
Synergizing Off-Target Predictions for In Silico Insights of CENH3 Knockout in Cannabis through CRISPR/Cas

 ,
,  ,
,  , and
, and
Abstract
:1. Introduction
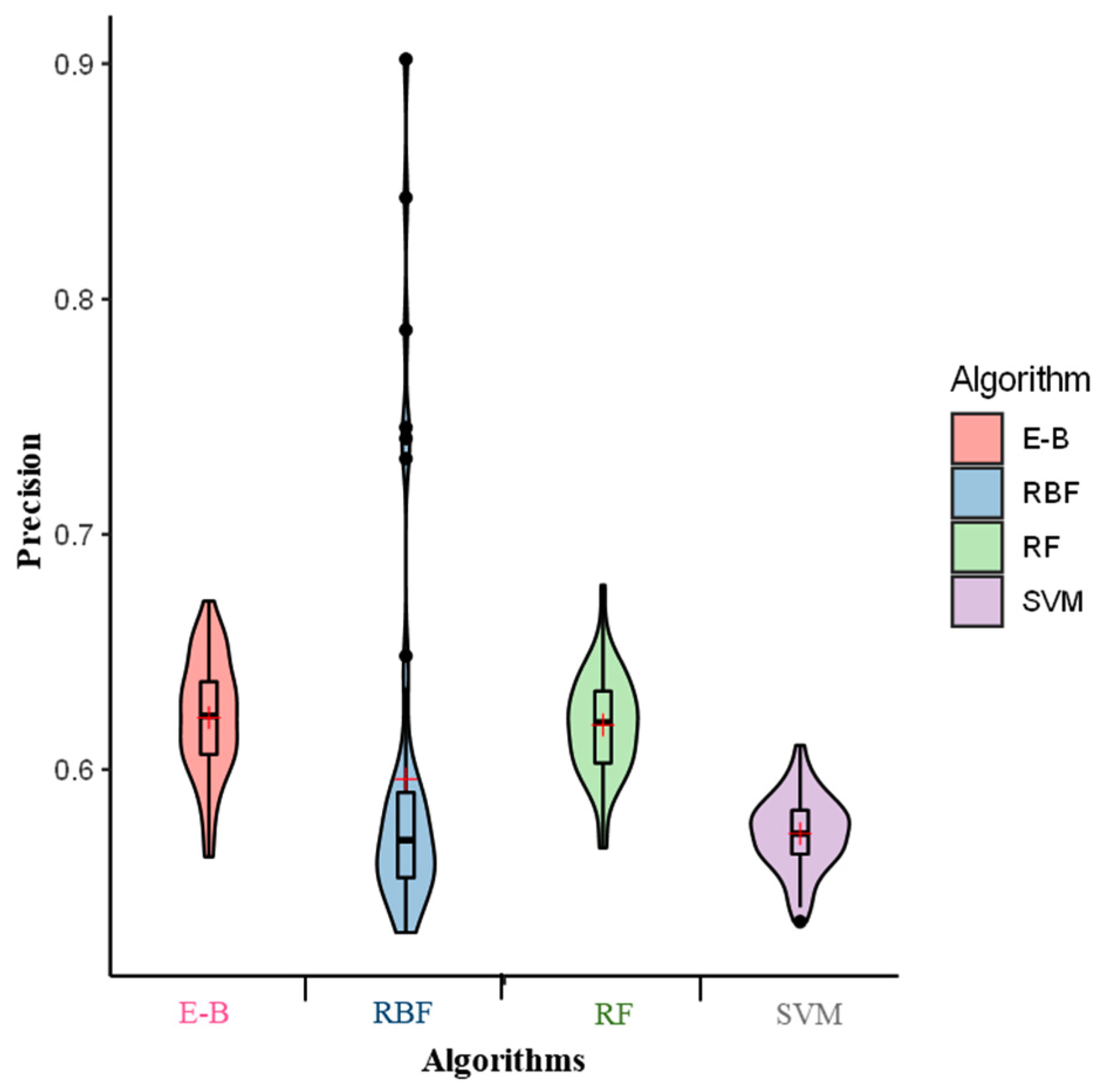
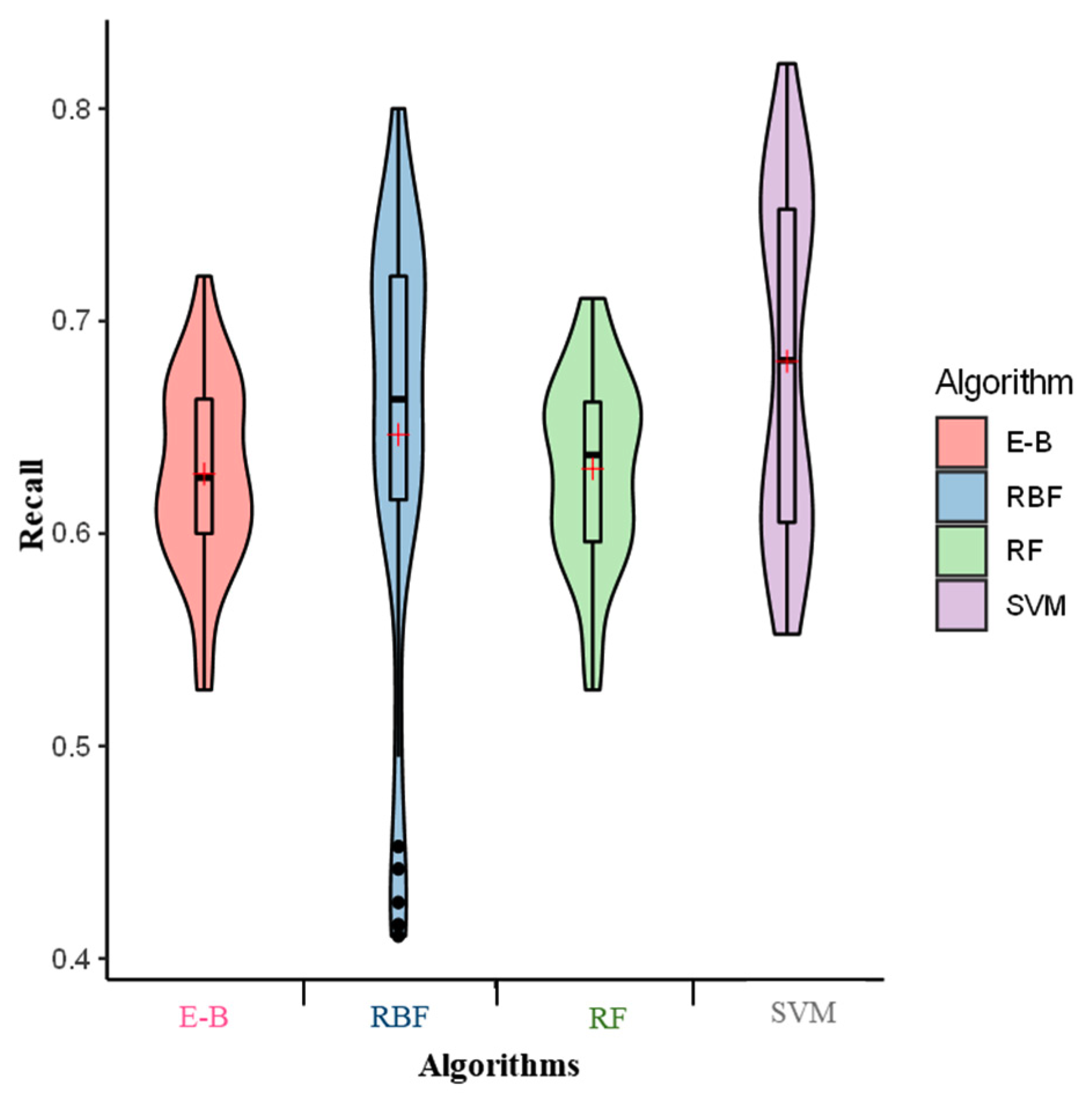
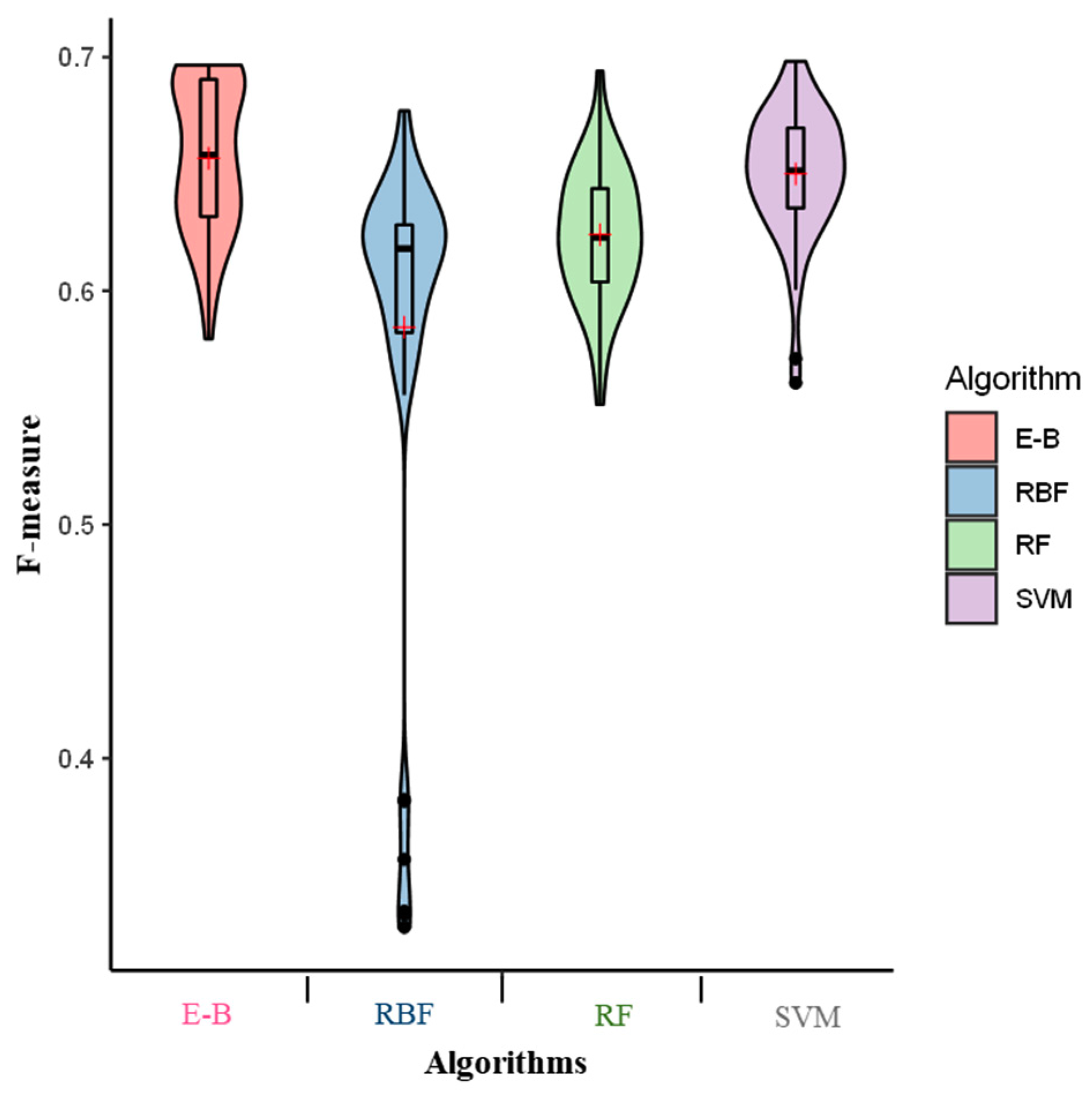
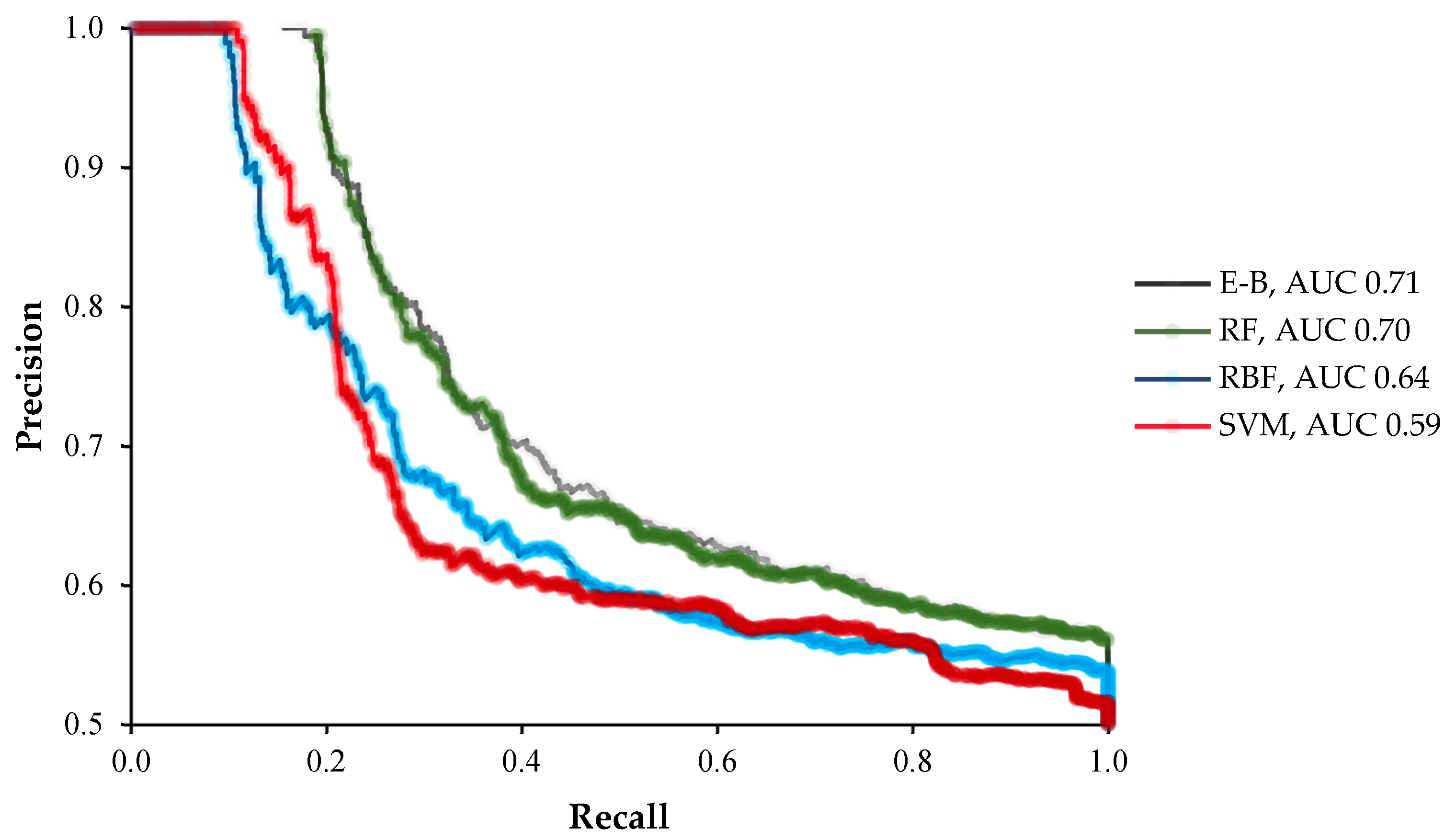
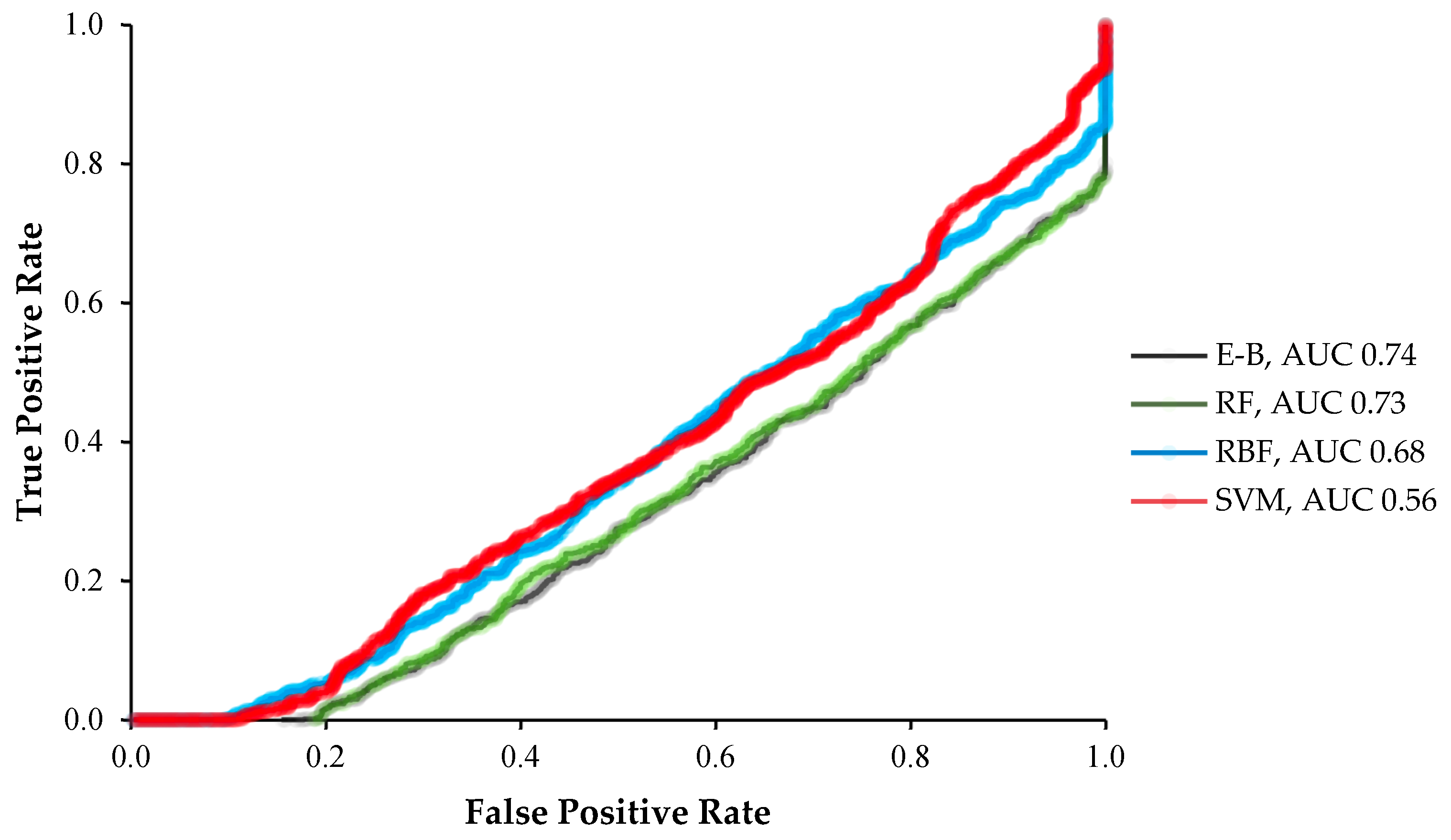
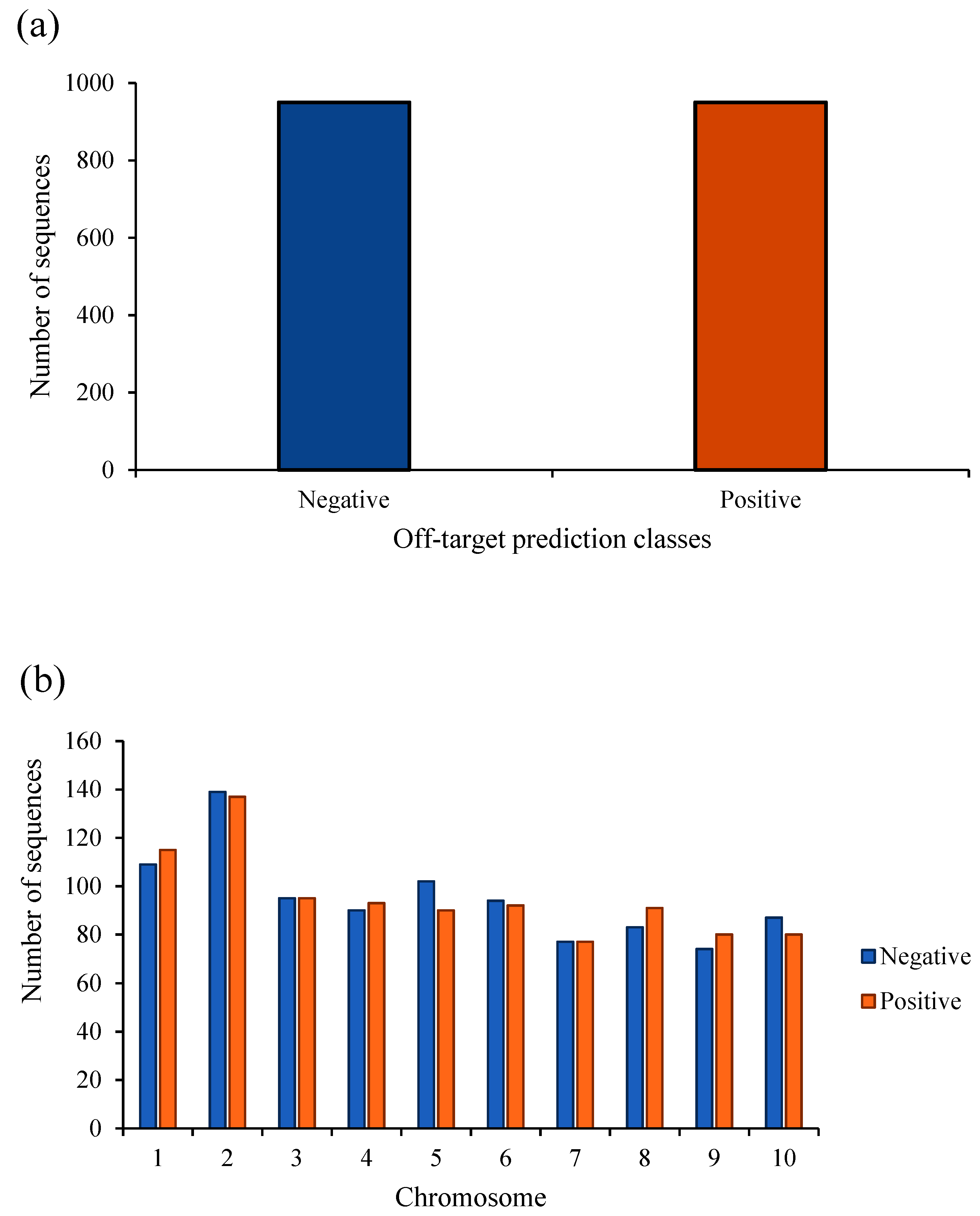
2. Results
3. Discussion
4. Materials and Methods
4.1. Datasets
4.2. Classification Models
4.3. Evaluation Criteria
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Hurgobin, B.; Tamiru-Oli, M.; Welling, M.T.; Doblin, M.S.; Bacic, A.; Whelan, J.; Lewsey, M.G. Recent advances in Cannabis sativa genomics research. New Phytol. 2021, 230, 73–89. [Google Scholar] [CrossRef]
- Hesami, M.; Pepe, M.; Alizadeh, M.; Rakei, A.; Baiton, A.; Phineas Jones, A.M. Recent advances in cannabis biotechnology. Ind. Crop. Prod. 2020, 158, 113026. [Google Scholar] [CrossRef]
- Monthony, A.S.; Page, S.R.G.; Hesami, M.; Jones, A.M.P. The Past, Present and Future of Cannabis sativa Tissue Culture. Plants 2021, 10, 185. [Google Scholar] [CrossRef]
- Barcaccia, G.; Palumbo, F.; Scariolo, F.; Vannozzi, A.; Borin, M.; Bona, S. Potentials and Challenges of Genomics for Breeding Cannabis Cultivars. Front. Plant Sci. 2020, 11, 1472. [Google Scholar] [CrossRef]
- Salentijn, E.M.J.; Petit, J.; Trindade, L.M. The Complex Interactions Between Flowering Behavior and Fiber Quality in Hemp. Front. Plant Sci. 2019, 10, 614. [Google Scholar] [CrossRef]
- Salami, S.A.; Martinelli, F.; Giovino, A.; Bachari, A.; Arad, N.; Mantri, N. It Is Our Turn to Get Cannabis High: Put Cannabinoids in Food and Health Baskets. Molecules 2020, 25, 4036. [Google Scholar] [CrossRef]
- Small, E.; Marcus, D. Tetrahydrocannabinol levels in hemp (Cannabis sativa) germplasm resources. Econ. Bot. 2003, 57, 545. [Google Scholar] [CrossRef]
