1.03 (s, 3H), 1.00 (s, 3H), 0.99 (s, 3H), 0.96 (s, 3H), 0.88 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 170.80, 166.65, 132.60, 129.92, 83.88, 73.24, 68.79, 55.34, 52.16, 51.68, 45.34, 45.27, 41.88, 40.61, 38.39, 37.41, 36.51, 34.28, 33.44, 31.97, 31.52, 31.12, 26.95, 25.44, 23.76, 22.66, 21.34, 20.47, 19.43, 18.34, 17.37, 16.94. ESI-HRMS (m/z) calcd for C32H50NO4 [M + H]+ 512.3734, found 512.3739.
3.2.5. Synthesis of Compound 15
To a solution of compound 14 (1.9 g, 3.62 mmol) in HOAc (170 mL), NaOAc (350 mg, 4.3 mmol) and Na2PdCl4 (1.3 g, 4.3 mmol) were added at room temperature. The mixture was stirred at room temperature for 3 d, and then ice water (300 mL) was added. The resulting mixture was stirred for an additional 20 min and then filtered. The filtered residue was washed with water several times, dried, and used without further purification.
The residue was dissolved in CH2Cl2 (20 mL) at room temperature, to which NEt3 (1 mL, 7.24 mmol), DMAP (88 mg, 0.72 mmol), and Ac2O (0.68 mL, 7.24 mmol) were added. The resulting mixture was stirred at room temperature for 3 h and then diluted with EtOAc. The mixture was thoroughly washed with water and brine, and the organic layer was dried with anhydrous Na2SO4. After filtration, the solution was concentrated under vacuum to give a residue. The residue was dissolved in THF (120 mL), and anhydrous pyridine (0.3 mL) was added dropwise. After the mixture was stirred for 40 min, a solution of Pb(OAc)4 (1.5 mg, 3.2 mmol) in HOAc (51 mL) was added dropwise at −78 °C. The mixture was warmed to room temperature and then stirred for 24 h. The reaction mixture was cooled to 0 °C, and a solution of NaBH4 (136 mg, 1.7 mmol) in NaOH (1M, 46 mL) was slowly added. The resulting solution was stirred at 0 °C for an additional 1 h and diluted with EtOAc. The mixture was washed with a saturated NaHCO3 solution and then brine, and the organic layer was dried with anhydrous Na2SO4. After filtration, the solution was concentrated to give a residue, which was purified by flash column chromatography on silica gel (petroleum/EtOAc = 7:1) to give compound 15 (1.4 g, 63% for 3 steps) as a white foamy solid. Rf = 0.3 (silica, PE/EtOAc = 4:1); [α = 98.9 (c = 1.0, CHCl3); 1H NMR (500 MHz, CDCl3): δ 5.81 (d, J = 10.3 Hz, 1H), 5.44–5.36 (m, 2H), 4.17–4.07 (m, 2H), 3.95 (d, J = 7.5 Hz, 1H), 3.16 (d, J = 7.5 Hz, 1H), 2.80–2.75 (m, 1H), 2.57–2.50 (m, 1H), 2.13 (s, 3H), 2.02 (d, J = 3.1 Hz, 6H), 1.12 (s, 3H), 1.08 (s, 3H), 1.00 (s, 3H), 0.93 (s, 6H), 0.86 (s, 3H). 13C NMR (126 MHz, CDCl3): δ 170.88, 170.67, 170.06, 169.77, 131.93, 130.23, 83.63, 73.20, 68.54, 67.63, 51.72, 51.63, 47.88, 45.24, 45.19, 44.09, 41.60, 37.36, 37.01, 35.91, 34.17, 33.37, 31.85, 31.45, 30.44, 25.34, 23.69, 21.26, 21.01, 20.40, 20.06, 19.95, 19.02, 18.71, 18.56, 17.44. ESI-HRMS (m/z) calcd for C36H53NNaO7 [M + Na]+ 634.3714, found 634.3717.
3.2.6. Synthesis of Compound 16
A solution of compound 15 (1.0 g, 1.6 mmol) and Na2CO3 (866 mg, 8.2 mmol) in MeOH (120 mL) was stirred at room temperature for 24 h. After removing MeOH in vacuo, the resulting colorless solid was dissolved in Et2O. The organic layer was washed with 1 N HCl solution, saturated NaHCO3 solution and brine, dried over anhydrous Na2SO4, and filtered. The filtrate was evaporated in vacuo to give a solid.
To a solution of the above product in THF (55 mL), NH4OAc (1.5 g, 20 mmol) and a buffered solution of TiCl3 (3.4 mL of HCl solution containing 15% TiCl3, 4 mmol) were added. The mixture was stirred at room temperature for 8 h. The mixture was washed with saturated NaHCO3 solution and brine, dried over anhydrous Na2SO4, filtered, and evaporated in vacuo to give a solid. The solid was purified by silica gel flash column chromatography (petroleum ether/EtOAc = 2/1) to give compound 16 (613 mg, 80%) as a white foam. Rf = 0.5 (silica, PE/EtOAc = 1:1); [α = 89.4 (c = 1.0, CHCl3); 1H NMR (500 MHz, CDCl3): δ 5.91–5.81 (m, 1H), 5.47 (dd, J = 10.3, 3.1 Hz, 1H), 4.21 (dd, J = 10.1, 5.9 Hz, 1H), 3.92 (d, J = 7.4 Hz, 1H), 3.70 (d, J = 11.4 Hz, 1H), 3.40 (d, J = 11.4 Hz, 1H), 3.13 (d, J = 7.5 Hz, 1H), 2.68 (m, 1H), 2.31 (m, 1H), 2.11 (m, 1H), 1.14 (s, 3H), 1.11 (s, 3H), 1.01 (s, 3H), 1.00 (s. 3H), 0.99 (s, 3H), 0.91 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 218.37, 131.71, 130.37, 72.28, 66.39, 64.99, 52.75, 51.80, 51.52, 48.41, 46.21, 45.51, 41.70, 38.73, 37.33, 35.91, 35.29, 35.01, 34.17, 33.53, 31.46, 30.67, 29.69, 24.94, 23.67, 20.61, 19.36, 18.31, 17.28, 16.25. ESI-HRMS (m/z) calcd for C30H46NaO4 [M + Na]+ 493.3288, found 493.3283.
3.2.7. Synthesis of Compound 17
To a stirred solution of compound 16 (90 mg, 0.2 mmol) in pyridine (4 mL), pivaloyl chloride (0.5 mL, 4 mmol) and DMAP (6 mg, 0.06 mmol) were added. The mixture was stirred at room temperature for 8 h before it was quenched with NaHCO3 and extracted with EtOAc. The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated under vacuum. The residue was purified by flash column chromatography on silica gel (petroleum ether/EtOAc = 8/1) to give the acylated product (115 mg, 94%) as a white solid. Rf = 0.5 (silica, PE/EtOAc = 3:1); [α = 113.9 (c = 1.0, CHCl3); 1H NMR (500 MHz, CDCl3): δ 5.90 (d, J = 10.3 Hz, 1H), 5.48 (dd, J = 10.3, 3.1 Hz, 1H), 5.39 (m, 1H), 4.13–4.03 (m, 2H), 4.00 (d, J = 7.4 Hz, 1H), 3.22 (d, J = 7.1 Hz, 1H), 2.60 (m, 1H), 2.46 (m, 1H), 1.20 (s, 9H), 1.17 (s, 9H), 1.15 (s, 3H), 1.06 (s, 3H), 1.05 (s, 3H), 1.00 (s, 3H), 0.97 (s, 3H), 0.90 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 213.52, 178.05, 177.81, 130.99, 83.74, 73.20, 68.03, 66.70, 51.75, 51.65, 50.54, 47.52, 45.34, 45.21, 41.61, 38.93, 38.77, 37.66, 37.33, 35.65, 34.58, 34.03, 33.31, 31.57, 31.39, 30.47, 27.20, 25.20, 23.65, 20.27, 19.18, 18.56, 16.97, 16.56. ESI-HRMS (m/z) calcd for C40H62NaO6 [M + Na]+ 661.4439, found 661.4445.
To a stirred solution of the above product (90 mg, 0.14 mmol) in MeOH (3 mL) at 0 °C, NaBH4 (23 mg, 0.5 mmol) was added. The stirring continued for 15 min, the solution was quenched by addition of saturated NH4Cl solution, then diluted with brine and extracted with CH2Cl2. The extract was dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The residue was purified by silica gel column chromatography (petroleum ether/EtOAc = 4/1) to give compound 17 (76 mg, 85%) as a white foam. Rf = 0.3 (silica, PE/EtOAc = 3:1); [α = 65.3 (c = 1.0, CHCl3); 1H NMR (500 MHz, CDCl3): δ 5.88 (d, J = 10.3 Hz, 1H), 5.42 (dd, J = 10.2, 3.0 Hz, 1H), 5.40–5.35 (m, 1H), 4.19 (d, J = 11.6 Hz, 1H), 3.99 (d, J = 7.4 Hz, 1H), 3.75 (d, J = 11.5 Hz, 1H), 3.36 (t, J = 8.2 Hz, 1H), 3.21 (d, J = 7.4 Hz, 1H), 2.33 (t, J = 7.5 Hz, 1H), 1.21 (s, 9H), 1.20 (s, 9H), 1.09 (s, 3H), 1.03 (s, 3H), 0.96 (s, 3H), 0.93 (s, 3H), 0.88 (s, 3H), 0.75 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 178.76, 178.14, 132.52, 129.64, 83.88, 73.14, 72.26, 68.19, 65.77, 52.75, 51.62, 47.73, 45.31, 45.17, 42.42, 41.68, 38.32, 37.32, 36.08, 34.05, 33.32, 31.56, 31.38, 31.05, 29.68, 27.31, 27.19, 25.91, 25.21, 23.64, 20.44, 19.43, 18.16, 17.37, 11.39. ESI-HRMS (m/z) calcd for C40H64NaO6 [M + Na]+ 663.4595, found 663.4598.
