
Strength of the [Z–I···Hal]− and [Z–Hal···I]− Halogen Bonds: Electron Density Properties and Halogen Bond Length as Estimators of Interaction Energy


Abstract
:1. Introduction
2. Computational Details
3. Computational Models
4. Results
4.1. Test of the Computational Method
4.2. Interaction Energies
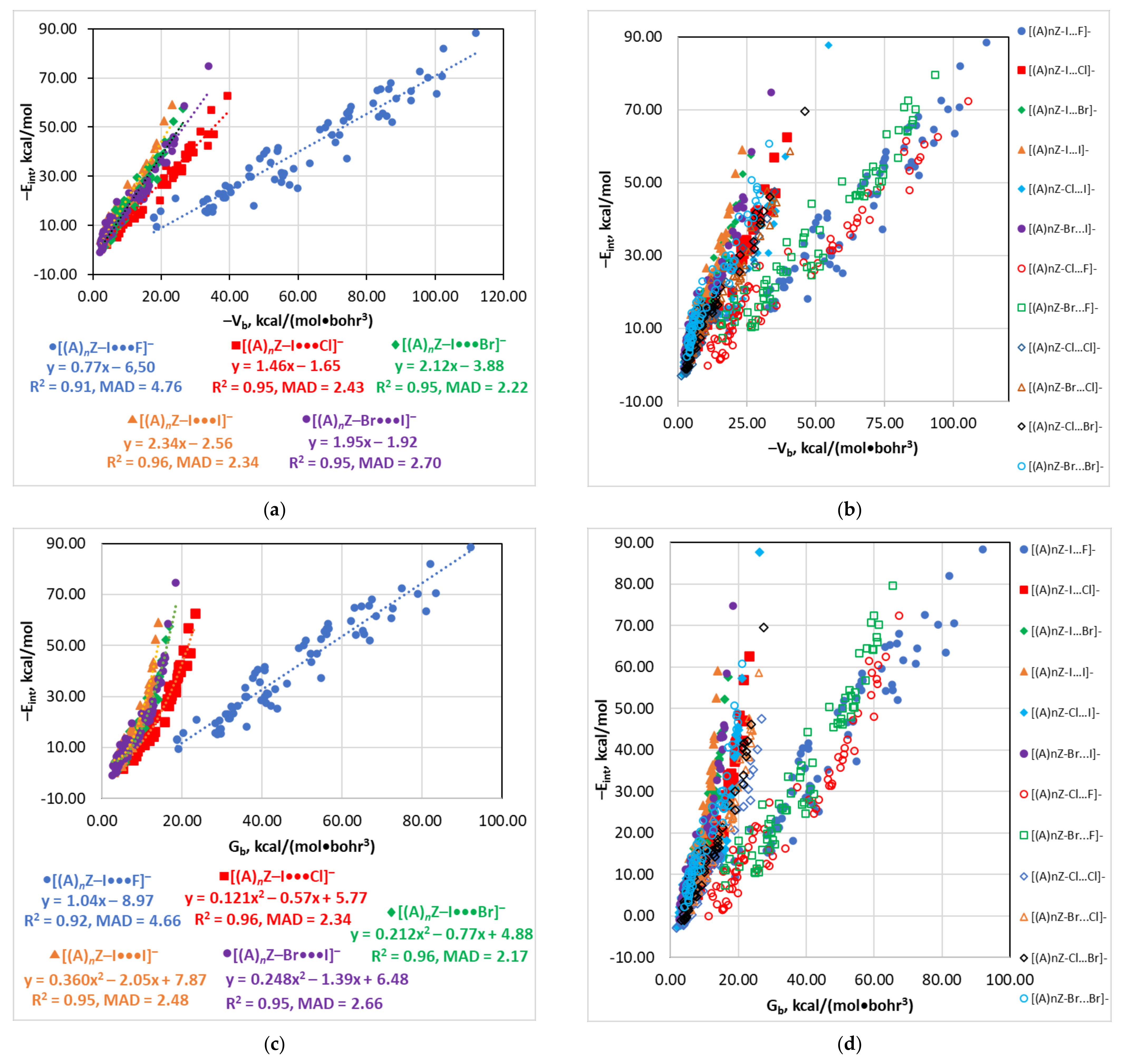
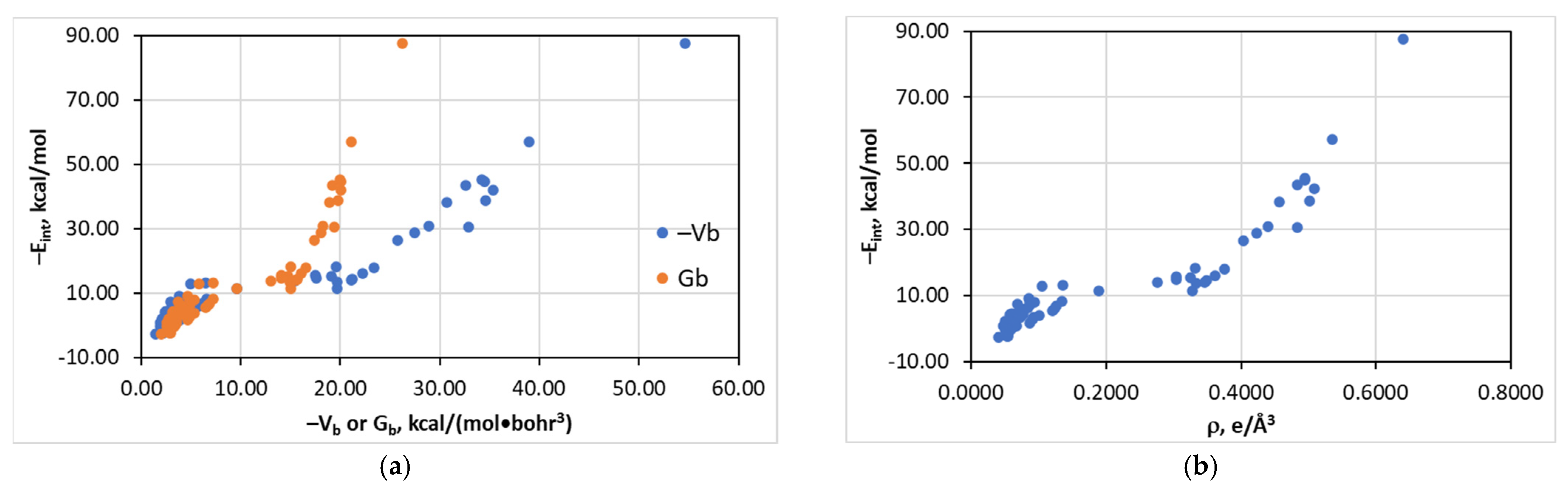
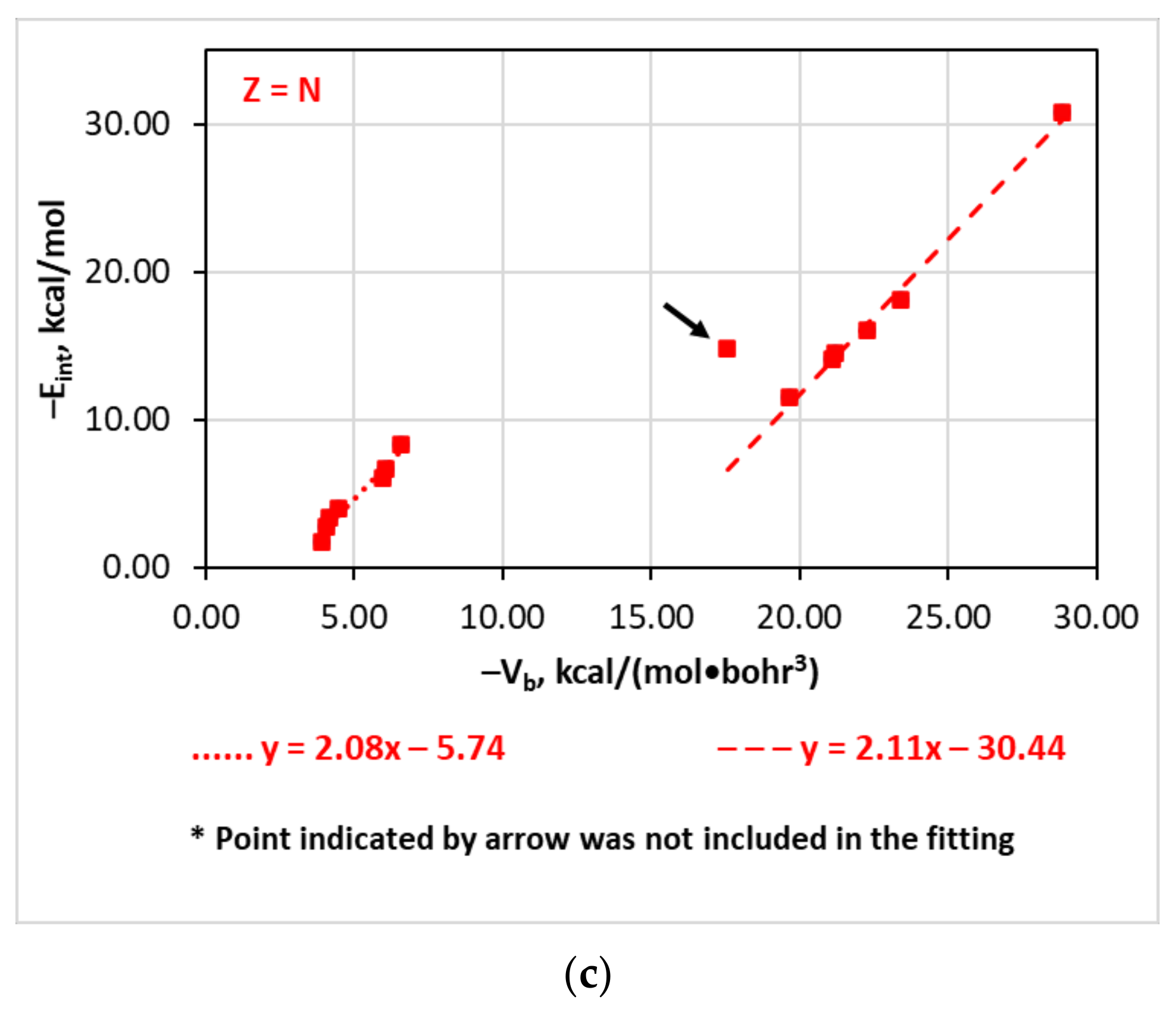
4.3. The Eint(Vb), Eint(Gb), and Eint(ρb) Relationships
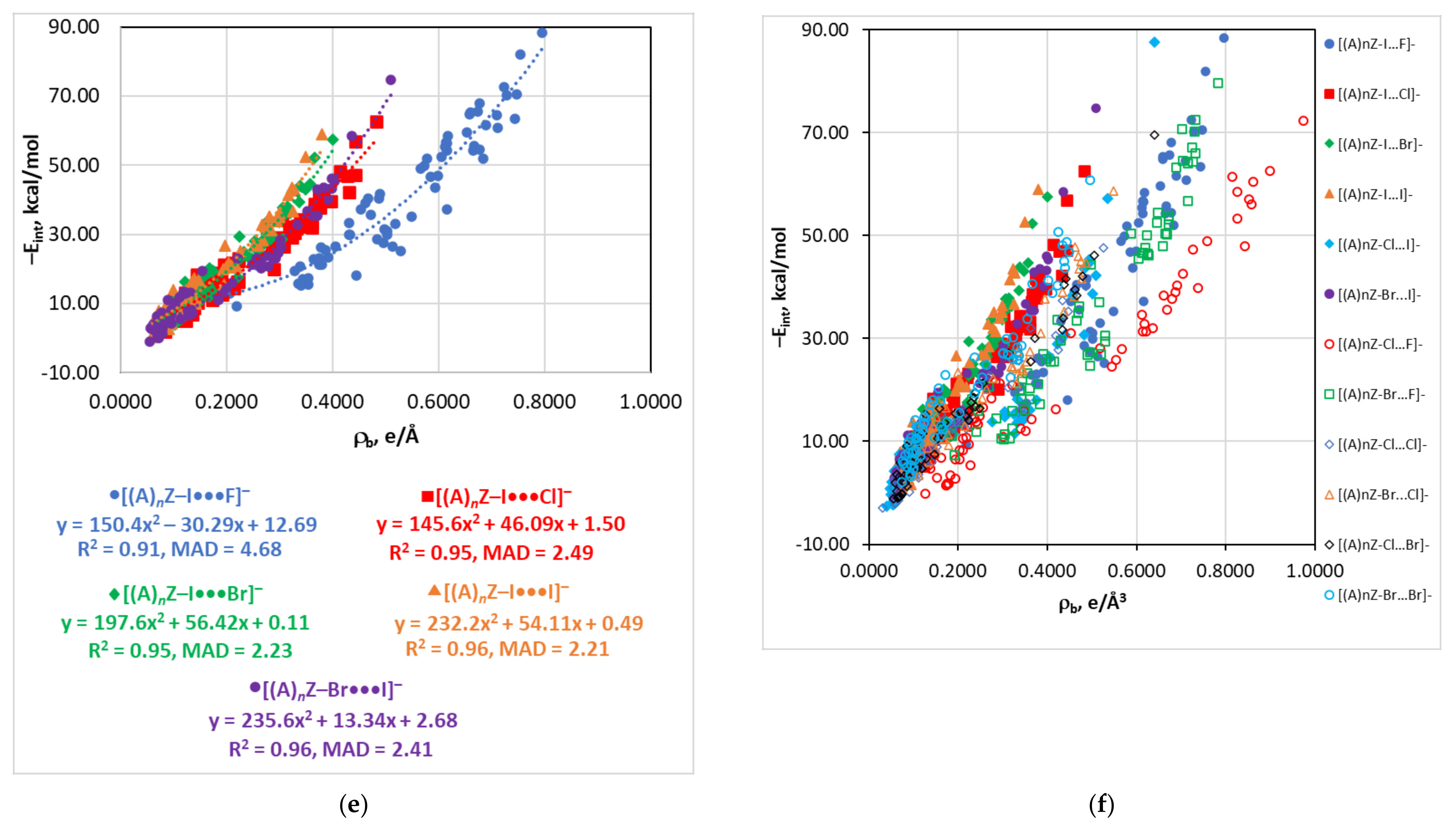
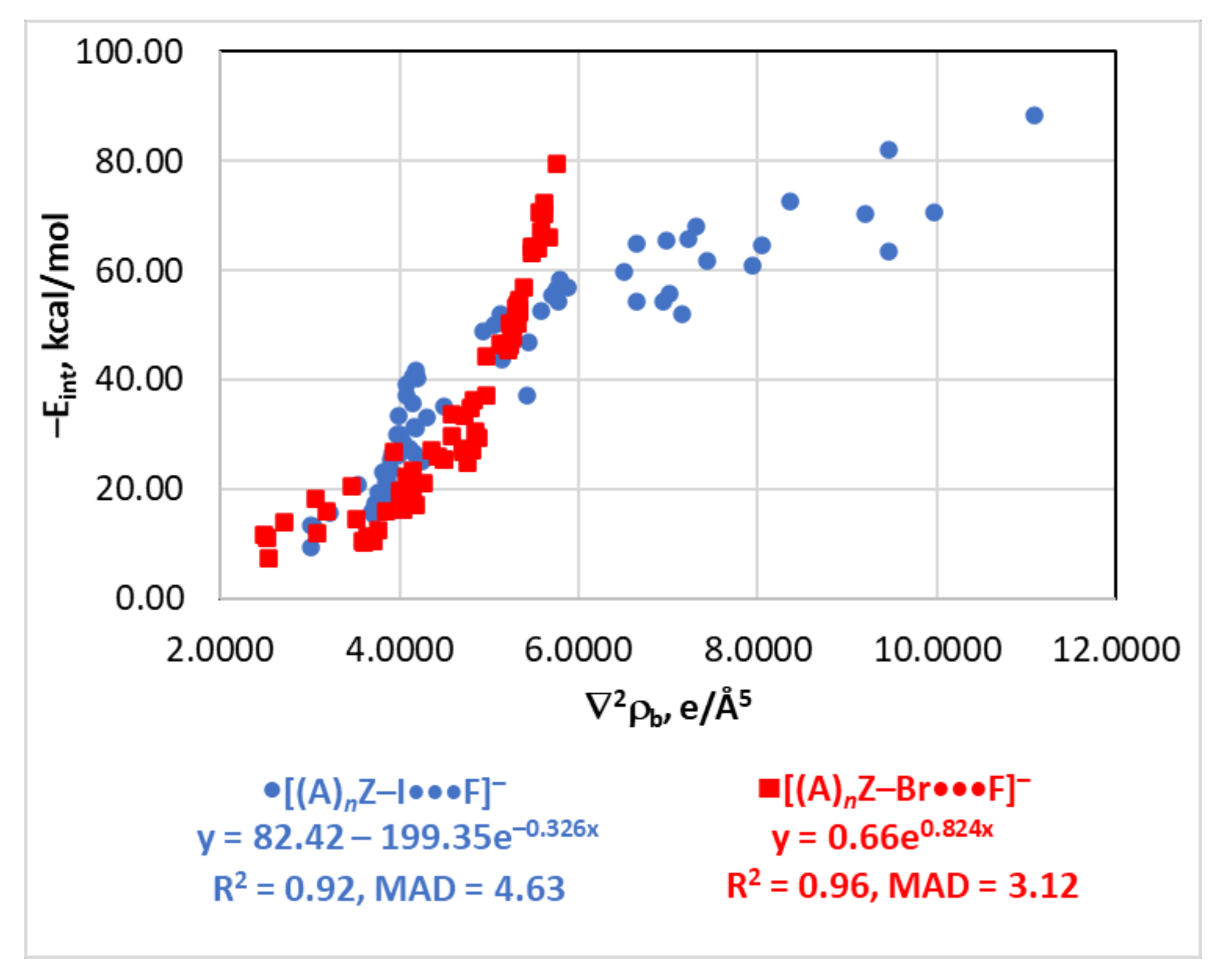
4.4. The Eint(∇2ρb) Relationship
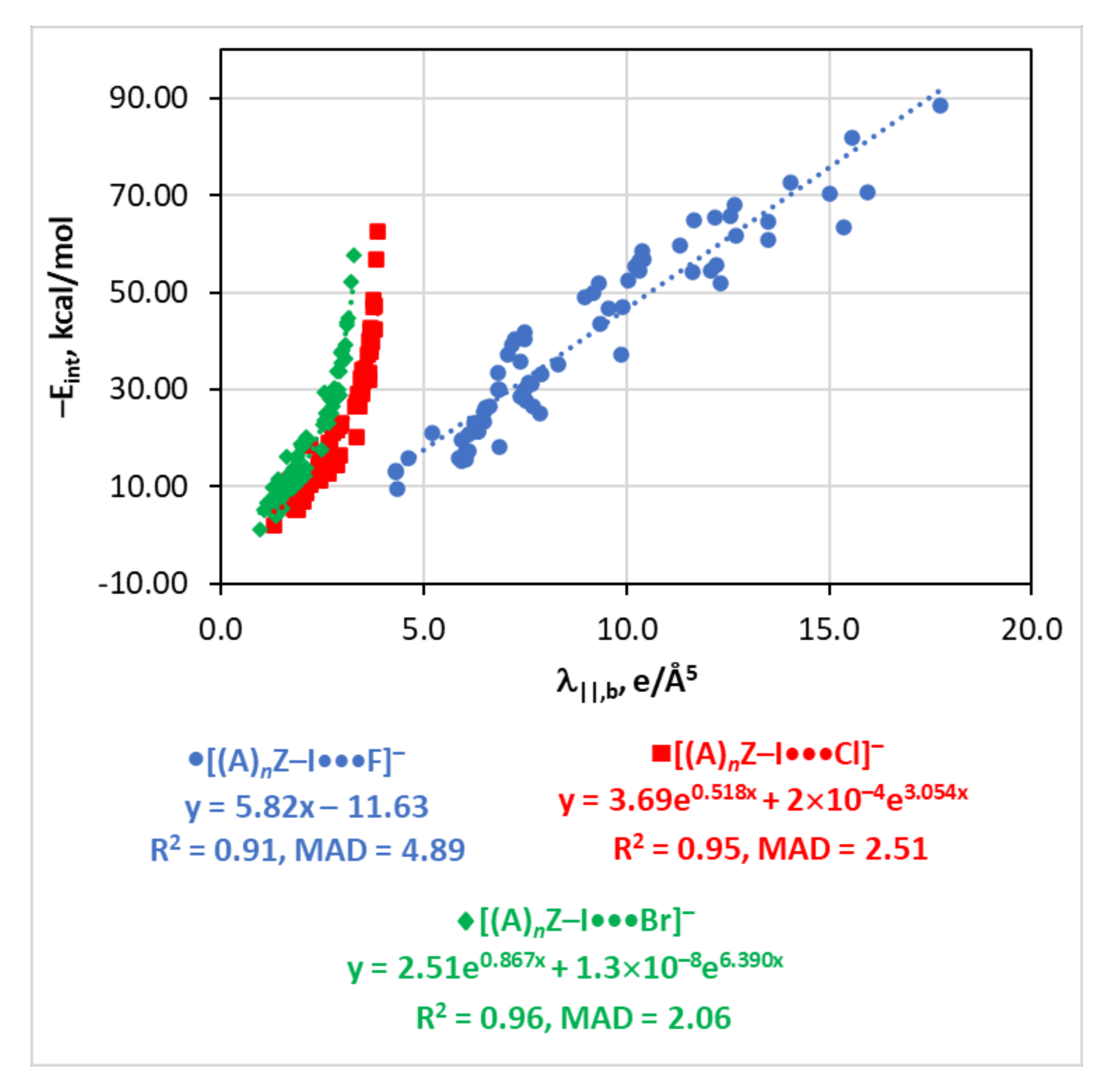
4.5. The Eint(λ||,b) Relationship
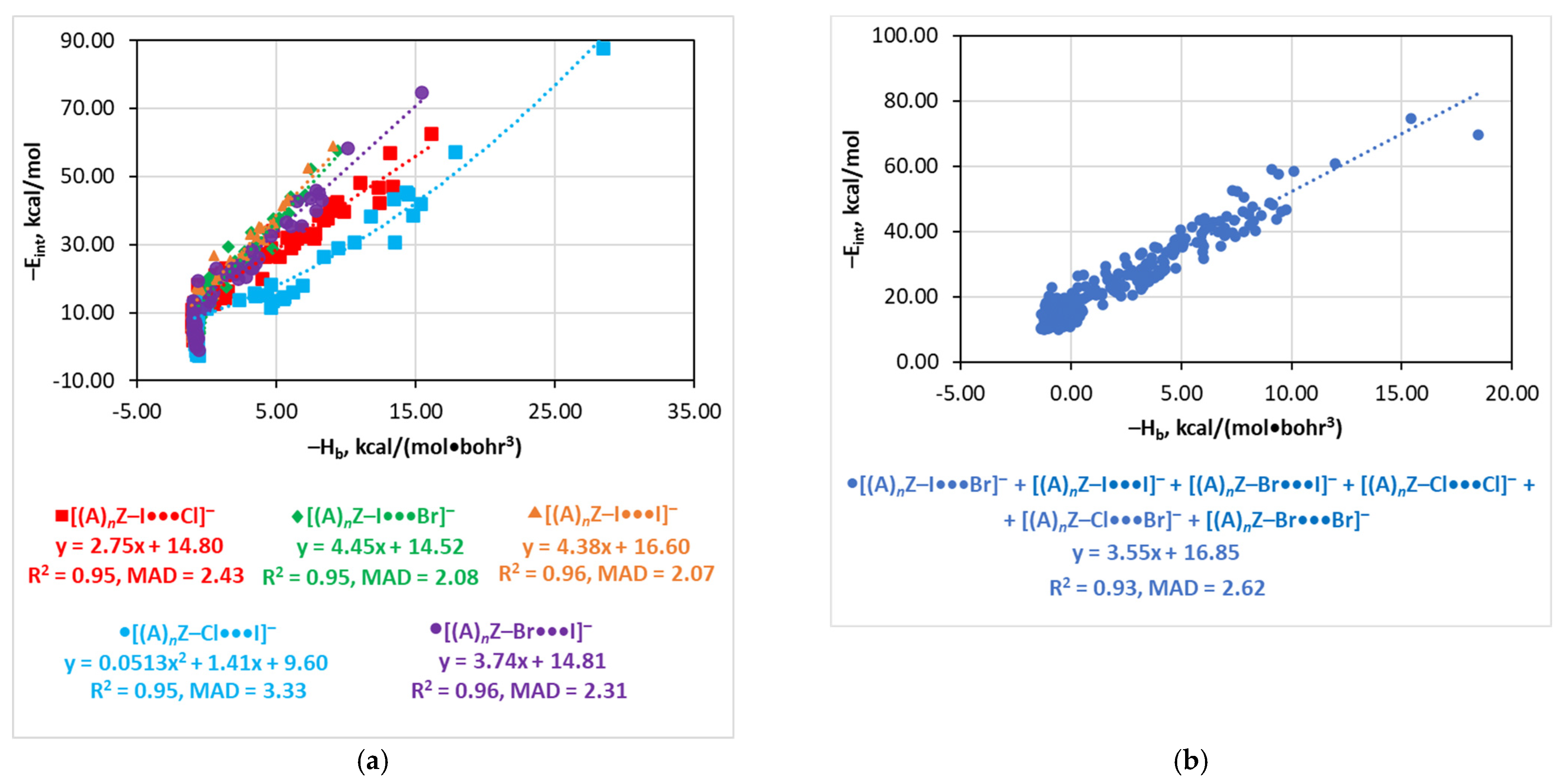
4.6. The Eint(Hb) Relationship
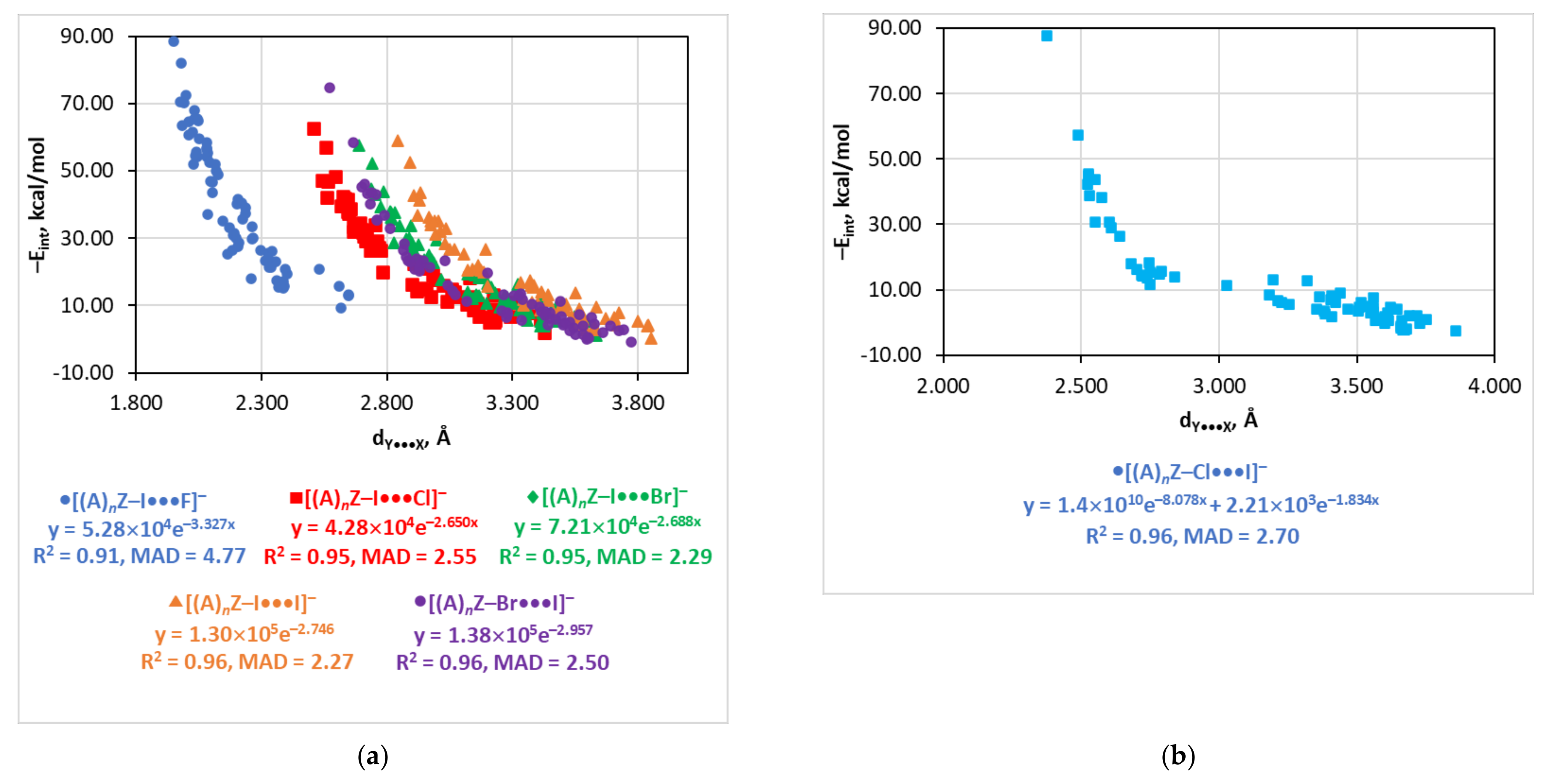
4.7. The Eint(dY…X) Relationship
4.8. The [(A)nZ–Cl···I]− Series
5. Discussion
6. Final Remarks
Supplementary Materials
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Desiraju, G.R.; Ho, P.S.; Kloo, L.; Legon, A.C.; Marquardt, R.; Metrangolo, P.; Politzer, P.; Resnati, G.; Rissanen, K. Definition of the Halogen Bond (IUPAC Recommendations 2013). Pure Appl. Chem. 2013, 85, 1711–1713. [Google Scholar] [CrossRef]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The Halogen Bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metrangolo, P.; Resnati, G. Halogen Bonding: Fundamentals and Applications; Springer: Berlin, Germany, 2008. [Google Scholar]
- Priimagi, A.; Cavallo, G.; Forni, A.; Gorynsztejn-Leben, M.; Kaivola, M.; Metrangolo, P.; Milani, R.; Shishido, A.; Pilati, T.; Resnati, R.; et al. Halogen Bonding versus Hydrogen Bonding in Driving Self-Assembly and Performance of Light-Responsive Supramolecular Polymers. Adv. Funct. Mater. 2012, 22, 2572–2579. [Google Scholar] [CrossRef] [Green Version]
- Natale, D.; Marequerivas, J.C. The combination of transition metal ions and hydrogen-bonding interactions. Chem. Commun. 2008, 425–437. [Google Scholar] [CrossRef] [PubMed]
- Feller, R.K.; Cheetham, A.K. Structural and chemical complexity in multicomponent inorganic–organic framework materials. CrystEngComm 2009, 11, 980–985. [Google Scholar] [CrossRef]
- Corradi, E.; Meille, S.V.; Messina, M.T.; Metrangolo, P.; Resnati, G. Halogen bonding versus hydrogen bonding in driving self-assembly processes. Angew. Chem. Int. Ed. 2000, 39, 1782–1786. [Google Scholar] [CrossRef]
- Desiraju, G.R. Crystal engineering: A holistic view. Angew. Chem. Int. Ed. 2007, 46, 8342–8356. [Google Scholar] [CrossRef]
- Cinčić, D.; Friščić, T.; Jones, W. Structural equivalence of Br and I halogen bonds: A route to isostructural materials with controllable properties. Chem. Mater. 2008, 20, 6623–6626. [Google Scholar] [CrossRef]
- Tepper, R.; Schubert, U.S. Halogen Bonding in Solution: Anion Recognition, Templated Self-Assembly, and Organocatalysis. Angew. Chem. Int. Ed. 2018, 57, 6004–6016. [Google Scholar] [CrossRef]
- Mahmudov, K.T.; Kopylovich, M.N.; Guedes da Silva, M.F.C.; Pombeiro, A.J.L. Non-covalent interactions in the synthesis of coordination compounds: Recent advances. Coord. Chem. Rev. 2017, 345, 54–72. [Google Scholar] [CrossRef]
- Li, B.; Zang, S.-Q.; Wang, L.-Y.; Mak, T.C.W. Halogen bonding: A powerful, emerging tool for constructing high-dimensional metal-containing supramolecular networks. Coord. Chem. Rev. 2016, 308, 1–21. [Google Scholar] [CrossRef]
- Saccone, M.; Cavallo, G.; Metrangolo, P.; Resnati, G.; Priimagi, A. Halogen-Bonded Photoresponsive Materials. In Halogen Bonding II. Topics in Current Chemistry; Metrangolo, P., Resnati, G., Eds.; Springer: Cham, Switzerland, 2014; Volume 359. [Google Scholar]
- Aakeröy, C.B.; Spartz, C.L. Halogen Bonding in Supramolecular Synthesis. In Halogen Bonding I. Topics in Current Chemistry; Metrangolo, P., Resnati, G., Eds.; Springer: Cham, Switzerland, 2014; Volume 358. [Google Scholar]
- Mukherjee, A.; Tothadi, S.; Desiraju, G.R. Halogen bonds in crystal engineering: Like hydrogen bonds yet different. Acc. Chem. Res. 2014, 47, 2514–2524. [Google Scholar] [CrossRef]
- Cariati, E.; Forni, A.; Biella, S.; Metrangolo, P.; Meyer, F.; Resnati, G.; Righetto, S.; Tordin, E.; Ugo, R. Tuning second-order NLO responses through halogen bonding. Chem. Commun. 2007, 2590–2592. [Google Scholar] [CrossRef] [PubMed]
- Christopherson, J.-C.; Topić, F.; Barrett, C.J.; Friščić, T. Halogen-Bonded Cocrystals as Optical Materials: Next-Generation Control over Light–Matter Interactions. Cryst. Growth Des. 2018, 18, 1245–1259. [Google Scholar] [CrossRef]
- Mahmudov, K.T.; Gurbanov, A.V.; Guseinov, F.I.; Guedes da Silva, M.F.C. Noncovalent interactions in metal complex catalysis. Coord. Chem. Rev. 2019, 387, 32–46. [Google Scholar] [CrossRef]