- Stack, G.M.; Toth, J.A.; Carlson, C.H.; Cala, A.R.; Marrero-González, M.I.; Wilk, R.L.; Gentner, D.R.; Crawford, J.L.; Philippe, G.; Rose, J.K.C.; et al. Season-long characterization of high-cannabinoid hemp (Cannabis sativa L.) reveals variation in cannabinoid accumulation, flowering time, and disease resistance. GCB Bioenergy 2021, 13, 1–16. [Google Scholar] [CrossRef]
- Niazian, M. Application of genetics and biotechnology for improving medicinal plants. Planta 2019, 249, 953–973. [Google Scholar] [CrossRef]
- Duvick, D.N. Biotechnology in the 1930s: The development of hybrid maize. Nat. Rev. Genet. 2001, 2, 69–74. [Google Scholar] [CrossRef]
- Niazian, M.; Shariatpanahi, M.E. In vitro-based doubled haploid production: Recent improvements. Euphytica 2020, 216, 69. [Google Scholar] [CrossRef]
- Niazian, M.; Nalousi, A.M. Artificial polyploidy induction for improvement of ornamental and medicinal plants. Plant Cell Tissue Organ Cult. 2020, 142, 11–23. [Google Scholar] [CrossRef]
- Lv, J.; Yu, K.; Wei, J.; Gui, H.; Liu, C.; Liang, D.; Wang, Y.; Zhou, H.; Carlin, R.; Rich, R.; et al. Generation of paternal haploids in wheat by genome editing of the centromeric histone CENH3. Nat. Biotechnol. 2020, 38, 1397–1401. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Zhu, L.; Zhao, B.; Zhao, Y.; Xie, Y.; Zheng, Z.; Li, Y.; Sun, J.; Wang, H. Development of a Haploid-Inducer Mediated Genome Editing System for Accelerating Maize Breeding. Mol. Plant 2019, 12, 597–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelliher, T.; Starr, D.; Su, X.; Tang, G.; Chen, Z.; Carter, J.; Wittich, P.E.; Dong, S.; Green, J.; Burch, E.; et al. One-step genome editing of elite crop germplasm during haploid induction. Nat. Biotechnol. 2019, 37, 287–292. [Google Scholar] [CrossRef]
- Zhu, H.; Li, C.; Gao, C. Applications of CRISPR–Cas in agriculture and plant biotechnology. Nat. Rev. Mol. Cell Biol. 2020, 21, 661–677. [Google Scholar] [CrossRef] [PubMed]