3.2.8. Synthesis of Compound 18
To a stirred mixture of compound 11 (180 mg, 0.24 mmol), compound 17 (120 mg, 0.18 mmol), and 4Å molecular sieves (400 mg) in CH2Cl2 (4 mL), PPh3AuNTf2 (26 mg, 0.04 mmol) was added. The mixture was stirred at room temperature for 1 h before it was quenched with NEt3. The mixture was filtered through a pad of celite and washed with EtOAc. The filtrate was concentrated under vacuum. The residue was purified by flash column chromatography (petroleum ether/EtOAc = 4:1) to give compound 18 (190 mg, 84%) as a white foam. Rf = 0.3 (silica, PE/EtOAc = 3:1); [α = 43.9 (c = 1.0, CHCl3); 1H NMR (600 MHz, CDCl3): δ 5.91–5.78 (m, 1H), 5.47–5.38 (m, 1H), 5.36 (dd, J = 9.6, 6.2 Hz, 1H), 5.18 (d, J = 3.5 Hz, 1H), 5.10 (m, 3H), 4.87 (t, J = 8.5 Hz, 1H), 4.58 (d, J = 7.9 Hz, 1H), 4.26 (m, 2H), 4.13 (m, 1H), 3.97 (d, J = 7.4 Hz, 1H), 3.81 (d, J = 11.3 Hz, 1H), 3.77–3.71 (m, 2H), 3.66–3.62 (m, 1H), 3.61 (d, J = 6.5 Hz, 1H), 3.36 (dd, J = 11.5, 4.7 Hz, 1H), 3.19 (d, J = 7.4 Hz, 1H), 2.11 (s, 3H), 2.10 (s, 3H), 2.09 (s, 3H), 2.00 (s, 3H), 1.99 (s, 3H), 1.97 (s, 3H), 1.24 (s, 9H), 1.19 (s, 9H), 1.17 (s, 3H), 1.07 (s, 3H), 1.00 (s, 3H), 0.95 (s, 3H), 0.90 (s, 3H), 0.88 (s, 3H), 0.71 (s, 3H). 13C NMR (150 MHz, CDCl3): δ 102.85, 100.52, 73.15, 72.60, 71.75, 71.68, 71.26, 71.10, 69.30, 68.31, 68.19, 65.00, 61.31, 52.72, 51.62, 47.66, 45.32, 45.17, 42.04, 41.69, 39.06, 38.96, 38.11, 37.35, 35.79, 34.08, 33.36, 31.55, 31.41, 30.98, 27.38, 27.32, 27.20, 25.28, 25.23, 23.65, 20.98, 20.76, 20.59, 20.40, 20.37, 19.44, 17.94, 17.22, 16.35, 12.17. ESI-HRMS (m/z) calcd for C64H96NaO21 [M + Na]+ 1223.6336, found 1223.6318.
3.2.9. Synthesis of Saikosaponin A (1)
To a stirred solution of compound 18 (110 mg, 0.1 mmol) in MeOH (3 mL), KOH (261 mg, 4.6 mmol) was added at room temperature. The mixture was stirred at reflux for 24 h before it was quenched with acetic acid. The mixture was concentrated under vacuum. The residue was purified by reversed-phase silica gel column chromatography (ODS RP-18) (MeOH/H2O = 5:1 to 7:1) to give saikosaponin A (1) (69 mg, 93%) as a white powder. Rf = 0.4 (silica, CHCl3/MeOH = 5:1); [α = −5.9 (c = 1.0, CH3OH); 1H NMR (600 MHz, pyridine-d5): δ 6.07–5.99 (m, 1H), 5.73–5.63 (m, 1H), 5.35 (d, J = 7.8 Hz, 1H), 5.00 (d, J = 7.8 Hz, 1H), 4.57 (m, 4H), 4.45–4.37 (m, 4H), 4.30 (m, 2H), 4.23 (m, 1H), 4.09–4.01 (m, 3H), 3.75–3.67 (m, 2H), 3.36 (d, J = 6.9 Hz, 1H), 2.52 (m, 1H), 2.35 (m, 1H), 1.45 (d, J = 6.3 Hz, 3H), 1.41 (s, 3H), 1.12 (s, 3H), 1.01 (s, 3H), 0.95 (s, 3H), 0.94 (s, 3H), 0.91 (s, 3H). 13C NMR (150 MHz, Pyridine-d5): δ 132.14, 131.11, 106.58, 105.95, 85.12, 83.94, 81.53, 78.71, 78.36, 75.76, 72.97, 72.09, 71.77, 71.49, 70.96, 64.00, 63.92, 62.64, 53.03, 52.09, 47.23, 46.94, 45.58, 43.68, 42.14, 38.60, 37.67, 36.20, 36.06, 34.62, 33.59, 31.55, 31.52, 26.06, 25.70, 23.77, 20.79, 19.99, 18.69, 17.49, 17.22, 13.00. ESI-HRMS (m/z) calcd for C42H68NaO13 [M + Na]+ 803.4552, found 803.4549.
3.2.10. Synthesis of Compound 19
Compound 16 (290 mg, 0.6 mmol) was dissolved in CH2Cl2 (10 mL), to which DMP (636 mg, 1.5 mmol) was added. After being stirred for 40 min at room temperature, the reaction mixture was quenched with a saturated Na2S2O3 solution and the aqueous phase was extracted with CH2Cl2. The combined organic extracts were dried over anhydrous Na2SO4 and concentrated in vacuo.
To a solution of the above product in MeOH (10 mL) under stirring at 0 °C, NaBH4 (95 mg, 2.5 mmol) was added. The mixture was stirred for 15 min before it was quenched by addition of saturated NH4Cl solution. The mixture was diluted with brine and extracted with CH2Cl2. The combined organic phases were dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The residue was purified by silica gel column chromatography (petroleum ether/EtOAc = 2/1) to give compound 19 (190 mg, 65%) as a white foam. Rf = 0.3 (silica, PE/EtOAc = 2:1); [α = 11.2 (c = 1.0, Pyridine); 1H NMR (500 MHz, Pyridine-d5): δ 6.05 (m, 1H), 5.74 (d, J = 8.8 Hz, 1H), 3.74 (d, J = 9.8 Hz, 1H), 3.62 (d, J = 5.6 Hz, 1H), 3.36 (d, J = 5.4 Hz, 1H), 2.69 (m, 1H), 2.52 (m, 1H), 1.64 (s, 3H), 1.41 (s, 3H), 1.10 (s, 6H), 1.06 (s, 3H), 1.00 (s, 3H). 13C NMR (125 MHz, Pyridine-d5): δ 132.46, 132.39, 85.30, 78.25, 77.58, 73.76, 68.15, 53.43, 51.80, 48.87, 45.79, 44.05, 43.50, 42.31, 39.10, 38.87, 37.25, 37.02, 35.88, 34.21, 32.34, 32.03, 31.74, 28.03, 24.89, 19.99, 19.14, 18.55, 18.44, 12.98. ESI-HRMS (m/z) calcd for C30H48NaO4 [M + Na]+ 495.3445, found 495.3437.
3.2.11. Synthesis of Compound 20
To a stirred solution of compound 19 (187 mg, 0.4 mmol) in CH2Cl2 (4 mL), pivaloyl chloride (58 μL, 0.48 mmol) and pyridine (0.57 mL, 7.2 mmol) were added. The mixture was stirred at −10 °C for 8 h before it was quenched with NaHCO3 and extracted with EtOAc. The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated under vacuum. The residue was purified by flash column chromatography on silica gel (petroleum ether/EtOAc = 8/1) to give compound 20 (178 mg, 81%) as a white solid. Rf = 0.3 (silica, PE/EtOAc = 3:1); [α = 41.7 (c = 1.0, CHCl3); 1H NMR (500 MHz, CDCl3): δ 5.87 (d, J = 10.4 Hz, 1H), 5.42 (dd, J = 10.3, 3.0 Hz, 1H), 4.17 (d, J = 11.5 Hz, 1H), 4.00 (d, J = 4.9 Hz, 1H), 3.78 (d, J = 11.5 Hz, 1H), 3.45 (d, J = 7.3 Hz, 1H), 3.40–3.36 (m, 1H), 3.17 (d, J = 7.3 Hz, 1H), 2.20 (m, 1H), 1.25 (s, 3H), 1.21 (s, 9H), 1.05 (s, 3H), 0.95 (s, 3H), 0.93 (s, 3H), 0.91 (s, 3H), 0.75 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 178.84, 132.14, 130.83, 84.74, 78.12, 72.49, 66.10, 52.88, 50.47, 47.92, 45.00, 42.98, 42.57, 41.46, 39.16, 38.41, 38.13, 36.79, 36.25, 35.10, 33.55, 31.72, 31.17, 30.47, 29.82, 27.47, 27.19, 26.13, 24.26, 19.11, 18.36, 18.00, 17.61, 11.52. ESI-HRMS (m/z) calcd for C35H56NaO5 [M + Na]+ 579.4020, found 579.4016.
3.2.12. Synthesis of Compound 21
A solution of tetramethylammonium triacetoxyborohydride (1.9 g, 7.2 mmol) in MeCN (27 mL) and AcOH (1.12 mL) was cooled to −40 °C. To the mixture was added a solution of the compound 16 (680 mg, 1.4 mmol) in CH2Cl2 (9 mL). The mixture was stirred at room temperature for 0.5 h before it was quenched with Rochelle salt (sat. aq.) and extracted with EtOAc. The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated under vacuum.
The above product was dissolved into DMF (5 mL), to which 2,6-lutidine (2.5 mL) was added, and the solution was cooled to 0 °C. Di-tert-butylsilylbis(trifluoromethansulfonate) (0. 6 mL, 1.7 mmol) was added. The mixture was stirred at room temperature for 2 h before it was quenched with NaHCO3 and extracted with EtOAc. The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated under vacuum.