- Szell, P.M.J.; Zablotny, S.; Bryce, D.L. Halogen bonding as a supramolecular dynamics catalyst. Nature Commun. 2019, 10, 916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schindler, S.; Huber, S.M. Halogen Bonds in Organic Synthesis and Organocatalysis. In Halogen Bonding II. Topics in Current Chemistry; Metrangolo, P., Resnati, G., Eds.; Springer: Cham, Switzerland, 2014; Volume 359. [Google Scholar]
- Xu, Z.; Yang, Z.; Liu, Y.; Lu, Y.; Chen, K.; Zhu, W. Halogen bond: Its role beyond drug-target binding affinity for drug discovery and development. J. Chem. Inf. Model. 2014, 54, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Shi, T.; Wang, Y.; Yang, H.; Yan, X.; Luo, X.; Jiang, H.; Zhu, W. Halogen bonding—A novel interaction for rational drug design? J. Med. Chem. 2009, 52, 2854–2862. [Google Scholar] [CrossRef] [PubMed]
- Mendez, L.; Henriquez, G.; Sirimulla, S.; Narayan, M. Looking back, looking forward at halogen bonding in drug discovery. Molecules 2017, 22, 1397. [Google Scholar] [CrossRef]
- Hua, Y.; Flood, A.H. Click chemistry generates privileged CH hydrogen-bonding triazoles: The latest addition to anion supramolecular chemistry. Chem. Soc. Rev. 2010, 39, 1262–1271. [Google Scholar] [CrossRef]
- Abate, A.; Biella, S.; Cavallo, G.; Meyer, F.; Neukirch, H.; Metrangolo, P.; Pilati, T.; Resnati, G.; Terraneo, G. Halide anions driven self-assembly of haloperfluoroarenes: Formation of one-dimensional non-covalent copolymers. J. Fluorine Chem. 2009, 130, 1171–1177. [Google Scholar] [CrossRef]
- Dey, B.; Choudhury, S.R.; Gamez, P.; Vargiu, A.V.; Robertazzi, A.; Chen, C.-Y.; Lee, H.M.; Jana, A.D.; Mukhopadhyay, S. Water−Chloride and Water−Bromide Hydrogen-Bonded Networks: Influence of the Nature of the Halide Ions on the Stability of the Supramolecular Assemblies. J. Phys. Chem. A 2009, 113, 8626–8634. [Google Scholar] [CrossRef] [PubMed]
- Cowan, J.A. Supramolecular Chemistry of Anions; Bianchi, A., Bowman-James, K., Garcoa-Espapa, E., Eds.; Wiley-VCH: New York, NY, USA, 1997. [Google Scholar]
- Bowman-James, K. Alfred Werner Revisited: The Coordination Chemistry of Anions. Acc. Chem. Res. 2005, 38, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Gale, P.A.; Quesada, R. Anion coordination and anion-templated assembly: Highlights from 2002 to 2004. Coord. Chem. Rev. 2006, 250, 3219–3244. [Google Scholar] [CrossRef]
- Feiters, M.C.; Meyer-Klaucke, W.; Kostenko, A.V.; Soldatov, A.V.; Leblanc, C.; Michel, G.; Potin, P.; Küpper, F.C.; Hollenstein, K.; Locher, K.P.; et al. Anion binding in biological systems. J. Phys. Conf. Ser. 2009, 190, 012196. [Google Scholar] [CrossRef]
- Yang, H.S.; Kim, E.; Lee, S.; Park, H.J.; Cooper, D.S.; Rajbhandari, I.; Choi, I. Mutation of Aspartate 555 of the Sodium/Bicarbonate Transporter SLC4A4/NBCe1 Induces Chloride Transport. J. Biol. Chem. 2009, 284, 15970–15979. [Google Scholar] [CrossRef] [Green Version]
- Okada, Y.; Sato, K.; Numata, T. Pathophysiology and puzzles of the volume-sensitive outwardly rectifying anion channel. J. Physiol. 2009, 587, 2141–2149. [Google Scholar]
- Desiraju, G.R.; Steiner, T. The Weak Hydrogen Bond in Structural Chemistry and Biology; Oxford University Press: Oxford, UK, 1999. [Google Scholar]
- Sarwar, M.G.; Dragisic, B.; Sagoo, S.; Taylor, M.S. A Tridentate Halogen-Bonding Receptor for Tight Binding of Halide Anions. Angew. Chem. Int. Ed. 2010, 49, 1674–1677. [Google Scholar] [CrossRef]
- Svec, J.; Necas, M.; Sindelar, V. Bambus [6]uril. Angew. Chem. Int. Ed. 2010, 49, 2378–2381. [Google Scholar] [CrossRef]
- Chang, K.-J.; Moon, D.; Lah, M.S.; Jeong, K.-S. Indole-Based Macrocycles as a Class of Receptors for Anions. Angew. Chem. Int. Ed. 2005, 44, 7926–7929. [Google Scholar] [CrossRef]
- Hisaki, I.; Sasaki, S.-I.; Hirose, K.; Tobe, Y. Synthesis and Anion-Selective Complexation of Homobenzylic Tripodal Thiourea Derivatives. Eur. J. Org. Chem. 2007, 607–615. [Google Scholar] [CrossRef]
- Li, Y.; Flood, A.H. Pure C–H Hydrogen Bonding to Chloride Ions: A Preorganized and Rigid Macrocyclic Receptor. Angew. Chem. Int. Ed. 2008, 47, 2649–2652. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.S.; Jacobsen, E.N. Asymmetric Catalysis by Chiral Hydrogen-Bond Donors. Angew. Chem. Int. Ed. 2006, 45, 1520–1543. [Google Scholar] [CrossRef]
- Lacour, J.; Moraleda, D. Chiral anion-mediated asymmetric ion pairing chemistry. Chem. Commun. 2009, 7073–7089. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, G.L.; Kanai, T.; Toste, F.D. Chiral Anion-Mediated Asymmetric Ring Opening of meso-Aziridinium and Episulfonium Ions. J. Am. Chem. Soc. 2008, 130, 14984–14986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamilton, G.L.; Kang, E.J.; Mba, M.; Toste, F.D. A Powerful Chiral Counterion Strategy for Asymmetric Transition Metal Catalysis. Science 2007, 317, 496–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coe, B.J.; Fielden, J.; Foxon, S.P.; Brunschwig, B.S.; Asselberghs, I.; Clays, K.; Samoc, A.; Samoc, M. Combining Very Large Quadratic and Cubic Nonlinear Optical Responses in Extended, Tris-Chelate Metallochromophores with Six π-Conjugated Pyridinium Substituents. J. Am. Chem. Soc. 2010, 132, 3496–3513. [Google Scholar] [CrossRef] [Green Version]