- Jacinto, F.V.; Link, W.; Ferreira, B.I. CRISPR/Cas9-mediated genome editing: From basic research to translational medicine. J. Cell. Mol. Med. 2020, 24, 3766–3778. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Zhang, X.; Cheng, L.; Luo, Y. An overview and metanalysis of machine and deep learning-based CRISPR gRNA design tools. RNA Biol. 2020, 17, 13–22. [Google Scholar] [CrossRef]
- Zhang, S.; Li, X.; Lin, Q.; Wong, K.-C. Synergizing CRISPR/Cas9 off-target predictions for ensemble insights and practical applications. Bioinformatics 2019, 35, 1108–1115. [Google Scholar] [CrossRef]
- Zhang, G.; Dai, Z.; Dai, X. C-RNNCrispr: Prediction of CRISPR/Cas9 sgRNA activity using convolutional and recurrent neural networks. Comput. Struct. Biotechnol. J. 2020, 18, 344–354. [Google Scholar] [CrossRef]
- Doench, J.G.; Hartenian, E.; Graham, D.B.; Tothova, Z.; Hegde, M.; Smith, I.; Sullender, M.; Ebert, B.L.; Xavier, R.J.; Root, D.E. Rational design of highly active sgRNAs for CRISPR-Cas9–mediated gene inactivation. Nat. Biotechnol. 2014, 32, 1262–1267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.; Cradick, T.J.; Brown, M.T.; Deshmukh, H.; Ranjan, P.; Sarode, N.; Wile, B.M.; Vertino, P.M.; Stewart, F.J.; Bao, G. CRISPR/Cas9 systems have off-target activity with insertions or deletions between target DNA and guide RNA sequences. Nucleic Acids Res. 2014, 42, 7473–7485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, S.Q.; Zheng, Z.; Nguyen, N.T.; Liebers, M.; Topkar, V.V.; Thapar, V.; Wyvekens, N.; Khayter, C.; Iafrate, A.J.; Le, L.P.; et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 2015, 33, 187–197. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Bae, S.; Park, J.; Kim, E.; Kim, S.; Yu, H.R.; Hwang, J.; Kim, J.-I.; Kim, J.-S. Digenome-seq: Genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nat. Methods 2015, 12, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, Y.; Wu, X.; Wang, J.; Wang, Y.; Qiu, Z.; Chang, T.; Huang, H.; Lin, R.-J.; Yee, J.-K. Unbiased detection of off-target cleavage by CRISPR-Cas9 and TALENs using integrase-defective lentiviral vectors. Nat. Biotechnol. 2015, 33, 175–178. [Google Scholar] [CrossRef]