The above product was dissolved in CH2Cl2 (5 mL), then the solution was added DMP (736 mg, 1.7 mmol). After being stirred for 40 min at room temperature, the reaction mixture was quenched with a saturated Na2S2O3 solution, and the aqueous phase was extracted with CH2Cl2. The combined organic extracts were dried over anhydrous Na2SO4 and concentrated in vacuo. The residue was purified by flash chromatography (petroleum ether/EtOAc = 15:1) to give compound 21 (507 mg, 74%) as a white solid. Rf = 0.5 (silica, PE/EtOAc = 6:1); [α = 12.9 (c = 1.0, CHCl3); 1H NMR (500 MHz, CDCl3): δ 5.95 (d, J = 10.3 Hz, 1H), 5.56–5.52 (m, 1H), 3.86 (m, 3H), 3.72 (d, J = 10.3 Hz, 1H), 3.44 (d, J = 7.9 Hz, 1H), 2.68 (m, 1H), 2.23 (m, 1H), 2.15 (m, 1H), 1.09 (s, 9H), 1.07 (s, 3H), 1.05 (s, 3H), 1.03 (s, 9H), 0.98 (s, 3H), 0.95 (s, 3H), 0.92 (s, 3H), 0.86 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 213.14, 213.12, 133.20, 128.84, 84.15, 80.53, 75.56, 56.35, 55.03, 52.37, 52.28, 49.63, 44.49, 41.78, 41.04, 38.89, 38.79, 36.64, 35.54, 33.40, 31.68, 31.09, 29.24, 27.94, 27.83, 26.68, 24.31, 23.70, 23.29, 20.50, 20.23, 19.52, 18.75, 17.14, 12.28. ESI-HRMS (m/z) calcd for C38H63O4Si [M + H]+ 611.4490, found 611.4488.
3.2.13. Synthesis of Compound 22
The compound 21 was dissolved in THF (5 mL), to which HF·pyr (1 mL) was added at room temperature. After stirring at room temperature for 1 h, the mixture was quenched with saturated NaHCO3 and extracted with EtOAc. The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated under vacuum. The residue was purified by flash column chromatography (petroleum ether/EtOAc = 2:1) to give the diol product (500 mg, 99%) as a white foam. Rf = 0.3 (silica, PE/EtOAc = 3:1); [α = 12.9 (c = 1.0, Pyridine); 1H NMR (500 MHz, Pyridine-d5): δ 6.11 (d, J = 10.4 Hz, 1H), 5.74 (m, 1H), 4.26 (dd, J = 11.2, 5.3 Hz, 1H), 4.22 (d, J = 10.4 Hz, 1H), 3.96 (d, J = 7.8 Hz, 1H), 3.74 (d, J = 10.4 Hz, 1H), 3.54 (d, J = 7.8 Hz, 1H), 2.87 (m, 1H), 2.36 (m, 1H), 1.42 (s, 3H), 1.07 (s, 3H), 1.05 (s, 3H), 1.03 (s, 3H), 0.87 (s, 3H), 0.83 (s, 3H). 13C NMR (125 MHz, pyridine-d5): δ 212.80, 133.74, 129.94, 84.73, 75.90, 73.18, 67.47, 56.85, 55.59, 53.25, 50.29, 48.50, 45.25, 43.62, 42.56, 39.47, 39.02, 36.97, 36.36, 33.78, 32.12, 31.86, 28.05, 24.97, 23.60, 20.70, 20.28, 18.98, 18.28, 13.08. ESI-HRMS (m/z) calcd for C30H47O4 [M + H]+ 471.3469, found 471.3463.
To a stirred solution of the above product (470 mg, 1 mmol) in CH2Cl2 (10 mL), pivaloyl chloride (0.15 mL, 1.2 mmol) and pyridine (0.8 mL, 12 mmol) were added. The mixture was stirred at −10 °C for 30 h before it was quenched with NaHCO3 and extracted with EtOAc. The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated under vacuum. The residue was purified by flash column chromatography on silica gel (petroleum ether/EtOAc = 9/1) to give compound 22 (487 mg, 81%) as a white solid. Rf = 0.4 (silica, PE/EtOAc = 3:1); [α = 12.9 (c = 1.0, CHCl3); 1H NMR (500 MHz, CDCl3): δ 5.94 (d, J = 10.3 Hz, 1H), 5.52 (dd, J = 10.3, 2.8 Hz, 1H), 4.15 (d, J = 11.5 Hz, 1H), 3.85 (d, J = 8.0 Hz, 1H), 3.75 (d, J = 11.5 Hz, 1H), 3.41 (t, J = 11.6 Hz, 1H), 3.37–3.28 (m, 1H), 2.75–2.56 (m, 1H), 1.18 (s, 9H), 1.16 (s, 3H), 0.95 (s, 3H), 0.92 (s, 3H), 0.88 (s, 3H), 0.83 (s, 3H), 0.73 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 213.04, 178.70, 133.13, 128.79, 84.14, 75.50, 72.23, 65.91, 56.26, 54.97, 52.70, 49.55, 47.87, 44.39, 42.47, 41.69, 39.09, 38.85, 38.35, 36.17, 35.47, 33.33, 31.57, 31.08, 27.37, 26.00, 24.21, 23.21, 20.11, 19.46, 18.16, 17.44, 11.50. ESI-HRMS (m/z) calcd for C35H54NaO5 [M + Na]+ 577.3863, found 577.3868.
3.2.14. Synthesis of Compound 23
To a stirred mixture of compound 22 (50 mg, 0.09 mmol), compound 12 (102 mg, 0.11 mmol), and 4Å molecular sieves (200 mg) in CH2Cl2 (2 mL), PPh3AuNTf2 (13 mg, 0.02 mmol) was added. The mixture was stirred at room temperature for 1 h before it was quenched with NEt3. The mixture was filtered through a pad of celite and washed with EtOAc. The filtrate was concentrated under vacuum. The residue was purified by flash column chromatography (petroleum ether/EtOAc = 3:1) to give compound 23 (107 mg, 95%) as a white foam. Rf = 0.3 (silica, PE/EtOAc = 4:1); [α = 53.3 (c = 1.0, CHCl3); 1H NMR (500 MHz, CDCl3): δ 8.09 (d, J = 7.3 Hz, 4H), 7.60–7.56 (m, 2H), 7.47 (t, J = 6.7 Hz, 4H), 5.93 (d, J = 10.4 Hz, 1H), 5.60–5.45 (m, 3H), 5.04–4.86 (m, 3H), 4.75 (m, 2H), 4.61 (d, J = 7.8 Hz, 1H), 4.50 (d, J = 7.9 Hz, 1H), 4.19 (d, J = 10.1 Hz, 2H), 4.14–4.09 (m, 2H), 4.04 (m, 1H), 3.85 (d, J = 7.8 Hz, 1H), 3.77 (t, J = 9.7 Hz, 2H), 3.59 (d, J = 9.2 Hz, 1H), 3.51 (d, J = 11.6 Hz, 1H), 3.45–3.37 (m, 2H), 2.66 (m, 1H), 2.22 (m, 1H), 2.13 (m, 1H), 1.98 (s, 3H), 1.94 (s, 3H), 1.83 (s, 3H), 1.44 (s, 3H), 1.25 (d, J = 6.0 Hz, 3H), 1.21 (s, 9H), 1.12 (s, 3H), 0.93 (s, 3H), 0.89 (s, 3H), 0.88 (s, 3H), 0.85 (s, 3H), 0.50 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 213.11, 177.70, 170.94, 170.33, 169.31, 168.99, 166.38, 165.05, 133.54, 133.21, 133.15, 130.27, 129.97, 129.56, 128.74, 128.46, 102.87, 101.00, 84.16, 81.63, 78.02, 75.55, 72.82, 71.74, 71.66, 71.08, 69.97, 68.10, 64.75, 61.43, 56.30, 55.01, 52.71, 49.60, 47.70, 44.42, 42.22, 41.70, 39.15, 38.89, 38.26, 35.86, 35.52, 33.38, 31.63, 31.00, 27.49, 27.43, 25.46, 24.27, 23.24, 20.78, 20.66, 20.59, 20.08, 19.78, 19.47, 17.86, 17.21, 16.89, 12.01. ESI-HRMS (m/z) calcd for C69H90NaO20 [M + Na]+ 1261.5918, found 1261.5925.
3.2.15. Synthesis of Saikosaponin D (2)
To a solution of compound 23 (102 mg, 0.08 mmol) in i-PrOH (2 mL) at −20 °C, NaBH4 (15 mg, 0.4 mmol) was added. The mixture was stirred for 3 h before it was quenched by addition of saturated NH4Cl solution and diluted with brine and extracted with CH2Cl2. The extract was dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The residue was purified by silica gel column chromatography (petroleum ether/EtOAc = 6/1) to give the 16-OH product (90 mg, 89%) as a white foam.