- Leventis, H.C.; O’Mahony, F.; Akhtar, J.; Afzaal, M.; O’Brien, P.; Haque, S.A. Transient Optical Studies of Interfacial Charge Transfer at Nanostructured Metal Oxide/PbS Quantum Dot/Organic Hole Conductor Heterojunctions. J. Am. Chem. Soc. 2010, 132, 2743–2750. [Google Scholar] [CrossRef]
- Zhao, C.; MacFarlane, D.R.; Bond, A.M. Modified Thermodynamics in Ionic Liquids for Controlled Electrocrystallization of Nanocubes, Nanowires, and Crystalline Thin Films of Silver−Tetracyanoquinodimethane. J. Am. Chem. Soc. 2009, 131, 16195–16205. [Google Scholar] [CrossRef]
- Wada, H.; de Caro, D.; Valade, L.; Ozawa, T.; Bando, Y.; Mori, T. Thin-film phases of organic charge-transfer complexes formed by chemical vapor deposition. Thin Solid Films 2009, 518, 299–304. [Google Scholar] [CrossRef]
- Uji, S.; Mori, T.; Takahashi, T. Focus on Organic Conductors. Sci. Technol. Adv. Mater. 2009, 10, 020301. [Google Scholar] [CrossRef]
- Hodgkiss, J.M.; Tu, G.; Albert-Seifried, S.; Huck, W.T.S.; Friend, R.H. Ion-Induced Formation of Charge-Transfer States in Conjugated Polyelectrolytes. J. Am. Chem. Soc. 2009, 131, 8913–8921. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.M.; Wang, H.H.; Emge, T.J.; Geiser, U.; Beno, M.A.; Carlson, R.J.; Thorn, K.D.; Schultz, A.J.; Whangbo, M.-H. Rational Design of Synthetic Metal Superconductors. Prog. Inorg. Chem. 1987, 35, 51–218. [Google Scholar]
- Poreba, T.; Ernst, M.; Zimmer, D.; Macchi, P.; Casati, N. Pressure-Induced Polymerization and Electrical Conductivity of a Polyiodide. Angew. Chem. Int. Ed. 2019, 58, 6625–6629. [Google Scholar] [CrossRef] [PubMed]
- Shaw, R.A.; Hill, J.G. A Simple Model for Halogen Bond Interaction Energies. Inorganics 2019, 7, 19. [Google Scholar] [CrossRef] [Green Version]
- Legon, A.C.; Millen, D.J. Hydrogen bonding as a probe of electron densities: Limiting gas-phase nucleophilicities and electrophilicities of B and HX. J. Am. Chem. Soc. 1987, 109, 356–358. [Google Scholar] [CrossRef]
- Legon, A.C. A reduced radial potential energy function for the halogen bond and the hydrogen bond in complexes B···XY and B···HX, where X and Y are halogen atoms. Phys. Chem. Chem. Phys. 2014, 16, 12415–12421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alkorta, I.; Legon, A.C. Nucleophilicities of Lewis Bases B and Electrophilicities of Lewis Acids A Determined from the Dissociation Energies of Complexes B···A Involving Hydrogen Bonds, Tetrel Bonds, Pnictogen Bonds, Chalcogen Bonds and Halogen Bonds. Molecules 2017, 22, 1786. [Google Scholar] [CrossRef] [Green Version]
- Afonin, A.V.; Vashchenko, A.V. Benchmark calculations of intramolecular hydrogen bond energy based on molecular tailoring and function-based approaches: Developing hybrid approach. Int. J. Quant. Chem. 2019, 119, e26001. [Google Scholar] [CrossRef]
- Vologzhanina, A.V.; Buikin, P.A.; Korlyukov, A.A. Peculiarities of Br··· Br bonding in crystal structures of polybromides and bromine solvates. CrystEngComm 2020, 22, 7361–7370. [Google Scholar] [CrossRef]
- Oliveira, V.P.; Marcial, B.L.; Machado, F.B.C.; Kraka, E. Metal-Halogen Bonding Seen through the Eyes of Vibrational Spectroscopy. Materials 2020, 13, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, C.T.; Chen, M.M.; Fang, Z.J.; Au, C.T.; Cao, C.Z. Relationship Investigation between C(sp2)–X and C(sp3)–X Bond Energies Based on Substituted Benzene and Methane. ACS Omega 2020, 5, 19304–19311. [Google Scholar] [CrossRef] [PubMed]
- Arkhipov, D.E.; Lyubeshkin, A.V.; Volodin, A.D.; Korlyukov, A.A. Molecular Structures Polymorphism the Role of F···F Interactions in Crystal Packing of Fluorinated Tosylates. Crystals 2019, 9, 242. [Google Scholar] [CrossRef] [Green Version]
- Bartashevich, E.; Matveychuk, Y.; Tsirelson, V. Identification of the Tetrel Bonds between Halide Anions and Carbon Atom of Methyl Groups Using Electronic Criterion. Molecules 2019, 24, 1083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mata, I.; Alkorta, I.; Espinosa, E.; Molins, E. Relationships between interaction energy, intermolecular distance and electron density properties in hydrogen bonded complexes under external electric fields. Chem. Phys. Lett. 2011, 507, 185–189. [Google Scholar] [CrossRef]
- Małecka, M. DFT studies and AIM analysis of intramolecular N–HO hydrogen bonds in 3-aminomethylene-2 methoxy-5,6-dimethyl-2-oxo-2,3-dihydro-2λ5-[1,2]oxaphosphinin-4-one and its derivatives. Struct. Chem. 2010, 21, 175–184. [Google Scholar] [CrossRef]
- Grabowski, S.J.; Bilewicz, E. Cooperativity halogen bonding effect—Ab initio calculations on H2CO⋯(ClF)n complexes. Chem. Phys. Lett. 2006, 427, 51–55. [Google Scholar] [CrossRef]
- Szatyłowicz, H. Structural aspects of the intermolecular hydrogen bond strength: H-bonded complexes of aniline, phenol and pyridine derivatives. J. Phys. Org. Chem. 2008, 21, 897–914. [Google Scholar] [CrossRef]