- Bae, S.; Park, J.; Kim, J.-S. Cas-OFFinder: A fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 2014, 30, 1473–1475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, A.; Cheng, Z.; Kong, L.; Zhu, Z.; Lin, S.; Gao, G.; Zhang, B. CasOT: A genome-wide Cas9/gRNA off-target searching tool. Bioinformatics 2014, 30, 1180–1182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, M.; Li, D.; Huan, X.; Manthey, J.; Lioutikova, E.; Zhou, H. Mathematical and computational analysis of CRISPR Cas9 sgRNA off-target homologies. Int. J. Biomath. 2017, 10, 1750085. [Google Scholar] [CrossRef]
- Naito, Y.; Hino, K.; Bono, H.; Ui-Tei, K. CRISPRdirect: Software for designing CRISPR/Cas guide RNA with reduced off-target sites. Bioinformatics 2015, 31, 1120–1123. [Google Scholar] [CrossRef]
- Ioannidis, K.; Dadiotis, E.; Mitsis, V.; Melliou, E.; Magiatis, P. Biotechnological Approaches on Two High CBD and CBG Cannabis sativa L. (Cannabaceae) Varieties: In Vitro Regeneration and Phytochemical Consistency Evaluation of Micropropagated Plants Using Quantitative 1H-NMR. Molecules 2020, 25, 5928. [Google Scholar] [CrossRef] [PubMed]
- Galán-Ávila, A.; García-Fortea, E.; Prohens, J.; Herraiz, F.J. Development of a Direct in vitro Plant Regeneration Protocol From Cannabis sativa L. Seedling Explants: Developmental Morphology of Shoot Regeneration and Ploidy Level of Regenerated Plants. Front. Plant Sci. 2020, 11, 645. [Google Scholar] [CrossRef] [PubMed]
- Wróbel, T.; Dreger, M.; Wielgus, K.; Słomski, R. Modified Nodal Cuttings and Shoot Tips Protocol for Rapid Regeneration of Cannabis sativa L. J. Nat. Fibers 2020, 8, 1–10. [Google Scholar] [CrossRef]
- Haeussler, M.; Schönig, K.; Eckert, H.; Eschstruth, A.; Mianné, J.; Renaud, J.-B.; Schneider-Maunoury, S.; Shkumatava, A.; Teboul, L.; Kent, J.; et al. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 2016, 17, 148. [Google Scholar] [CrossRef] [PubMed]