To a solution of the above product (49 mg, 0.04 mmol) in MeOH (3 mL), KOH (110 mg, 2 mmol) was added at room temperature. The mixture was stirred at 55 °C for 24 h before it was quenched with acetic acid. The mixture was concentrated under vacuum. The residue was purified by reversed-phase silica gel column chromatography (ODS RP-18) (CHCl3/MeOH = 5:1 to 7:1) to give saikosaponin D (2) (26 mg, 83%) as a white powder. Rf = 0.6 (silica, CHCl3/MeOH = 3:1); [α = 40.4 (c = 1.0, CH3OH); 1H NMR (500 MHz, pyridine-d5): δ 6.31 (d, J = 4.1 Hz, 1H), 6.06 (d, J = 10.3 Hz, 1H), 5.73 (d, J = 2.8 Hz, 1H), 5.71 (d, J = 2.6 Hz, 1H), 5.36 (d, J = 7.8 Hz, 1H), 5.00 (d, J = 7.8 Hz, 1H), 4.57 (m, 2H), 4.46–4.37 (m, 2H), 4.28 (m, 5H), 4.09–4.02 (m, 3H), 3.73 (d, J = 10.8 Hz, 1H), 3.71–3.67 (m, 1H), 3.62 (d, J = 3.2 Hz, 1H), 3.60 (d, J = 7.0 Hz, 1H), 3.35 (d, J = 7.0 Hz, 1H), 2.74–2.64 (m, 1H), 1.65 (s, 3H), 1.46 (s, 3H), 1.45 (s, 3H), 1.38 (s, 3H), 1.04 (s, 3H), 0.98 (s, 3H), 0.96 (s,3H). 13C NMR (126 MHz, pyridine-d5): δ 150.59, 150.46, 150.25, 150.03, 136.22, 136.13, 135.93, 135.73, 132.41, 132.35, 124.20, 124.10, 123.90, 123.70, 107.09, 106.40, 85.63, 85.28, 82.03, 79.20, 78.83, 78.20, 77.52, 76.22, 72.55, 72.22, 71.93, 71.42, 64.43, 63.08, 53.42, 51.74, 50.07, 47.75, 45.73, 44.14, 43.99, 42.27, 39.05, 38.76, 37.21, 36.68, 35.85, 34.15, 32.29, 31.93, 31.69, 26.53, 24.80, 19.95, 19.24, 18.50, 17.96, 17.65, 13. ESI-HRMS (m/z) calcd for C42H69NaO13 [M + H]+ 781.4733, found 781.4736.
3.2.16. Synthesis of Saikosaponin Y (5)
To a stirred solution of the compound 23 (85 mg, 0.07 mmol) in MeOH (3 mL), KOH (307 mg, 5.5 mmol) was added at room temperature. The mixture was stirred at 55 °C for 24 h before it was quenched with acetic acid. The mixture was concentrated under vacuum. The residue was purified by reversed-phase silica gel column chromatography (ODS RP-18) (CHCl3/MeOH = 5:1 to 7:1) to give saikosaponin Y (5) (50 mg, 94%) as a white powder. Rf = 0.3 (silica, CHCl3/MeOH = 4:1); [α = 26.6 (c = 1.0, CH3OH); 1H NMR (500 MHz, CD3OD): δ 6.02 (d, J = 10.3 Hz, 1H), 5.52 (dd, J = 10.2, 3.1 Hz, 1H), 4.53 (d, J = 7.7 Hz, 1H), 4.37 (d, J = 7.4 Hz, 1H), 3.89(d, J = 7.4 Hz, 1H), 3.97–3.71 (m, 2H), 3.76–3.53 (m, 7H), 3.43 (d, J = 7.9 Hz, 1H), 3.40–3.15 (m, 7H), 2.77 (m, 1H), 2.26 (m, 1H), 2.09 (m, 1H), 1.25 (d, J = 6.2 Hz, 3H), 1.19 (s, 3H), 1.00 (s, 3H), 0.96 (s, 3H), 0.91 (s, 3H), 0.89 (s, 3H), 0.70 (s, 3H). 13C NMR (125 MHz, CD3OD): δ 214.74, 134.56, 129.61, 105.65, 85.59, 85.12, 82.97, 77.88, 77.66, 76.29, 75.33, 72.33, 71.82, 71.32, 71.18, 62.35, 57.35, 56.24, 53.73, 50.84, 47.97, 45.39, 44.05, 42.98, 39.98, 39.18, 37.07, 36.64, 33.84, 32.45, 32.03, 26.14, 25.08, 23.52, 20.75, 20.16, 18.88, 18.12, 16.98, 12.80. ESI-HRMS (m/z) calcd for C42H67O13 [M + H]+ 779.4576, found 779.4569.
3.2.17. Synthesis of Compound 24
To a stirred solution of compound 9 (290 mg, 1.1 mmol) in pyridine (5 mL), benzoyl chloride (0.8 mL, 4.9 mmol) and DMAP (39 mg, 0.3 mmol) were added. The mixture was stirred at room temperature for 8 h before it was quenched with NaHCO3 and extracted with EtOAc. The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated under vacuum. The residue was purified by flash column chromatography on silica gel (petroleum ether/EtOAc = 9/1) to give corresponding benzoylated product (625 mg, 99%) as a white solid.
To a stirred solution of the above product (582 mg, 1.0 mmol) in CH3CN/H2O (8 mL/2 mL), ceric ammonium nitrate (1.2 g, 2.1 mmol) was added. The mixture was stirred at room temperature for 2 h before it was quenched with Na2S2O3 and extracted with EtOAc. The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated under vacuum. The residue was purified by flash column chromatography (petroleum ether/EtOAc = 2:1) to give the lactol (375 mg, 81%) as an orange foam. The α/β anomers were difficult to separate.
The lactol (298 mg, 0.62 mmol), ortho-(cyclopropylethynyl) benzoic acid (138 mg, 0.82 mmol), EDCI (156 mg, 0.82 mmol), and DMAP (100 mg, 0.82 mmol) were dissolved in CH2Cl2 (5 mL). The mixture was stirred at room temperature for 4 h and concentrated under vacuum. The residue was purified by flash chromatography (petroleum ether/EtOAc = 3:1) to give compound 24 (401 mg, 99%; α/β = 1:2.1) as a white foam.
24α: Rf = 0.3 (silica, PE/EtOAc = 4:1); [α = 205.6 (c = 1.0, CHCl3); 1H NMR (500 MHz,CDCl3): δ 8.14 (d, J = 7.3 Hz, 2H), 8.01 (d, J = 7.6 Hz, 1H), 7.89 (d, J = 7.4 Hz, 2H), 7.82 (d, J = 7.4 Hz, 2H), 7.64 (t, J = 7.4 Hz, 1H), 7.50 (m, 6H), 7.37 (t, J = 7.2 Hz, 1H), 7.28 (m, 4H), 6.94 (d, J = 3.6 Hz, 1H), 6.12 (dd, J = 10.7, 3.3 Hz, 1H), 6.02 (dd, J = 10.7, 3.6 Hz, 1H), 5.93 (d, J = 2.7 Hz, 1H), 4.82 (q, J = 6.4 Hz, 1H), 1.60 (m, 1H), 1.35 (d, J = 6.5 Hz, 3H), 0.95–0.82 (m, 4H). 13C NMR (125 MHz, CDCl3): δ 166.05, 165.76, 164.52, 135.08, 133.67, 133.44, 133.34, 132.41, 130.98, 130.54, 130.03, 129.88, 129.77, 129.26, 129.11, 128.95, 128.76, 128.46, 128.40, 127.41, 125.06, 99.97, 91.16, 74.95, 71.56, 69.31, 68.30, 67.70, 16.38, 9.13, 9.10.
24β: Rf = 0.25 (silica, PE/EtOAc = 4:1); [α = 216.5 (c = 1.0, CHCl3); 1H NMR (500 MHz,CDCl3): δ 8.16–8.12 (m, 2H), 8.01–7.98 (m, 1H), 7.92–7.88 (m, 2H), 7.81 (dd, J = 8.2, 7.1 Hz, 2H), 7.63 (t, J = 7.4 Hz, 1H), 7.50 (t, J = 7.7 Hz, 2H), 7.47–7.38 (m, 4H), 7.32 (t, J = 7.8 Hz, 2H), 7.29–7.24 (m, 3H), 6.26 (d, J = 8.4 Hz, 1H), 6.03 (dd, J = 10.3, 8.4 Hz, 1H), 5.81 (d, J = 3.0 Hz, 1H), 5.70 (dd, J = 10.4, 3.5 Hz, 1H), 4.33 (q, J = 6.2 Hz, 1H), 1.54 (m, 1H), 1.39 (d, J = 6.4 Hz, 3H), 0.90 (m, 4H). 13C NMR (125 MHz, CDCl3): δ 166.07, 165.69, 165.51, 163.75, 134.49, 133.65, 133.48, 133.42, 132.53, 131.08, 130.15, 129.89, 129.85, 129.50, 129.29, 129.04, 128.92, 128.72, 128.51, 128.43, 127.20, 125.84, 100.53, 92.90, 74.44, 72.22, 71.14, 71.01, 68.89, 16.33, 9.15, 9.11. ESI-HRMS (m/z) calcd for C39H32NaO9 [M + Na]+ 667.1339, found 667.1934.
3.2.18. Synthesis of Compound 25
To a stirred mixture of compound 24 (108 mg, 0.17 mmol), compound 17 (90mg, 0.14 mmol), and 4Å molecular sieves (300 mg) in CH2Cl2 (3 mL), PPh3AuNTf2 (21 mg, 0.03 mmol) was added. The mixture was stirred at room temperature for 1 h before it was quenched with NEt3. The mixture was filtered through a pad of celite and washed with EtOAc. The filtrate was concentrated under vacuum. The residue was purified by flash column chromatography (petroleum ether/EtOAc = 3:1) to give compound 25 (140 mg, 91%) as a white foam. Rf = 0.3 (silica, PE/EtOAc = 4:1); [α = 216.5 (c = 1.0, CHCl3); 1H NMR (500 MHz, CDCl3): δ 8.10 (d, J = 7.5 Hz, 2H), 7.97 (d, J = 7.5 Hz, 2H), 7.76 (d, J = 7.6 Hz, 2H), 7.60 (t, J = 7.4 Hz, 1H), 7.49 (m, 3H), 7.43–7.34 (m, 3H), 7.22 (t, J = 7.7 Hz, 2H), 5.89 (d, J = 10.4 Hz, 1H), 5.76 (dd, J = 10.2, 8.2 Hz, 1H), 5.70 (d, J = 3.0 Hz, 1H), 5.48 (dd, J = 10.5, 3.4 Hz, 1H), 5.42 (dd, J = 10.2, 2.2 Hz, 1H), 5.36 (dd, J = 9.2, 6.1 Hz, 1H), 4.69 (d, J = 7.9 Hz, 1H), 4.04–3.92 (m, 2H), 3.72 (d, J = 10.3 Hz, 1H), 3.65 (d, J = 11.6 Hz, 1H), 3.54 (dd, J = 11.6, 4.6 Hz, 1H), 3.20 (d, J = 7.2 Hz, 1H), 1.33 (d, J = 6.2 Hz, 3H), 1.24 (s, 9H), 1.19 (s, 9H), 1.05 (s, 3H), 1.00 (s, 3H), 0.96 (s, 3H), 0.90 (s, 3H), 0.88 (s, 3H), 0.57 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 178.27, 177.40, 166.23, 165.83, 165.44, 133.51, 133.39, 133.29, 132.67, 130.17, 129.95, 129.85, 129.71, 129.37, 129.18, 128.97, 128.62, 128.54, 128.36, 103.35, 83.95, 82.76, 73.26, 72.31, 71.17, 69.92, 69.65, 68.27, 64.49, 52.81, 51.70, 47.54, 45.42, 45.26, 42.25, 41.77, 39.12, 39.06, 38.28, 37.42, 35.91, 34.17, 33.48, 31.64, 31.53, 31.04, 29.83, 27.54, 27.32, 25.47, 25.34, 23.78, 20.50, 19.58, 18.08, 17.19, 16.59, 12.07. ESI-HRMS (m/z) calcd for C67H86NaO13 [M + Na]+ 1121.5961 found, 1121.5967.