- Hugas, D.; Simon, S.; Duran, M. Electron Density Topological Properties Are Useful To Assess the Difference between Hydrogen and Dihydrogen Complexes. J. Chem. Phys. A 2007, 111, 4506–4512. [Google Scholar] [CrossRef] [PubMed]
- Rozas, I.; Alkorta, I.; Elguero, J. Behavior of Ylides Containing N, O, and C Atoms as Hydrogen Bond Acceptors. J. Am. Chem. Soc. 2000, 122, 11154–11161. [Google Scholar] [CrossRef]
- Lipkowski, P.; Grabowski, S.J.; Robinson, T.L.; Leszczynski, J. Properties of the C−H···H Dihydrogen Bond: An ab Initio and Topological Analysis. J. Phys. Chem. A 2004, 108, 10865–10872. [Google Scholar] [CrossRef]
- Grabowski, S.J. Hydrogen bonding strength—measures based on geometric and topological parameters. J. Phys. Org. Chem. 2004, 17, 18–31. [Google Scholar] [CrossRef]
- Lyssenko, K.A. Analysis of supramolecular architectures: Beyond molecular packing diagrams. Mendeleev Commun. 2012, 22, 1–7. [Google Scholar] [CrossRef]
- Lyssenko, K.A.; Barzilovich, P.Y.; Nelyubina, Y.V.; Astaf’ev, E.A.; Antipin, M.Y.; Aldoshin, S.M. Charge transfer and hydrogen bond energy in glycinium salts. Russ. Chem. Bull. Int. Ed. 2009, 58, 31–40. [Google Scholar] [CrossRef]
- Gálvez, O.; Gómez, P.C.; Pacios, L.F. Variation with the intermolecular distance of properties dependent on the electron density in cyclic dimers with two hydrogen bonds. J. Chem. Phys. 2003, 118, 4878. [Google Scholar] [CrossRef]
- Zou, J.W.; Jiang, Y.-J.; Guo, M.; Hu, G.-X.; Zhang, B.; Liu, H.-C.; Yu, Q.-S. Ab Initio Study of the Complexes of Halogen-Containing Molecules RX (X = Cl, Br, and I) and NH3: Towards Understanding the Nature of Halogen Bonding and the Electron-Accepting Propensities of Covalently Bonded Halogen Atoms. Chem. Eur. J. 2005, 11, 740–751. [Google Scholar] [CrossRef] [PubMed]
- Rozenberg, M.; Loewenschuss, A.; Marcus, Y. An empirical correlation between stretching vibration redshift and hydrogen bond length. Phys. Chem. Chem. Phys. 2000, 2, 2699–2702. [Google Scholar] [CrossRef]
- Pacios, L.F. Change with the Intermolecular Distance of Electron Properties of Hydrogen Bond Dimers at Equilibrium and Non-equilibrium Geometries. Struct. Chem. 2005, 16, 223–241. [Google Scholar] [CrossRef]
- Raissi, H.; Nadim, E.S.; Yoosefian, M.; Farzad, F.; Ghiamati, E.; Nowroozi, A.R.; Fazli, M.; Amoozadeh, A. The effects of substitutions on structure, electron density, resonance and intramolecular hydrogen bonding strength in 3-mercapto-propenethial. J. Mol. Struct. Theochem. 2010, 960, 1–9. [Google Scholar] [CrossRef]
- Roohi, H.; Bagheri, S. Influence of substitution on the strength and nature of CH···N hydrogen bond in XCCH···NH3 complexes. Int. J. Quant. Chem. 2011, 111, 961–969. [Google Scholar] [CrossRef]
- Hayashi, S.; Matsuiwa, K.; Kitamoto, M.; Nakanishi, W. Dynamic Behavior of Hydrogen Bonds from Pure Closed Shell to Shared Shell Interaction Regions Elucidated by AIM Dual Functional Analysis. J. Phys. Chem. A 2013, 117, 1804–1816. [Google Scholar] [CrossRef] [PubMed]
- D’Oria, E.; Novoa, J.J. The strength–length relationship at the light of ab initio computations: Does it really hold? CrystEngComm 2004, 6, 368–376. [Google Scholar] [CrossRef]
- Buralli, G.J.; Petelski, A.N.; Peruchena, N.M.; Sosa, G.L.; Duarte, D.J.R. Multicenter (FX)n/NH3 Halogen Bonds (X = Cl, Br and n = 1–5). QTAIM Descriptors of the Strength of the X…N Interaction. Molecules 2017, 22, 2034. [Google Scholar] [CrossRef] [Green Version]
- Bartashevich, E.V.; Matveychuk, Y.V.; Mukhitdinova, S.E.; Sobalev, S.A.; Khrenova, M.G.; Tsirelson, V.G. The common trends for the halogen, chalcogen, and pnictogen bonds via sorting principles and local bonding properties. Theor. Chem. Acc. 2020, 139, 26. [Google Scholar] [CrossRef]
- Boyd, R.J.; Choi, S.C. Hydrogen bonding between nitriles and hydrogen halides and the topological properties of molecular charge distributions. Chem. Phys. Lett. 1986, 129, 62–65. [Google Scholar] [CrossRef]
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]
- Mata, I.; Molins, E.; Alkorta, I.; Espinosa, E. Effect of an external electric field on the dissociation energy and the electron density properties: The case of the hydrogen bonded dimer HF···HF. J. Chem. Phys. 2009, 130, 044104. [Google Scholar] [CrossRef] [Green Version]
- Espinosa, E.; Alkorta, I.; Elguero, J.; Molins, E. From weak to strong interactions: A comprehensive analysis of the topological and energetic properties of the electron density distribution involving X-H···F–Y systems. J. Chem. Phys. 2002, 117, 5529–5542. [Google Scholar] [CrossRef]
- Parthasarathi, R.; Subramanian, V.; Sathyamurthy, N. Hydrogen bonding without borders: An atoms-in-molecules perspective. J. Phys. Chem. A 2006, 110, 3349–3351. [Google Scholar] [CrossRef]