- Feng, C.; Yuan, J.; Bai, H.; Liu, Y.; Su, H.; Liu, Y.; Shi, L.; Gao, Z.; Birchler, J.A.; Han, F. The deposition of CENH3 in maize is stringently regulated. Plant J. 2020, 102, 6–17. [Google Scholar] [CrossRef] [PubMed]
- Evtushenko, E.V.; Elisafenko, E.A.; Gatzkaya, S.S.; Lipikhina, Y.A.; Houben, A.; Vershinin, A.V. Conserved molecular structure of the centromeric histone CENH3 in Secale and its phylogenetic relationships. Sci. Rep. 2017, 7, 17628. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.M.; Cradick, T.J.; Fine, E.J.; Bao, G. Nuclease Target Site Selection for Maximizing On-target Activity and Minimizing Off-target Effects in Genome Editing. Mol. Ther. 2016, 24, 475–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.-H.; Tee, L.Y.; Wang, X.-G.; Huang, Q.-S.; Yang, S.-H. Off-target Effects in CRISPR/Cas9-mediated Genome Engineering. Mol. Ther. Nucleic Acids 2015, 4, e264. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Zheng, Y.; Zhao, Z.; Liu, T.; Li, J. Recognition of CRISPR/Cas9 off-target sites through ensemble learning of uneven mismatch distributions. Bioinformatics 2018, 34, i757–i765. [Google Scholar] [CrossRef] [Green Version]
- Doench, J.G.; Fusi, N.; Sullender, M.; Hegde, M.; Vaimberg, E.W.; Donovan, K.F.; Smith, I.; Tothova, Z.; Wilen, C.; Orchard, R.; et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 2016, 34, 184–191. [Google Scholar] [CrossRef] [Green Version]
- Hsu, P.D.; Scott, D.A.; Weinstein, J.A.; Ran, F.A.; Konermann, S.; Agarwala, V.; Li, Y.; Fine, E.J.; Wu, X.; Shalem, O.; et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013, 31, 827–832. [Google Scholar] [CrossRef]
- Anderson, K.R.; Haeussler, M.; Watanabe, C.; Janakiraman, V.; Lund, J.; Modrusan, Z.; Stinson, J.; Bei, Q.; Buechler, A.; Yu, C.; et al. CRISPR off-target analysis in genetically engineered rats and mice. Nat. Methods 2018, 15, 512–514. [Google Scholar] [CrossRef] [PubMed]