3.2.19. Synthesis of Compound Prosaikogenin F (3)
To a stirred solution of compound 25 (105 mg, 0.1 mmol) in MeOH (3 mL), KOH (272 mg, 5.0 mmol) was added. The mixture was stirred at 55 °C for 24 h before it was quenched with acetic acid. The mixture was concentrated under vacuum. The residue was purified by reversed-phase silica gel column chromatography (ODS RP-18) (CHCl3/MeOH = 5:1 to 7:1) to give prosaikogenin F (3) (51 mg, 87%) as a white powder. Rf = 0.6 (silica, CHCl3/MeOH = 4:1); [α = 78.6 (c = 1.0, CH3OH); 1H NMR (500 MHz, pyridine-d5): δ 6.03 (d, J = 10.4 Hz, 1H), 5.69 (dd, J = 10.3, 2.9 Hz, 1H), 4.98 (d, J = 7.7 Hz, 1H), 4.56–4.50 (m, 1H), 4.44–4.35 (m, 3H), 4.31 (dd, J = 11.9, 4.5 Hz, 1H), 4.02 (d, J = 6.3 Hz, 2H), 3.82–3.75 (m, 1H), 3.71 (d, J = 10.7 Hz, 1H), 3.36 (d, J = 6.9 Hz, 1H), 2.51 (d, J = 13.3 Hz, 1H), 2.38 (m, 1H), 1.55 (d, J = 6.4 Hz, 3H), 1.42 (s, 3H), 1.12 (s, 3H), 1.02 (s, 3H), 0.95 (s, 3H), 0.94 (s, 3H), 0.92 (s, 3H). 13C NMR (125 MHz, pyridine-d5): δ 132.68, 131.67, 106.83, 84.48, 82.12, 75.99, 73.52, 73.46, 73.32, 71.77, 64.75, 64.54, 53.59, 52.65, 47.88, 47.50, 46.14, 44.19, 42.71, 39.15, 38.23, 36.81, 36.63, 35.19, 34.15, 32.11, 26.51, 26.26, 24.33, 21.36, 20.57, 19.29, 18.08, 18.00, 13.53. ESI-HRMS (m/z) calcd for C67H86NaO13 [M + Na]+ 641.4024, found 641.4021.
3.2.20. Synthesis of Compound 26
To a stirred mixture of compound 24 (97 mg, 0.15 mmol), compound 22 (70 mg, 0.13 mmol), and 4Å molecular sieves (300 mg) in CH2Cl2 (3 mL), PPh3AuNTf2 (19 mg, 0.03 mmol) was added. The mixture was stirred at room temperature for 1 h before it was quenched with NEt3.The mixture was filtered through a pad of celite and washed with EtOAc. The filtrate was concentrated under vacuum. The residue was purified by flash column chromatography (petroleum ether/EtOAc = 5:1) to give compound 26 (106 mg, 84%) as a white foam. Rf = 0.3 (silica, PE/EtOAc = 4:1); [α = 125.0 (c = 1.0, CHCl3); 1H NMR (500 MHz, CDCl3): δ 8.09 (d, J = 7.3 Hz, 2H), 7.97 (d, J = 7.3 Hz, 2H), 7.76 (d, J = 7.4 Hz, 2H), 7.59 (t, J = 7.4 Hz, 1H), 7.53–7.43 (m,3H), 7.38 (m, 3H), 7.21 (t, J = 7.7 Hz, 2H), 5.97 (d, J = 10.4 Hz, 1H), 5.77 (dd, J = 10.4, 8.0 Hz, 1H), 5.69 (d, J = 3.3 Hz, 1H), 5.54 (dd, J = 10.3, 2.7 Hz, 1H), 5.49 (dd, J = 10.5, 3.4 Hz, 1H), 4.71 (d, J = 7.9 Hz, 1H), 4.00 (dd, J = 12.7, 6.2 Hz, 1H), 3.86 (d, J = 7.9 Hz, 1H), 3.73 (d, J = 11.5 Hz, 1H), 3.66 (d, J = 11.6 Hz, 1H), 3.55 (dd, J = 11.5, 4.6 Hz, 1H), 3.44 (d, J = 8.0 Hz, 1H), 2.68 (m, 1H), 1.33 (d, J = 6.3 Hz, 3H), 1.22 (s, 9H), 1.15 (s, 3H), 0.95 (s, 3H), 0.93 (s, 3H), 0.91 (s, 3H), 0.86 (s, 3H), 0.59 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 212.98, 177.29, 166.18, 165.78, 165.40, 133.47, 133.34, 133.25, 133.15, 130.12, 129.90, 129.80, 129.38, 129.20, 128.97, 128.79, 128.59, 128.50, 128.32, 103.28, 84.14, 82.61, 75.52, 72.28, 71.17, 69.96, 69.64, 64.51, 56.27, 54.99, 52.70, 49.57, 47.63, 44.38, 42.25, 41.70, 39.07, 38.87, 38.26, 35.91, 35.49, 33.36, 31.61, 31.00, 27.47, 25.43, 24.25, 23.23, 20.06, 19.49, 17.96, 17.17, 16.56, 12.04. ESI-HRMS (m/z) calcd for C62H76NaO12 [M + Na]+ 1035.5229, found 1035.5219.
3.2.21. Synthesis of Prosaikogenin G (4)
To a stirred solution of compound 26 (90 mg, 0.09 mmol) in i-PrOH (3 mL) at −20 °C, NaBH4 (7 mg, 0.18 mmol) was added. The mixture was stirred for 3 h, the solution was quenched by addition of saturated NH4Cl solution and diluted with brine and extracted with CH2Cl2. The extract was dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The residue was purified by silica gel column chromatography (petroleum ether/EtOAc = 8:1) to give the 16-OH product (86 mg, 95%) as a white foam.
To a stirred solution of the above product (70 mg, 0.07 mmol) in MeOH (3 mL), KOH (193 mg, 3.5 mmol) was added at room temperature. The mixture was stirred at 55 °C for 24 h before it was quenched with acetic acid. The mixture concentrated under vacuum. The residue was purified by reversed-phase silica gel column chromatography (ODS RP-18) (CHCl3/MeOH = 5:1) to give prosaikogenin G (4) (35 mg, 83%) as a white powder. Rf = 0.6 (silica, CHCl3/MeOH = 4:1); [α = 54.1 (c = 1.0, CH3OH); 1H NMR (500 MHz, pyridine-d5): δ 6.30 (d, J = 4.0 Hz, 1H), 6.07 (d, J = 10.4 Hz, 1H), 5.72 (dd, J = 10.2, 2.6 Hz, 1H), 4.99 (d, J = 7.7 Hz, 1H), 4.38 (t, J = 8.4 Hz, 2H), 4.32 (dd, J = 11.8, 4.5 Hz, 1H), 3.79 (q, J = 6.3 Hz, 1H), 3.72 (d, J = 9.5 Hz, 1H), 3.60(d, J = 7.0 Hz, 1H), 3.35 (d, J = 7.0 Hz, 1H), 2.72–2.65 (m, 1H), 2.53 (m, 1H), 2.40 (m, 1H), 1.64 (s, 3H), 1.56 (d, J = 6.4 Hz, 3H), 1.38 (s, 3H), 1.06 (s, 3H), 1.04 (s, 3H), 0.98 (s, 3H), 0.96 (s, 3H). 13C NMR (125 MHz, pyridine-d5): δ 132.52, 132.45, 106.80, 85.38, 82.20, 78.30, 77.61, 75.99, 73.47, 73.32, 71.77, 64.84, 53.52, 51.84, 47.94, 45.83, 44.20, 44.09, 42.38, 39.14, 38.87, 37.31, 36.82, 35.94, 34.25, 32.39, 32.04, 31.79, 26.52, 24.90, 20.06, 19.37, 18.60, 18.08, 17.97, 13.55. ESI-HRMS (m/z) calcd for C36H58NaO8 [M + Na]+ 641.4024, found 641.4050.