- Vener, M.V.; Egorova, A.N.; Churakov, A.V.; Tsirelson, V.G. Intermolecular hydrogen bond energies in crystals evaluated using electron density properties: DFT computations with periodic boundary conditions. J. Comput. Chem. 2012, 33, 2303–2309. [Google Scholar] [CrossRef]
- Levina, E.O.; Chernyshov, I.Y.; Voronin, A.P.; Alekseiko, L.N.; Stash, A.I.; Vener, M.V. Solving the enigma of weak fluorine contacts in the solid state: A periodic DFT study of fluorinated organic crystals. RSC Adv. 2019, 9, 12520–12537. [Google Scholar] [CrossRef] [Green Version]
- Ivanov, D.M.; Novikov, A.S.; Ananyev, I.V.; Kirina, Y.V.; Kukushkin, V.Y. Halogen bonding between metal centers and halocarbons. Chem. Commun. 2016, 52, 5565–5568. [Google Scholar] [CrossRef] [Green Version]
- Romanova, A.; Lyssenko, K.; Ananyev, I. Estimations of Energy of Noncovalent Bonding from Integrals over Interatomic Zero-Flux Surfaces: Correlation Trends and Beyond. J. Comput. Chem. 2018, 39, 1607–1616. [Google Scholar] [CrossRef]
- Saleh, G.; Gatti, C.; Presti, L.L.; Contreras-Garca, J. Revealing Non-covalent Interactions in Molecular Crystals through Their Experimental Electron Densities. Chem. Eur. J. 2012, 18, 15523–15536. [Google Scholar] [CrossRef]
- Esrafili, M.D.; Ahmadi, B. A theoretical investigation on the nature of Cl···N and Br···N halogen bonds in F–Ar–X···NCY complexes (X = Cl, Br and Y = H, F, Cl, Br, OH, NH2, CH3 and CN). Comput. Theor. Chem. 2012, 997, 77–82. [Google Scholar] [CrossRef]
- Mata, I.; Alkorta, I.; Molins, E.; Espinosa, E. Universal Features of the Electron Density Distribution in Hydrogen-Bonding Regions: A Comprehensive Study Involving H···X (X = H, C, N, O, F, S, Cl, π) Interactions. Chem. Eur. J. 2010, 16, 2442–2452. [Google Scholar] [CrossRef] [PubMed]
- Emamian, S.; Lu, T.; Kruse, H.; Emamian, H. Exploring Nature and Predicting Strength of Hydrogen Bonds: A Correlation Analysis between Atoms-in-Molecules Descriptors, Binding Energies, and Energy Components of Symmetry-Adapted Perturbation Theory. J. Comput. Chem. 2019, 40, 2868–2881. [Google Scholar] [CrossRef]
- de Oliveira, B.G.; Zabardasti, A.; do Rego, D.G.; Pour, M.M. The formation of H···X hydrogen bond, C···X carbon-halide or Si···X tetrel bonds on the silylene-halogen dimers (X = F or Cl): Intermolecular strength, molecular orbital interactions and prediction of covalency. Theor. Chem. Acc. 2020, 139, 131. [Google Scholar] [CrossRef]
- Boyd, R.J.; Choi, S.C. A bond-length-bond-order relationship for intermolecular interactions based on the topological properties of molecular charge distributions. Chem. Phys. Lett. 1985, 120, 80–85. [Google Scholar] [CrossRef]
- Mó, O.; Yáñez, M.; Elguero, J. Cooperative (nonpairwise) effects in water trimers: An ab initio molecular orbital study. J. Chem. Phys. 1992, 97, 6628. [Google Scholar] [CrossRef]
- Ebrahimi, A.; Khorassani, S.M.H.; Delarami, H. Estimation of individual binding energies in some dimers involving multiple hydrogen bonds using topological properties of electron charge density. Chem. Phys. 2009, 365, 18–23. [Google Scholar] [CrossRef]
- Koch, U.; Popelier, P.L.A. Characterization of C-H-O Hydrogen Bonds on the Basis of the Charge Density. J. Phys. Chem. 1995, 99, 9747–9754. [Google Scholar] [CrossRef]
- Amezaga, N.J.M.; Pamies, S.C.; Peruchena, N.M.; Sosa, G.L. Halogen Bonding: A Study based on the Electronic Charge Density. J. Phys. Chem. A 2010, 114, 552–562. [Google Scholar] [CrossRef]
- Grabowski, S.J. What Is the Covalency of Hydrogen Bonding? Chem. Rev. 2011, 111, 2597–2625. [Google Scholar] [CrossRef]
- Nikolaienko, T.Y.; Bulavina, L.A.; Hovorun, D.M. Bridging QTAIM with vibrational spectroscopy: The energy of intramolecular hydrogen bonds in DNA-related biomolecules. Phys. Chem. Chem. Phys. 2012, 14, 7441–7447. [Google Scholar] [CrossRef]
- Porta, P.D.; Zanasi, R.; Monaco, G. Hydrogen–hydrogen bonding: The current density perspective. J. Comput. Chem. 2015, 36, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Poater, J.; Fradera, X.; Solà, M.; Duran, M.; Simon, S. On the electron-pair nature of the hydrogen bond in the framework of the atoms in molecules theory. Chem. Phys. Lett. 2003, 369, 248–255. [Google Scholar] [CrossRef]
- D’Oria, E.; Novoa, J.J. Cation–Anion Hydrogen Bonds: A New Class of Hydrogen Bonds That Extends Their Strength beyond the Covalent Limit. A Theoretical Characterization. J. Phys. Chem. A 2011, 115, 13114–13123. [Google Scholar] [CrossRef] [PubMed]
- Shi, F.-Q.; An, J.-J.; Yu, J.-Y. Theoretical Study on Measure of Hydrogen Bonding Strength: R–CN···pyrrole Complexes. Chin. J. Chem. 2005, 23, 400–403. [Google Scholar]
- Cubero, E.; Orozco, M.; Hobza, P.; Luque, F.J. Hydrogen Bond versus Anti-Hydrogen Bond: A Comparative Analysis Based on the Electron Density Topology. J. Phys. Chem. A 1999, 103, 6394–6401. [Google Scholar] [CrossRef]