- Niu, M.; Lin, Y.; Zou, Q. sgRNACNN: Identifying sgRNA on-target activity in four crops using ensembles of convolutional neural networks. Plant Mol. Biol. 2021, 105, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Mahood, E.H.; Kruse, L.H.; Moghe, G.D. Machine learning: A powerful tool for gene function prediction in plants. Appl. Plant Sci. 2020, 8, e11376. [Google Scholar] [CrossRef]
- Wang, H.; Joshi, P.; Hong, S.H.; Maye, P.F.; Rowe, D.W.; Shin, D.G. cTAP: A Machine Learning Framework for Predicting Target Genes of a Transcription Factor using a Cohort of Gene Expression Data Sets. In Proceedings of the 2020 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Seoul, Korea, 16–19 December 2020; pp. 164–167. [Google Scholar]
- Yoosefzadeh Najafabadi, M.; Earl, H.J.; Tulpan, D.; Sulik, J.; Eskandari, M. Application of Machine Learning Algorithms in Plant Breeding: Predicting Yield from Hyperspectral Reflectance in Soybean. Front. Plant Sci. 2021, 11, 624273. [Google Scholar] [CrossRef]
- Niazian, M.; Niedbała, G. Machine Learning for Plant Breeding and Biotechnology. Agriculture 2020, 10, 436. [Google Scholar] [CrossRef]
- Jafari, M.; Shahsavar, A. The application of artificial neural networks in modeling and predicting the effects of melatonin on morphological responses of citrus to drought stress. PLoS ONE 2020, 15, e0240427. [Google Scholar] [CrossRef] [PubMed]
- Hesami, M.; Jones, A.M.P. Application of artificial intelligence models and optimization algorithms in plant cell and tissue culture. Appl. Microbiol. Biotechnol. 2020, 104, 9449–9485. [Google Scholar] [CrossRef]
- Hesami, M.; Condori-Apfata, J.A.; Valderrama Valencia, M.; Mohammadi, M. Application of Artificial Neural Network for Modeling and Studying In Vitro Genotype-Independent Shoot Regeneration in Wheat. Appl. Sci. 2020, 10, 5370. [Google Scholar] [CrossRef]
- Hesami, M.; Naderi, R.; Tohidfar, M.; Yoosefzadeh-Najafabadi, M. Application of Adaptive Neuro-Fuzzy Inference System-Non-dominated Sorting Genetic Algorithm-II (ANFIS-NSGAII) for Modeling and Optimizing Somatic Embryogenesis of Chrysanthemum. Front. Plant Sci. 2019, 10, 869. [Google Scholar] [CrossRef]