3.2.22. Synthesis of Prosaikogenin (6)
To a stirred solution of the compound 26 (100 mg, 0.1 mmol) in MeOH (3 mL), KOH (360 mg, 5 mmol) was added at room temperature. The mixture was stirred at 55 °C for 24 h before it was quenched with acetic acid. The mixture was concentrated under vacuum. The residue was purified by reversed-phase silica gel column chromatography (ODS RP-18) (MeOH/H2O = 5:1) to give compound prosaikogenin (6) (57 mg, 93%) as a white powder. Rf = 0.6 (silica, MeOH/H2O = 4:1); [α = 13.3 (c = 1.0, MeOH); 1H NMR (500 MHz, pyridine-d5): δ 6.06 (t, J = 17.8 Hz, 1H), 5.72 (dd, J = 10.3, 2.8 Hz, 1H), 4.99 (d, J = 7.7 Hz, 1H), 4.43–4.35 (m, 1H), 4.32 (dd, J = 11.9, 4.6 Hz, 1H), 4.08–3.99 (m, 1H), 3.94 (d, J = 7.8 Hz, 1H), 3.78 (q, J = 6.4 Hz, 1H), 3.71 (d, J = 10.8 Hz, 1H), 3.53 (d, J = 7.9 Hz, 1H), 2.85 (d, J = 14.4 Hz, 1H), 1.55 (d, J = 6.4 Hz, 1H), 1.39 (s, 1H), 1.03 (s, 1H), 1.00 (s, 1H), 0.93 (s, 1H), 0.87 (s, 1H), 0.82 (s, 1H). 13C NMR (125 MHz, pyridine-d5): δ 212.79, 133.70, 129.96, 106.87, 84.74, 81.88, 75.90, 73.38, 73.25, 71.75, 64.55, 56.85, 55.57, 53.27, 50.27, 47.74, 45.25, 44.16, 42.57, 39.44, 39.00, 36.72, 36.36, 33.80, 32.13, 31.82, 26.49, 24.98, 23.60, 20.71, 20.29, 19.13, 18.01, 17.90, 13.52. ESI-HRMS (m/z) calcd for C42H67O13 [M]+ 639.3867, found 639.3869.
3.2.23. Synthesis of Compound 27
To a stirred solution of the compound 10 (225 mg, 0.4 mmol) in MeOH (3 mL), CH3ONa (12 mg, 0.2 mmol) was added. The mixture was stirred at room temperature for 8 h before it was quenched with acid resin until pH = 7. The mixture was filtered through a pad of celite and washed with MeOH. The filtrate was concentrated under vacuum.
To a stirred mixture of the above product in pyridine (5 mL), triphenylmethyl chloride (1.04 g, 3.8 mmol) and DMAP (45 mg, 0.4 mmol) were added. The mixture was stirred for 8 h at 75 °C, then benzoyl chloride (0.43 mL, 3.8 mmol) was added in the flask at 0 °C. The mixture was stirred for 4 h at room temperature, the solution was quenched by addition of saturated NaHCO3 solution, diluted with brine and extracted with CH2Cl2. The combined organic phases were dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The residue was purified by silica gel column chromatography (petroleum ether/EtOAc = 5:1) to give compound 27 (431 mg, 97%) as a white foam. Rf = 0.3 (silica, PE/EtOAc = 4:1); [α = 50.0 (c = 1.0, CHCl3); 1H NMR (500 MHz, CDCl3): δ 8.19 (d, J = 8.0 Hz, 2H), 8.14 (d, J = 7.4 Hz, 1H), 7.99 (d, J = 8.1 Hz, 2H), 7.73 (t, J = 6.2 Hz, 3H), 7.63 (m, 2H), 7.57–7.46 (m, 11H), 7.43–7.28 (m, 6H), 7.25–7.19 (m, 4H), 7.14 (m, 3H), 7.01–6.92 (m, 4H), 6.81 (d, J = 8.4 Hz, 2H), 5.99 (s, 1H), 5.80 (d, J = 3.4 Hz, 1H), 5.72 (m, 1H), 5.54 (m, 2H), 5.43 (m, 1H), 5.20 (d, J = 7.6 Hz, 1H), 4.96–4.88 (m, 1H), 4.48 (d, J = 6.0 Hz, 1H), 3.96–3.90 (m, 1H), 3.76 (s, 3H), 3.62–3.54 (m, 1H), 3.30 (d, J = 10.5 Hz, 1H), 1.23 (d, J = 6.0 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ 166.30, 165.85, 165.60, 164.97, 164.63, 155.40, 151.01, 143.74, 133.71, 133.53, 133.21, 133.17, 133.09, 132.62, 130.26, 130.23, 130.06, 129.80, 129.77, 129.74, 129.38, 129.28, 129.10, 128.99, 128.89, 128.70, 128.66, 128.56, 128.47, 128.30, 128.27, 128.00, 127.97, 127.13, 118.54, 114.72, 102.05, 96.66, 87.00, 74.24, 74.13, 73.26, 73.23, 72.06, 71.17, 70.04, 66.39, 62.90, 55.72, 16.50. ESI-HRMS (m/z) calcd for C73H62NaO16 [M + Na]+ 1217.3930, found 1217.3938.
3.2.24. Synthesis of Compound 29
To a stirred solution of compound 27 (420 mg, 0.4 mmol), 28 (438 mg, 0.7 mmol), and 4Å molecular sieves (400 mg) in CH2Cl2 (4 mL), TfOH (62 μL, 0.7 mmol) was added at −20 °C. The mixture was stirred at the same temperature for 1.5 h before it was quenched with NEt3 (5.0 mL). The mixture was filtered through a pad of celite and washed with CH2Cl2 (3 × 2 mL). The filtrate was concentrated under vacuum. The residue was purified by flash column chromatography (petroleum ether/EtOAc = 3:1) to give compound 29 (400 mg, 74%) as an orange foam. Rf = 0.3 (silica, PE/EtOAc = 4:1); [α = 21.2 (c = 0.25, CHCl3); 1H NMR (500 MHz, CDCl3): δ 8.17 (d, J = 8.0 Hz, 2H), 8.14–8.07 (m, 4H), 7.89 (d, J = 7.9 Hz, 2H), 7.83 (m, 4H), 7.78 (d, J = 7.9 Hz, 2H), 7.66–7.61 (m, 3H), 7.61–7.28 (m, 23H), 7.20 (t, J = 7.5 Hz, 1H), 7.16 (t, J = 7.8 Hz, 2H), 7.04 (d, J = 8.9 Hz, 2H), 6.87 (t, J = 7.7 Hz, 2H), 6.77 (d, J = 9.0 Hz, 2H), 6.00–5.90 (m, 2H), 5.82 (d, J = 3.6 Hz, 1H), 5.74 (t, J = 9.5 Hz, 1H), 5.66 (t, J = 9.8 Hz, 1H), 5.57 (dd, J = 11.6, 6.1 Hz, 1H), 5.48–5.42 (m, 2H), 5.24 (m, 2H), 4.89 (d, J = 6.1 Hz, 1H), 4.83 (d, J = 7.7 Hz, 1H), 4.73 (m, 2H), 4.58–4.47 (m, 2H), 4.09 (m, 1H), 4.00 (m, 1H), 3.94 (d, J = 8.1 Hz, 1H), 3.72 (s, 3H), 1.39 (d, J = 6.2 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ 166.68, 166.30, 165.88, 165.74, 165.52, 165.40, 165.32, 165.20, 164.58, 155.65, 150.88, 133.60, 133.55, 133.51, 133.41, 133.32, 133.24, 133.20, 132.64, 130.35, 129.99, 129.92, 129.86, 129.80, 129.67, 129.40, 129.27, 129.03, 128.86, 128.80, 128.68, 128.65, 128.59, 128.56, 128.52, 128.47, 128.42, 128.26, 127.93, 119.74, 114.71, 102.25, 101.31, 97.14, 75.51, 75.12, 73.91, 73.00, 72.50, 72.34, 71.66, 71.16, 69.41, 69.31, 65.85, 63.14, 55.66, 16.66. ESI-HRMS (m/z) calcd for C88H74NaO25 [M + Na]+ 1553.4411, found 1553.4409.
3.2.25. Synthesis of Compound 30
To a stirred solution of compound 29 (350 mg, 0.2 mmol) in CH3CN/H2O (2 mL/0.5 mL), ceric ammonium nitrate (275 mg, 0.5 mmol) was added. The mixture was stirred at 0 °C for 2 h before it was quenched with NaHCO3 and extracted with EtOAc. The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated under vacuum. The residue was purified by flash column chromatography (petroleum ether/EtOAc = 2:1) to give the lactol (266 mg, 81%) as an orange foam. The α/β anomers were difficult to separate.
The lactol (230 mg, 0.16 mmol), ortho-(cyclopropylethynyl) benzoic acid (42 mg, 0.24 mmol), EDCI (44 mg, 0.2 mmol) and DMAP (6 mg, 0.05 mmol) were dissolved in CH2Cl2 (5 mL). The mixture was stirred at room temperature for 4 h and concentrated under vacuum. The residue was purified by flash chromatography (petroleum/EtOAc = 3:1) to give compound 30 (250 mg, 99%) as a white foam. Rf = 0.3 (silica, PE/EtOAc = 2:1); The α/β anomers were difficult to separate.