- Parthasarathi, R.; Subramanian, V.; Sathyamurthy, N. Hydrogen Bonding in Phenol, Water, and Phenol−Water Clusters. J. Phys. Chem. A 2005, 109, 843–850. [Google Scholar] [CrossRef]
- Wojtulewski, S.; Grabowski, S.J. Unconventional F–H⋯π hydrogen bonds—ab initio and AIM study. J. Molec. Struct. 2002, 605, 235–240. [Google Scholar] [CrossRef]
- Bagheri, S.; Masoodi, H.R.; Abadi, M.N. Estimation of individual NH···X (X = N, O) hydrogen bonding energies in some complexes involving multiple hydrogen bonds using NBO calculations. Theor. Chem. Acc. 2015, 134, 127. [Google Scholar] [CrossRef]
- Jabłonski, M.; Solà, M. Influence of Confinement on Hydrogen Bond Energy. The Case of the FH···NCH Dimer. J. Phys. Chem. A 2010, 114, 10253–10260. [Google Scholar] [CrossRef]
- Ayoub, A.T.; Tuszynski, J.; Klobukowski, M. Estimating hydrogen bond energies: Comparison of methods. Theor. Chem. Acc. 2014, 133, 1520. [Google Scholar] [CrossRef]
- Brovarets, O.O.; Yurenko, Y.P.; Hovorun, D.M. Intermolecular CH···O/N H-bonds in the biologically important pairs of natural nucleobases: A thorough quantum-chemical study. J. Biomol. Struct. Dyn. 2014, 32, 993–1022. [Google Scholar] [CrossRef]
- Martyniak, A.; Majerz, I.; Filarowski, A. Peculiarities of quasi-aromatic hydrogen bonding. RSC Adv. 2012, 2, 8135–8144. [Google Scholar] [CrossRef]
- Iogansen, A.V. Direct Proportionality of the Hydrogen Bonding Energy and the Intensification of the Stretching ν(XH) Vibration in Infrared Spectra. Spectrochim. Acta A 1999, 55, 1585–1612. [Google Scholar] [CrossRef]
- Alkorta, I.; Legon, A.C. An Ab Initio Investigation of the Geometries and Binding Strengths of Tetrel-, Pnictogen-, and Chalcogen-Bonded Complexes of CO2, N2O, and CS2 with Simple Lewis Bases: Some Generalizations. Molecules 2018, 23, 2250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuznetsov, M.L. Can halogen bond energy be reliably estimated from electron density properties at bond critical point? The case of the (A)nZ Y···X− (X, Y = F, Cl, Br) interactions. Int. J. Quant. Chem. 2019, 119, e25869. [Google Scholar] [CrossRef]
- Kuznetsov, M.L. Relationships between Interaction Energy and Electron Density Properties for Homo Halogen Bonds of the [(A)nY–X···X–Z(B)m] Type (X = Cl, Br, I). Molecules 2019, 24, 2733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spackman, M.A. How Reliable Are Intermolecular Interaction Energies Estimated from Topological Analysis of Experimental Electron Densities? Cryst. Growth Des. 2015, 15, 5624–5628. [Google Scholar] [CrossRef]
- Bartashevich, E.V.; Tsirelson, V.G. Interplay between non-covalent interactions in complexes and crystals with halogen bonds. Russ. Chem. Rev. 2014, 83, 1181–1203. [Google Scholar] [CrossRef]
- Bauzá, A.; Frontera, A. Halogen and Chalcogen Bond Energies Evaluated Using Electron Density Properties. ChemPhysChem 2020, 21, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Gaussian 09; Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. (Eds.) Revision D.01; Gaussian: Wallingford, CT, USA, 2013. [Google Scholar]
- Pritchard, B.P.; Altarawy, D.; Didier, B.; Gibson, T.D.; Windus, T.L. A New Basis Set Exchange: An Open, Up-to-date Resource for the Molecular Sciences Community. J. Chem. Inf. Model. 2019, 59, 4814–4820. [Google Scholar] [CrossRef]
- Jorge, F.E.; Canal Neto, A.; Camiletti, G.G.; Machado, S.F. Contracted Gaussian basis sets for Douglas−Kroll−Hess calculations: Estimating scalar relativistic effects of some atomic and molecular properties. J. Chem. Phys. 2009, 130, 064108. [Google Scholar] [CrossRef]
- Neto, A.C.; Muniz, E.P.; Centoducatte, R.; Jorge, F.E. Gaussian basis sets for correlated wave functions. Hydrogen, helium, first-and second-row atoms. J. Mol. Struct. Theochem. 2005, 718, 219–224. [Google Scholar] [CrossRef]
- Camiletti, G.G.; Machado, S.F.; Jorge, F.E. Gaussian basis set of double zeta quality for atoms K through Kr: Application in DFT calculations of molecular properties. J. Comp. Chem. 2008, 29, 2434–2444. [Google Scholar] [CrossRef]
- Barros, C.L.; De Oliveira, P.J.P.; Jorge, F.E.; Neto, A.C.; Campos, M. Gaussian basis set of double zeta quality for atoms Rb through Xe: Application in non-relativistic and relativistic calculations of atomic and molecular properties. Mol. Phys. 2010, 108, 1965–1972. [Google Scholar] [CrossRef]
- Kozuch, S.; Martin, J.M.L. Halogen bonds: Benchmarks and theoretical analysis. J. Chem. Theory Comput. 2013, 9, 1918–1931. [Google Scholar] [CrossRef] [PubMed]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Simon, S.; Duran, M.; Dannenberg, J.J.J. How does basis set superposition error change the potential surfaces for hydrogen-bonded dimers? Chem. Phys. 1996, 105, 11024–11031. [Google Scholar] [CrossRef] [Green Version]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Keith, T.A.; Gristmill, T.K. AIMAll; Version 14.10.27; Gristmill Software: Overland Park, KS, USA, 2014; Available online: Aim.tkgristmill.com (accessed on 1 September 2015).