- Salehi, M.; Farhadi, S.; Moieni, A.; Safaie, N.; Hesami, M. A hybrid model based on general regression neural network and fruit fly optimization algorithm for forecasting and optimizing paclitaxel biosynthesis in Corylus avellana cell culture. Plant Methods 2021, 17, 13. [Google Scholar] [CrossRef]
- Silva, J.C.F.; Teixeira, R.M.; Silva, F.F.; Brommonschenkel, S.H.; Fontes, E.P.B. Machine learning approaches and their current application in plant molecular biology: A systematic review. Plant Sci. 2019, 284, 37–47. [Google Scholar] [CrossRef]
- Yeom, S.; Giacomelli, I.; Menaged, A.; Fredrikson, M.; Jha, S. Overfitting, robustness, and malicious algorithms: A study of potential causes of privacy risk in machine learning. J. Comput. Secur. 2020, 28, 35–70. [Google Scholar] [CrossRef] [Green Version]
- Hesami, M.; Naderi, R.; Tohidfar, M. Introducing a hybrid artificial intelligence method for high-throughput modeling and optimizing plant tissue culture processes: The establishment of a new embryogenesis medium for chrysanthemum, as a case study. Appl. Microbiol. Biotechnol. 2020, 104, 10249–10263. [Google Scholar] [CrossRef] [PubMed]
- Hesami, M.; Alizadeh, M.; Naderi, R.; Tohidfar, M. Forecasting and optimizing Agrobacterium-mediated genetic transformation via ensemble model- fruit fly optimization algorithm: A data mining approach using chrysanthemum databases. PLoS ONE 2020, 15, e0239901. [Google Scholar] [CrossRef]
- Hesami, M.; Naderi, R.; Tohidfar, M. Modeling and Optimizing Medium Composition for Shoot Regeneration of Chrysanthemum via Radial Basis Function-Non-dominated Sorting Genetic Algorithm-II (RBF-NSGAII). Sci. Rep. 2019, 9, 18237. [Google Scholar] [CrossRef] [Green Version]
- Hesami, M.; Naderi, R.; Tohidfar, M.; Yoosefzadeh-Najafabadi, M. Development of support vector machine-based model and comparative analysis with artificial neural network for modeling the plant tissue culture procedures: Effect of plant growth regulators on somatic embryogenesis of chrysanthemum, as a case study. Plant Methods 2020, 16, 112. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.; Frank, E.; Holmes, G.; Pfahringer, B.; Reutemann, P.; Witten, I.H. The WEKA data mining software: An update. ACM SIGKDD Explor. Newsl. 2009, 11, 10–18. [Google Scholar] [CrossRef]