3.2.26. Synthesis of Compound 31
To a stirred mixture of compound 30 (220 mg, 0.14 mmol), compound 17 (68 mg, 0.1 mmol), and 4Å molecular sieves (300 mg) in CH2Cl2 (3 mL), PPh3AuNTf2 (16 mg, 0.03 mmol) was added. The mixture was stirred at 0 °C for 1 h before it was quenched with NEt3. The mixture was filtered through a pad of celite and washed with EtOAc. The filtrate was concentrated under vacuum. The residue was purified by flash column chromatography (petroleum ether/EtOAc = 4:1) to give compound 31 (171 mg, 79%) as a white foam. Rf = 0.3 (silica, PE/EtOAc = 4:1); [α = 36.3 (c = 1, CHCl3); 1H NMR (500 MHz, CDCl3): δ 8.14 (d, J = 7.4 Hz, 2H), 8.05 (m, 4H), 7.89 (d, J = 7.6 Hz, 2H), 7.85 (d, J = 7.5 Hz, 2H), 7.81 (d, J = 7.5 Hz, 2H), 7.68–7.51 (m, 9H), 7.44 (m, 10H), 7.38–7.27 (m, 10H), 7.13 (t, J = 7.8 Hz, 2H), 7.01 (t, J = 7.7 Hz, 2H), 5.92 (d, J = 9.6 Hz, 1H), 5.90–5.84 (m, 1H), 5.68 (m, 2H), 5.53 (t, J = 9.7 Hz, 1H), 5.47 (m, 2H), 5.42–5.38 (m, 1H), 5.35 (d, J = 7.9 Hz, 2H), 5.14 (td, J = 9.9, 4.5 Hz, 2H), 4.69–4.61 (m, 2H), 4.54–4.46 (m, 2H), 4.45–4.40 (m, 1H), 4.08 (dd, J = 9.9, 3.7 Hz, 1H), 3.96 (d, J = 6.9 Hz, 4H), 3.56 (d, J = 11.5 Hz, 1H), 3.46 (m, 2H), 3.19 (d, J = 7.2 Hz, 1H), 1.37 (d, J = 6.2 Hz, 3H), 1.25 (s, 3H), 1.18 (s, 9H), 1.10 (s, 9H), 1.03 (s, 3H), 0.97 (s, 3H), 0.94 (s, 3H), 0.87 (s, 3H), 0.85 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 178.28, 177.34, 166.86, 166.28, 165.96, 165.68, 165.32, 165.23, 164.61, 164.46, 133.70, 133.56, 133.23, 132.72, 130.49, 129.96, 129.91, 129.85, 129.75, 129.65, 129.53, 129.27, 128.94, 128.90, 128.73, 128.68, 128.56, 128.52, 128.45, 128.34, 128.22, 127.96, 102.90, 101.81, 101.15, 83.94, 81.91, 78.18, 75.11, 73.41, 73.11, 72.71, 72.39, 72.31, 71.87, 71.59, 69.64, 69.47, 69.38, 68.30, 63.13, 52.78, 51.70, 47.46, 45.42, 45.27, 42.13, 41.75, 39.06, 38.99, 37.43, 35.85, 34.18, 33.47, 31.64, 31.52, 30.96, 29.83, 27.48, 27.32, 25.34, 23.77, 20.48, 19.56, 17.93, 17.22, 17.03, 12.01. ESI-HRMS (m/z) calcd for C121H130NaO29 [M + Na]+ 2069.8595, found 2069.8590.
3.2.27. Synthesis of Clinoposaponin I (7)
To a stirred solution of the compound 31 (51mg, 0.02 mmol) in MeOH (3 mL), KOH (111 mg, 2.0 mmol) was added. The mixture was stirred at 55 °C for 24 h before it was quenched with acetic acid. The mixture concentrated under vacuum. The residue was purified by reversed-phase silica gel column chromatography (ODS RP-18) (MeOH/H2O = 2:1) to give compound 7 (21 mg, 89%) as a white powder. Rf = 0.5 (reverse silica, MeOH/H2O = 2:1); [α = 28.5 (c = 0.25, CH3OH); 1H NMR (500 MHz, pyridine-d5): δ 6.01 (m, 1H), 5.69 (dd, J = 10.3, 2.8 Hz, 1H), 5.28 (d, J = 7.8 Hz, 1H), 5.09 (d, J = 7.7 Hz, 3H), 5.00 (d, J = 7.8 Hz, 1H), 4.86 (d, J = 10.9 Hz, 1H), 4.53 (m, 1H), 4.45–4.37 (m, 3H), 4.28 (dt, J = 11.9, 5.9 Hz, 3H), 4.22 (t, J = 8.0 Hz, 2H), 4.19–3.98 (m, 5H), 3.89 (dd, J = 11.7, 5.1 Hz, 2H), 3.72 (d, J = 10.6 Hz, 1H), 3.37 (d, J = 6.9 Hz, 1H), 2.52 (m, 1H), 2.32 (d, J = 9.8 Hz, 1H), 1.52 (d, J = 6.3 Hz, 3H), 1.41 (s, 3H), 1.12 (s, 3H), 1.00 (s, 3H), 0.94 (s, 3H), 0.93 (s, 3H), 0.91 (s, 3H). 13C NMR (125 MHz, pyridine-d5): δ 132.68, 131.67, 106.73, 106.59, 105.98, 85.42, 84.47, 82.12, 78.88, 77.81, 75.99, 75.79, 73.52, 72.59, 72.18, 71.97, 71.52, 64.52, 63.06, 53.59, 52.62, 50.18, 47.82, 47.49, 46.11, 44.20, 42.68, 39.14, 38.20, 36.75, 34.13, 32.10, 26.26, 24.31, 21.34, 20.56, 19.24, 17.82, 13.53. ESI-HRMS (m/z) calcd for C48H78NaO18 [M + Na]+ 965.5080, found 965.5077.
4. Conclusions
Here, we report the facile synthesis of seven saikosaponins; namely, saikosaponin A (1), D (2), prosaikosaponin F (3), G (4), saikosaponin Y (5), prosaikogenin (6), and clinoposaponin I (7), which have been identified from Radix Bupleuri, a common traditional Chinese medicine and relevant plants. These mono-, di-, and trisaccharide saponins feature triterpene aglycones of high oxidation states, which bear 13,28-epoxy-ether moiety and oxo groups at C16 and C23. The methods previously developed for the selective hydroxylation of the triterpenes of low oxidation states, such as oleanolic acid, have enabled the present synthesis. In addition, the gold(I)-catalyzed glycosylation with o-alkynylbenzoates as donors has been applied successfully in the present synthesis. Given the conserved nature of the structures of saikosaponins, the work reported herein offers the prospect of being able to access many more members of saikosaponins and their natural and synthetic analogs, thus facilitating in-depth studies on biological and pharmacological activities of these components of folk medicines.
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/article/10.3390/molecules26071941/s1. Copies of NMR spectra of compounds 1–7, 10–12, 14–27, 29 and 31.
Author Contributions
Conceptualization, P.X. and B.Y.; methodology, Z.W. and B.W.; formal analysis, Z.W. and T.M.; investigation, Z.W.; resources, B.Y.; data curation, Z.W.; writing—original draft preparation, Z.W. and P.X.; writing—review and editing, B.Y.; visualization, Z.W.; supervision, B.Y.; project administration, P.X. and B.Y.; funding acquisition, B.Y. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Shanghai Municipal Science and Technology Major Project, National Natural Science Foundation of China (22031011 & 21621002), Key Research Program of Frontier Sciences of CAS (ZDBS-LY-SLH030), Strategic Priority Research Program of CAS (XDB20020000), and Youth Innovation Promotion Association of CAS (2020258).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available in the article and Supplementary Material.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds 1–7 are available from the authors.
References
- Yuan, B.; Yang, R.; Ma, Y.; Zhou, S.; Zhang, X.; Liu, Y. A systematic review of the active saikosaponins and extracts isolated from Radix Bupleuri and their applications. Pharm. Biol. 2017, 55, 620–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, F.; Dong, X.; Yin, X.; Wang, W.; You, L.; Ni, J. Radix Bupleuri: A review of traditional uses, botany, phytochemistry, pharmacology, and toxicology. Biomed. Res. Int. 2017, 2017, 7597596. [Google Scholar] [CrossRef] [Green Version]
- National Pharmacopoeia Committee. Pharmacopoeia of People’s Republic of China. Part 1; Chemical Industry Press: Beijing, China, 2010. [Google Scholar]
- Li, X.Q.; Song, Y.N.; Wang, S.J.; Rahman, K.; Zhu, J.Y.; Zhang, H. Saikosaponins: A review of pharmacological effects. J. Asian Nat. Prod. Res. 2018, 20, 399–411. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, K.; Amagaya, S.; Ogihara, Y. New derivatives of saikosaponins. Chem. Pharm. Bull. 1985, 33, 3349–3355. [Google Scholar] [CrossRef] [Green Version]
- Nose, M.; Amagaya, S.; Ogihara, Y. Effects of saikosaponin metabolites on the hemolysis of red blood cells and their adsorbability on the cell membrane. Chem. Pharm. Bull. 1989, 37, 3306–3310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, P.; Qiu, H.; Wang, M.; Tian, Y.; Zhang, Z.; Song, R. In vitro metabolism study of saikosaponin d and its derivatives in rat liver microsomes. Xenobiotica 2017, 47, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Li, D.Q.; Wu, J.; Liu, L.Y.; Wu, Y.Y.; Li, L.Z.; Huang, X.X.; Liu, Q.B.; Yang, J.Y.; Song, S.J.; Wu, C.F. Cytotoxic triterpenoid glycosides (saikosaponins) from the roots of Bupleurum chinense. Bioorg. Med. Chem. Lett. 2015, 25, 3887–3892. [Google Scholar] [CrossRef]
- Fang, W.; Yang, Y.J.; Guo, B.L.; Cen, S. Anti-influenza triterpenoid saponins (saikosaponins) from the roots of Bupleurum marginatum var. stenophyllum. Bioorg. Med. Chem. Lett. 2017, 27, 1654–1659. [Google Scholar] [CrossRef]
- Liu, X.; Latkolik, S.; Atanasov, A.G.; Kunert, O.; Pferschy-Wenzig, E.M.; Heiss, E.H.; Malainer, C.; Schinkovitz, A.; Kollroser, M.; Dirsch, V.M.; et al. Bupleurum chinense roots: A bioactivity-guided approach toward saponin-type NF-ĸB inhibitors. Planta Med. 2017, 83, 1242–1250. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, A.; Miyase, T.; Ueno, A.; Maeda, T. Clinoposaponins I-V, new oleanane-triterpene saponins from Clinopodium gracile O. Kuntze. Chem. Pharm. Bull. 1993, 41, 1270–1274. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Ma, G.; Zhong, M.; Yu, S.; Xu, X.; Hu, Y.; Zhang, Y.; Wei, H.; Yang, J. Triterpene saponins from Tabellae Clinopodii. Fitoterapia 2013, 90, 14–19. [Google Scholar] [CrossRef]