- Burns, L.A.; Marshall, M.S.; Sherrill, C.D. Comparing Counterpoise-Corrected, Uncorrected, and Averaged Binding Energies for Benchmarking Noncovalent Interactions. J. Chem. Theor. Comput. 2014, 10, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Hiraoka, K.; Mizuno, T.; Iino, T.; Eguchi, D.; Yamabe, S. Characteristic Changes of Bond Energies for Gas-Phase Cluster Ions of Halide Ions with Methane and Chloromethanes. J. Phys. Chem. A 2001, 105, 4887–4893. [Google Scholar] [CrossRef]
- Wenthold, P.G.; Squires, R.R. Bond dissociation energies of F2− and HF2−. A gas-phase experimental and G2 theoretical study. J. Chem. Phys. 1995, 99, 2002–2005. [Google Scholar]
- Keesee, R.G.; Castleman, A.W., Jr. Gas-phase studies of hydration complexes of Cl− and I− and comparison to electrostatic calculations in the gas phase. Chem. Phys. Lett. 1980, 74, 139–142. [Google Scholar] [CrossRef]
- Do, K.; Klein, T.P.; Pommerening, C.A.; Sunderlin, L.S. A new flowing afterglow-guided ion beam tandem mass spectrometer. Applications to the thermochemistry of polyiodide ions. J. Am. Soc. Mass Spectrom. 1997, 8, 688–696. [Google Scholar] [CrossRef] [Green Version]
- Medvedev, M.G.; Bushmarinov, I.S.; Sun, J.; Perdew, J.P.; Lyssenko, K.A. Density functional theory is straying from the path toward the exact functional. Science 2017, 355, 49–52. [Google Scholar] [CrossRef]
- Bertolotti, F.; Shishkina, A.V.; Forni, A.; Gervasio, G.; Stash, A.I.; Tsirelson, V.G. Intermolecular Bonding Features in Solid Iodine. Cryst. Growth Des. 2014, 14, 3587–3595. [Google Scholar] [CrossRef]
- Wang, R.; Dols, T.S.; Lehmann, C.W.; Englert, U. Charge Density of Intra- and Intermolecular Halogen Contacts. Z. Anorg. Allg. Chem. 2013, 639, 1933–1939. [Google Scholar] [CrossRef]
- Hathwar, V.R.; Row, T.N.G. Nature of Cl···Cl Intermolecular Interactions via Experimental and Theoretical Charge Density Analysis: Correlation of Polar Flattening Effects with Geometry. J. Phys. Chem. A 2010, 114, 13434–13441. [Google Scholar] [CrossRef]
- Wang, A.; Wang, R.; Kalf, I.; Dreier, A.; Lehmann, C.W.; Englert, U. Charge-Assisted Halogen Bonds in Halogen-Substituted Pyridinium Salts: Experimental Electron Density. Cryst. Growth Des. 2017, 17, 2357–2364. [Google Scholar] [CrossRef]
- Hathwar, V.R.; Gonnade, R.G.; Munshi, P.; Bhadbhade, M.M.; Row, T.N.G. Halogen Bonding in 2,5-Dichloro-1,4-benzoquinone: Insights from Experimental and Theoretical Charge Density Analysis. Cryst. Growth Des. 2011, 11, 1855–1862. [Google Scholar] [CrossRef] [Green Version]
- Brezgunova, M.E.; Aubert, E.; Dahaoui, S.; Fertey, P.; Lebègue, S.; Jelsch, C.; Ángyán, J.G.; Espinosa, E. Charge Density Analysis and Topological Properties of Hal3-Synthons and Their Comparison with Competing Hydrogen Bonds. Cryst. Growth Des. 2012, 12, 5373–5386. [Google Scholar] [CrossRef]
- Merkens, C.; Pan, F.; Englert, U. 3-(4-Pyridyl)-2,4-pentanedione—A bridge between coordinative, halogen, and hydrogen bonds. CrystEngComm 2013, 15, 8153–8158. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Interaction | Estimator | Relationship | Reference |
|---|---|---|---|
| H···O | Vb | Eint ~ 0.5Vb | [82] |
| FH···FR | Gb | Eint ~ −0.429Gb | [83,86] |
| Vb | Eint ~ 0.37Vb − 3.1 | [83] | |
| Cl···X a | Vb | Eint ~ 0.49Vb | [119] |
| Gb | Eint ~ −0.47Gb | [119] | |
| Br···X a | Vb | Eint ~ 0.58Vb | [119] |
| Vb | Eint ~ 0.375Vb − 0.57 | [120] | |
| Gb | Eint ~ −0.57Gb | [119] | |
| I···X a | Vb | Eint ~ 0.68Vb | [119] |
| Vb | Eint ~ 0.556Vb + 0.64 | [120] | |
| Gb | Eint ~ −0.67Gb | [119] | |
| F···F | Gb | Eint ~ −0.129Gb | [87] |
| Cl···Cl | Vb | Eint ~ −0.1006Vb2 − 0.218Vb − 0.55 | [117] |
| Gb | Eint ~ −0.0841Gb2 + 0.367Gb − 0.84 | [117] | |
| Br···Br | Vb | Eint ~ −0.0926Vb2 − 0.173Vb − 0.16 | [117] |
| Gb | Eint ~ −0.1178Gb2 + 0.73Gb − 1.5 | [117] | |
| I···I | Vb | Eint ~ −0.0635Vb2 − 0.217Vb − 0.25 | [117] |
| Gb | Eint ~ −0.1564Gb2 + 1.138Gb − 2.25 | [117] |
| Structure | Method | ΔH a CP/no-CP b |
|---|---|---|
| [H3C–H···F]− | M06-2X/6-31+G* | −6.8/−7.1 |
| CCSD/6-31+G* | −4.2/−5.1 | |
| CCSD(T)//CCSD/6-31+G* c | −4.3/−5.4 | |
| CCSD/6-311+G** | −4.7/−5.8 | |
| CCSD/aug-cc-pVTZ//6-31+G* d | −6.0/−6.4 | |
| CCSD(T)/aug-cc-pVTZ//CCSD/6-31+G* e | −6.3/−6.8 | |
| exp. f | −6.7 ± 0.2 | |
| [F–H–F]− | M06-2X/6-31+G* | −50.2/−50.7 |
| CCSD/6-31+G* | −42.7/−44.8 | |
| CCSD(T)//CCSD/6-31+G* c | −42.8/−45.2 | |
| CCSD/6-311+G** | −42.3/−46.5 | |
| CCSD/aug-cc-pVTZ//6-31+G* d | −43.7/−45.1 | |
| CCSD(T)/aug-cc-pVTZ//CCSD/6-31+G* e | −43.9/−45.4 | |
| CCSD/aug-cc-pVTZ | −44.8/−46.3 | |
| CCSD(T)//CCSD/aug-cc-pVTZ c | −45.0/−46.6 | |
| exp. g | −45.8 ± 1.6 | |
| [HO–H···Cl]− | M06-2X/6-31+G* | −15.0/−15.2 |
| CCSD/6-31+G* | −12.3/−14.2 | |
| CCSD(T)//CCSD/6-31+G* c | −12.5/−14.5 | |
| CCSD/aug-cc-pVTZ//6-31+G* d | −13.3/−13.9 | |
| CCSD(T)/aug-cc-pVTZ//CCSD/6-31+G* e | −13.8/−14.4 | |
| CCSD/aug-cc-pVTZ | −13.5/−14.2 | |
| CCSD(T)//CCSD/aug-cc-pVTZ c | −14.0/−14.8 | |