- Siegmann, B.; Jarmer, T. Comparison of different regression models and validation techniques for the assessment of wheat leaf area index from hyperspectral data. Int. J. Remote Sens. 2015, 36, 4519–4534. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| sgRNA | Putative Off-Target DNA Sequences | Chromosome | Start | End | MIT | CFD |
|---|---|---|---|---|---|---|
| TTAGCAGTGTCCAAGTCTTCTGG | TCAGCAGCGTCTAAATCTTCAGG | 7 | 638 | 660 | 0.199 | 0.434 |
| TTAGCAGTGTCCAAGTCTTCTGG | CTAGAGGTGTCCATGTCTTCAGG | 5 | 21,767 | 21,789 | 0.135 | 0.187 |
| AGCTTTAGTTGCACTTCAGGAGG | AGCTTTAATTGAATTTCATGGGG | 8 | 2079 | 2101 | 0.033 | 0.349 |
| CACGTCGACTTGGAGGGAAAGGG | CAGGTCGACGTCGAGGAAAAAGG | 3 | 3689 | 3711 | 0.259 | 0.123 |
| AGCCTGGAACAAAGGCTCTCCGG | AGACTGCAACAAAGCATCTCCGG | 5 | 1624 | 1646 | 0.047 | 0.162 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hesami, M.; Yoosefzadeh Najafabadi, M.; Adamek, K.; Torkamaneh, D.; Jones, A.M.P. Synergizing Off-Target Predictions for In Silico Insights of CENH3 Knockout in Cannabis through CRISPR/Cas. Molecules 2021, 26, 2053. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26072053
Hesami M, Yoosefzadeh Najafabadi M, Adamek K, Torkamaneh D, Jones AMP. Synergizing Off-Target Predictions for In Silico Insights of CENH3 Knockout in Cannabis through CRISPR/Cas. Molecules. 2021; 26(7):2053. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26072053
Chicago/Turabian StyleHesami, Mohsen, Mohsen Yoosefzadeh Najafabadi, Kristian Adamek, Davoud Torkamaneh, and Andrew Maxwell Phineas Jones. 2021. "Synergizing Off-Target Predictions for In Silico Insights of CENH3 Knockout in Cannabis through CRISPR/Cas" Molecules 26, no. 7: 2053. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26072053