- Cui, B.S.; Qiao, Y.Q.; Yuan, Y.; Tang, L.; Chen, H.; Li, Y.; Li, S. Hepatoprotective saikosaponin homologs from Comastoma pedunculatum. Planta Med. 2014, 80, 1647–1656. [Google Scholar] [CrossRef]
- Huang, W.; Sun, R.; Zhang, Z. “Dose-time-toxicity” relationship study on hepatotoxicity caused by multiple dose of total Bupleurum saponin crude extracts to rats. Zhongguo Zhong Yao Za Zhi (China J. Chin. Mater. Med.) 2010, 35, 3344–3347. [Google Scholar]
- Chen, L.; Zhang, F.; Kong, D.; Zhu, X.; Chen, W.; Wang, A.; Zheng, S. Saikosaponin D disrupts platelet-derived growth factor-beta receptor/p38 pathway leading to mitochondrial apoptosis in human LO2 hepatocyte cells: A potential mechanism of hepatotoxicity. Chem. Biol. Interact. 2013, 206, 76–82. [Google Scholar] [CrossRef]
- Yang, Y.; Laval, S.; Yu, B. Chemical synthesis of saponins. Adv. Carbohydr. Chem. Biochem. 2014, 71, 137–226. [Google Scholar]
- Ge, S.-J.; Tu, Y.-H.; Xia, J.-H.; Sun, J.-S. Synthetic investigation toward the D-ring-functionalized cytotoxic oleanane-type saponins pithedulosides D and E. Eur. J. Org. Chem. 2017, 2017, 3929–3934. [Google Scholar] [CrossRef]
- Zhu, D.; Yu, B. Synthesis of the diverse glycosides in traditional Chinese medicine. Chin. J. Chem. 2018, 36, 681–691. [Google Scholar] [CrossRef]
- Zeng, Z.Y.; Liao, J.X.; Hu, Z.N.; Liu, D.Y.; Zhang, Q.J.; Sun, J.S. Synthetic investigation toward QS-21 analogues. Org. Lett. 2020, 22, 8613–8617. [Google Scholar] [CrossRef]
- Mu, T.; Wei, B.; Zhu, D.; Yu, B. Site-selective C-H hydroxylation of pentacyclic triterpenoids directed by transient chiral pyridine-imino groups. Nat. Commun. 2020, 11, 4371. [Google Scholar] [CrossRef]
- Sun, L.; Zhu, D.; Beverborg, L.O.G.; Wang, R.; Dang, Y.; Ma, M.; Li, W.; Yu, B. Synthesis and antiproliferative activities of OSW-1 analogues bearing 2″-O-p-acylaminobenzoyl residues. Chin. J. Chem. 2020, 38, 1091–1097. [Google Scholar] [CrossRef]
- Hu, Z.; Xu, P.; Wei, B.; Yu, B. Total synthesis of phenylpropanoid glycosides, acteoside, isoacteoside and ligupurpuroside J. Chem. J. Chin. Univ. 2020, 41, 1708–1720. [Google Scholar]
- Gouliaras, C.; Lee, D.; Chan, L.; Taylor, M.S. Regioselective activation of glycosyl acceptors by a diarylborinic acid-derived catalyst. J. Am. Chem. Soc. 2011, 133, 13926–13929. [Google Scholar] [CrossRef]
- Mancini, R.S.; McClary, C.A.; Anthonipillai, S.; Taylor, M.S. Organoboron-promoted regioselective glycosylations in the synthesis of a saponin-derived pentasaccharide from Spergularia ramosa. J. Org. Chem. 2015, 80, 8501–8510. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yang, Y.; Yu, B. An efficient glycosylation protocol with glycosyl ortho-alkynylbenzoates as donors under the catalysis of Ph3PAuOTf. Tetrahedron Lett. 2008, 49, 3604–3608. [Google Scholar] [CrossRef]
- Li, Y.; Yang, X.; Liu, Y.; Zhu, C.; Yang, Y.; Yu, B. Gold(I)-catalyzed glycosylation with glycosyl ortho-alkynylbenzoates as donors: General scope and application in the synthesis of a cyclic triterpene saponin. Chem. Eur. J. 2010, 16, 1871–1882. [Google Scholar] [CrossRef]
- Wulff, G.; Schmidt, W. Über die orthoesterbildung als konkurrenzreaktion zur glykosylierung. Carbohydr. Res. 1977, 53, 33–46. [Google Scholar] [CrossRef]
- Ma, Y.; Lian, G.; Li, Y.; Yu, B. Identification of 3,6-di-O-acetyl-1,2,4-O-orthoacetyl-α-d-glucopyranose as a direct evidence for the 4-O-acyl group participation in glycosylation. Chem. Commun. 2011, 47, 7515–7517. [Google Scholar] [CrossRef] [PubMed]
- Bérces, A.; Whitfield, D.M.; Nukada, T.; do Santos, Z.I.; Obuchowska, A.; Krepinsky, J.J. Is acyl migration to the aglycon avoidable in 2-acyl assisted glycosylation reactions? Can. J. Chem. 2004, 82, 1157–1171. [Google Scholar] [CrossRef]
- Baldwin, J.E.; Jones, R.H.; Najera, C.; Yus, M. Functionalisation of unactivated methyl groups through cyclopalladation reactions. Tetrahedron 1985, 41, 699–711. [Google Scholar] [CrossRef]
- Carr, K.; Saxton, H.M.; Sutherland, J.K. The 4α-demethylation of lanostenone. J. Chem. Soc. Perkin Trans. 1 1988, 1599–1601. [Google Scholar] [CrossRef]
- Bore, L.; Honda, T.; Gribble, G.W. Synthesis of β-boswellic acid analogues with a carboxyl group at C-17 isolated from the bark of Schefflera octophylla. J. Org. Chem. 2000, 65, 6278–6282. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Granados, A.; Lopez, P.E.; Melguizo, E.; Parra, A.; Simeo, Y. Remote hydroxylation of methyl groups by regioselective cyclopalladation. Partial synthesis of hyptatic acid-A. J. Org. Chem. 2007, 72, 3500–3509. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Tang, P.; Yu, B. Total synthesis of lobatoside E, a potent antitumor cyclic triterpene saponin. J. Am. Chem. Soc. 2008, 130, 5872–5873. [Google Scholar] [CrossRef]
- Pyo, S.H.; Cho, J.S.; Choi, H.J.; Han, B.H. Evaluation of paclitaxel rearrangement involving opening of the oxetane ring and migration of acetyl and benzoyl groups. J. Pharm. Biomed. Anal. 2007, 43, 1141–1145. [Google Scholar] [CrossRef]
- Lassfolk, R.; Rahkila, J.; Johansson, M.P.; Ekholm, F.S.; Warna, J.; Leino, R. Acetyl group migration across the saccharide units in oligomannoside model compound. J. Am. Chem. Soc. 2019, 141, 1646–1654. [Google Scholar] [CrossRef] [Green Version]
- Roslund, M.U.; Aitio, O.; Warna, J.; Maaheimo, H.; Murzin, D.Y.; Leino, R. Acyl group migration and cleavage in selectively protected β-d-galactopyranosides as studied by NMR spectroscopy and kinetic calculations. J. Am. Chem. Soc. 2008, 130, 8769–8772. [Google Scholar] [CrossRef]
- Tang, Y.; Li, J.; Zhu, Y.; Li, Y.; Yu, B. Mechanistic insights into the gold(I)-catalyzed activation of glycosyl ortho-alkynylbenzoates for glycosidation. J. Am. Chem. Soc. 2013, 135, 18396–18405. [Google Scholar] [CrossRef]
- Nose, M.; Amagaya, S.; Takeda, T.; Ogihara, Y. New derivatives of saikosaponin c. Chem. Pharm. Bull. 1989, 37, 1293–1296. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Sun, J.; Niu, Y.; Li, R.; Liao, J.; Zhang, F.; Yu, B. Synthetic access toward the diverse ginsenosides. Chem. Sci. 2013, 4, 3899–3905. [Google Scholar] [CrossRef]
- Aliotta, G.; de Napoli, L.; Giordano, F.; Piccialli, G.; Piccialli, V.; Santacroce, C. An oleanane triterpene from Anagallis arvensis. Phytochemistry 1992, 31, 929–933. [Google Scholar] [CrossRef]
- Nagamitsu, T.; Sunazuka, T.; Obata, R.; Tomoda, H.; Tanaka, H.; Harigaya, Y.; Omura, S.; Smith, A.B. Total synthesis of (+)-pyripyropene A, a potent, orally bioavailable inhibitor of Acyl-CoA: Cholesterol acyltransferase. J. Org. Chem. 1995, 60, 8126–8127. [Google Scholar] [CrossRef]
- Li, T.; Wu, G.; Feng, S.; Hu, X.; Zhang, W.; Tang, S.; Xie, X.; She, X. Concise formal synthesis of (+)-pyripyropene A. Tetrahedron 2019, 75, 3939–3942. [Google Scholar] [CrossRef]
Figure 1.
Saikosaponin A (1), D (2), and congeners (3–7).

Scheme 1.
Retrosynthetic plan for saikosaponins 1–7.

Scheme 2.
Preparation of disaccharide o-alkynylbenzoates 11 and 12.

Scheme 3.
Preparation of aglycone derivative 17.

Scheme 4.
Gold(I)-catalyzed glycosylation and synthesis of saikosaponin A (1).

Scheme 5.
An attempt at glycosylation of diol 20.

Scheme 6.
Synthesis of saikosaponin D (2) and saikosaponin Y (5).

Scheme 7.
Synthesis of prosaikosaponin F (3), G (4), and prosaikogenin (6).

Scheme 8.
Synthesis of clinoposaponin I (7).

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Wang, Z.; Wei, B.; Mu, T.; Xu, P.; Yu, B. Facile Synthesis of Saikosaponins. Molecules 2021, 26, 1941. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26071941
AMA Style
Wang Z, Wei B, Mu T, Xu P, Yu B. Facile Synthesis of Saikosaponins. Molecules. 2021; 26(7):1941. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26071941
Chicago/Turabian StyleWang, Ziqiang, Bingcheng Wei, Tong Mu, Peng Xu, and Biao Yu. 2021. "Facile Synthesis of Saikosaponins" Molecules 26, no. 7: 1941. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26071941