| exp. h | −14.9 ± 0.2 | |
| [HO–H···I]− | M06-2X/ADZP–DKH//ADZP i | −10.3/−14.0 |
| CCSD(T)/aug-cc-pVTZ(PP)//CCSD/ADZP j | −8.1/−11.0 | |
| exp. h | −11.1 ± 0.1 | |
| [H3C–H···I]− | M06-2X/ADZP–DKH//ADZP i | −1.6/−3.1 |
| CCSD(T)/aug-cc-pVTZ(PP)//CCSD/ADZP j | −0.4/−2.4 | |
| exp. f | −2.6 ± 0.2 | |
| [I–I–I]− | M06-2X/ADZP–DKH//ADZP k | −29.3/−36.6 |
| CCSD(T)/aug-cc-pVTZ(PP)//CCSD/ADZP k | −27.3/−30.6 | |
| exp. l,m | −30.1 ± 1.4 |
| Ref. Code | Contact | ρb,theor (M06-2X) | ρb,theor (PBE0-D3BJ) | ρb,exp [Ref.] |
|---|---|---|---|---|
| ICSD 194468 | I···I | 0.097 | 0.102 | 0.101 [139] |
| ETUDUT01 | Cl···Cl | 0.052 | 0.050 | 0.048(2) [140] |
| IJIGOU | Cl···Cl | 0.033 | 0.032 | 0.03 [141] |
| FUFNOJ02 | Cl···Cl | 0.053 | 0.051 | 0.05 [141] |
| IJIHAL | Cl···Cl | 0.045 | 0.044 | 0.03 [141] |
| CIHBAX01 | Br···Cl | 0.079 | 0.078 | 0.081(2) [142] |
| BZQDCL11 | Cl···O | 0.055 | 0.054 | 0.054(1) [143] |
| PCPHOL01 | Cl···Cl | 0.050 | 0.048 | 0.058 [144] |
| ROFKAZ01 | Br···Br | 0.061 | 0.060 | 0.063 [144] |
| XIPRUL | I···N | 0.174 | 0.176 | 0.154(12) [145] |
| XIPRUL | I···O | 0.099 | 0.105 | 0.092(7) [145] |
| Series | Vb | Gb | ρb | d(Y···X) | ||||
|---|---|---|---|---|---|---|---|---|
| R2 | MAD | R2 | MAD | R2 | MAD | R2 | MAD | |
| [(A)nZ–I···F]− | 0.91 | 4.76 | 0.92 | 4.66 | 0.91 | 4.68 | 0.91 | 4.77 |
| [(A)nZ–Cl···F]− a | 0.95 | 3.36 | 0.95 | 3.53 | 0.94 b | 3.59 b | 0.94 | 3.88 |
| [(A)nZ–Br···F]− a | 0.94 | 3.83 | 0.95 | 3.29 | 0.94 b | 3.73 b | 0.93 | 4.00 |
| [(A)nZ–I···Cl]− | 0.95 | 2.43 | 0.96 | 2.34 | 0.95 | 2.49 | 0.95 | 2.55 |
| [(A)nZ–Cl···Cl]− a | 0.95 | 2.06 | 0.95 | 2.08 | 0.95 | 2.15 | 0.95 | 2.17 |
| [(A)nZ–Br···Cl]− a | 0.94 | 2.73 | 0.94 | 2.63 | 0.93 | 3.01 | 0.94 | 2.78 |
| [(A)nZ–Cl···Br]− a | 0.96 | 2.17 | 0.95 | 2.31 | 0.95 | 2.35 | 0.96 | 2.33 |
| [(A)nZ–Br···Br]− a | 0.94 | 2.75 | 0.94 | 2.77 | 0.93 | 3.01 | 0.93 | 2.89 |
| [(A)nZ–I···Br]− | 0.95 | 2.22 | 0.96 | 2.17 | 0.95 | 2.23 | 0.95 | 2.29 |
| [(A)nZ–I···I]− | 0.96 | 2.34 | 0.95 | 2.48 | 0.96 | 2.21 | 0.96 | 2.27 |
| [(A)nZ–Br···I]− | 0.95 | 2.70 | 0.95 | 2.66 | 0.96 | 2.41 | 0.96 | 2.50 |
| Series | Estimator | Equation | R2 | MAD |
|---|---|---|---|---|
| [(A)nZ–I···F]− | Vb | −Eint = −0.77Vb − 6.50 | 0.91 | 4.76 |
| Gb | −Eint = 1.04Gb − 8.97 | 0.92 | 4.66 | |
| ρb | −Eint = 150.4ρb2 − 30.29ρb + 12.69 | 0.91 | 4.68 | |
| dY···X | −Eint = 5.28 × 104e−3.327d | 0.91 | 4.77 | |
| [(A)nZ–I···Cl]− | Vb | −Eint = −1.46Vb − 1.65 | 0.95 | 2.43 |
| Gb | −Eint = 0.121Gb2 − 0.57Gb + 5.77 | 0.96 | 2.34 | |
| Hb | −Eint = −2.75Hb + 14.80 a | 0.95 | 2.43 | |
| ρb | −Eint = 145.6ρb2 + 46.09ρb + 1.50 | 0.95 | 2.49 | |
| λ||,b | −Eint = 3.69e0.518λ||,b + 2 × 10−4e3.054λ||,b | 0.95 | 2.51 | |
| dY···X | −Eint = 4.28 × 104e−2.650d | 0.95 | 2.55 | |
| [(A)nZ–I···Br]− | Vb | −Eint = −2.12Vb − 3.88 | 0.95 | 2.22 |
| Gb | −Eint = 0.212Gb2 − 0.77Gb + 4.88 | 0.96 | 2.17 | |
| Hb | −Eint = −4.45Hb + 14.52 a | 0.95 | 2.08 | |
| ρb | −Eint = 197.6ρb2 + 56.42ρb + 0.11 | 0.95 | 2.23 | |
| λ||,b | −Eint = 2.51e0.867λ||,b + 1.3 × 10−8e6.390λ||,b | 0.96 | 2.06 | |
| dY···X | −Eint = 7.21 × 104e−2.688d | 0.95 | 2.29 | |
| [(A)nZ–I···I]− | Vb | −Eint = −2.34Vb − 2.56 | 0.96 | 2.34 |
| Gb | −Eint = 0.360Gb2 − 2.05Gb + 7.87 | 0.95 | 2.48 | |
| Hb | −Eint = −4.38Hb + 16.60 a | 0.96 | 2.07 | |
| ρb | −Eint = 232.2ρb2 + 54.11ρb + 0.49 | 0.96 | 2.21 | |
| dY···X | −Eint = 1.30 × 105e−2.746d | 0.96 | 2.27 | |
| [(A)nZ–Cl···I]− | Hb | −Eint = 0.0513Hb2 − 1.41Hb + 9.60 a | 0.95 | 3.33 |
| dY···X | −Eint = 1.4 × 1010e−8.078d + 2.21 × 103e−1.834d | 0.96 | 2.70 | |
| [(A)nZ–Br···I]− | Vb | −Eint = −1.95Vb − 1.92 | 0.95 | 2.70 |
| Gb | −Eint = 0.248Gb2 − 1.39Gb + 6.48 | 0.95 | 2.66 | |
| Hb | −Eint = −3.74Hb + 14.81 a | 0.96 | 2.31 | |
| ρb | −Eint = 235.6ρb2 + 13.34ρb + 2.68 | 0.96 | 2.41 | |
| dY···X | −Eint = 1.38 × 105e−2.957d | 0.96 | 2.50 | |
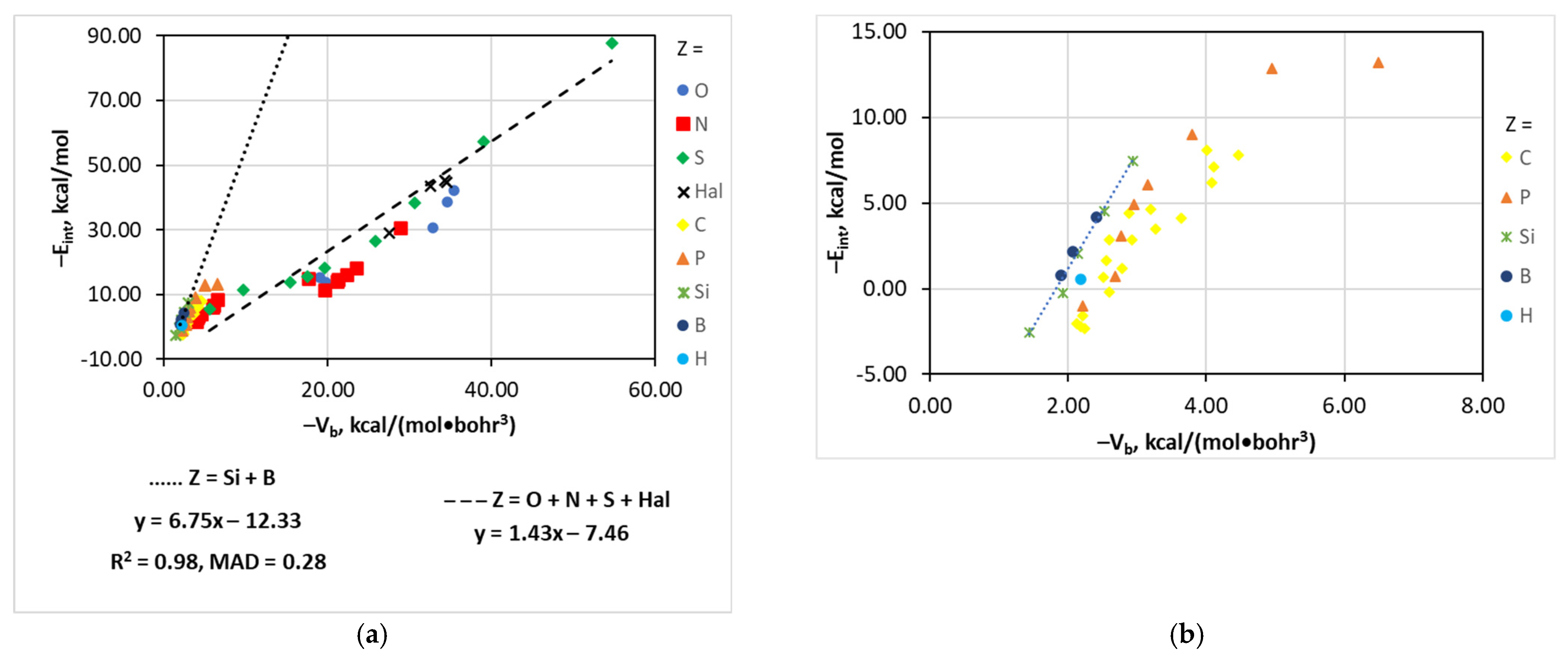
| [(A)nP–Cl···I]− | Vb | −Eint = 14.92 − 68.71e0.639Vb | 0.97 | 0.64 |
| [(A)nC–Cl···I]− | Vb | −Eint = 9.84 − 57.41e0.725Vb | 0.92 | 0.77 |
| [(A)nSi,B–Cl···I]− | Vb | −Eint = −6.75Vb − 12.33 | 0.98 | 0.28 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuznetsov, M.L. Strength of the [Z–I···Hal]− and [Z–Hal···I]− Halogen Bonds: Electron Density Properties and Halogen Bond Length as Estimators of Interaction Energy. Molecules 2021, 26, 2083. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26072083
Kuznetsov ML. Strength of the [Z–I···Hal]− and [Z–Hal···I]− Halogen Bonds: Electron Density Properties and Halogen Bond Length as Estimators of Interaction Energy. Molecules. 2021; 26(7):2083. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26072083
Chicago/Turabian StyleKuznetsov, Maxim L. 2021. "Strength of the [Z–I···Hal]− and [Z–Hal···I]− Halogen Bonds: Electron Density Properties and Halogen Bond Length as Estimators of Interaction Energy" Molecules 26, no. 7: 2083. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26072083