
Ursolic Acid Lactone Obtained from Eucalyptus tereticornis Increases Glucose Uptake and Reduces Inflammatory Activity and Intracellular Neutral Fat: An In Vitro Study

 , , and
, , and
Abstract
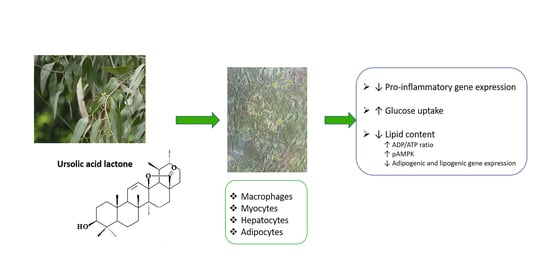
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Effect of UAL on J774.A1 Macrophage Cell Line
2.2. Effect of UAL on C2C12 Myocyte Cell Line
2.3. Effect of UAL on HepG2 Hepatocyte Cell Line
2.4. Effect of UAL on 3T3-L1 Adipocyte Cell Line
3. Discussion
4. Materials and Methods
4.1. Ursolic Acid Lactone (3β-hydroxy-urs-11-en-28,13β-olide) Purification
4.2. Cell Cultures
4.3. Macrophage Cell Culture and Activation
4.4. Adipocyte Cell Culture and Differentiation
4.5. Myotube Cell Culture and Differentiation
4.6. Hepatocyte Cell Culture and Steatosis Induction
4.7. Cell Viability
4.8. Determination of Oxidative Stress in J774.A1 and 3T3-L1 Cells
4.9. RNA Extraction and Real-Time PCR in J774.A1 and 3T3-L1 Cells
4.10. Measurement of Glucose Concentration in C2C12 Cells
4.11. Measurement of Triacylglycerol Concentration in 3T3-L1 and HepG2 Cells
4.12. Quantification of Adipokines in 3T3-L1 Cells
4.13. Mitochondrial Membrane Potential in 3T3-L1
4.14. Determination of 3T3-L1 Cellular ADP/ATP Ratio
4.15. Western Blot Analysis
4.16. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- de Freitas Junior, L.M.; de Almeida, E.B. Medicinal plants for the treatment of obesity: Ethnopharmacological approach and chemical and biological studies. Am. J. Transl. Res. 2017, 9, 2050. [Google Scholar] [PubMed]
- International Diabetes Federation. IDF Diabetes Atla, 9th ed.; International Diabetes Federation: Brussels, Belgium, 2019; Volume 2019. [Google Scholar]
- Schnurr, T.M.; Jakupović, H.; Carrasquilla, G.D.; Ängquist, L.; Grarup, N.; Sørensen, T.I.A.; Tjønneland, A.; Overvad, K.; Pedersen, O.; Hansen, T.; et al. Obesity, unfavourable lifestyle and genetic risk of type 2 diabetes: A case-cohort study. Diabetologia 2020. [Google Scholar] [CrossRef] [PubMed]
- Smith, U.; Kahn, B.B. Adipose tissue regulates insulin sensitivity: Role of adipogenesis, de novo lipogenesis and novel lipids. J. Intern. Med. 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ríos, J.L.; Francini, F.; Schinella, G.R. Natural products for the treatment of type 2 diabetes mellitus. Planta Med. 2015, 81, 975–994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.; Li, Y.; Dai, Y.; Peng, J. Natural products for the treatment of type 2 diabetes mellitus: Pharmacology and mechanisms. Pharmacol. Res. 2018, 130, 451–465. [Google Scholar] [CrossRef] [PubMed]
- Alqahtani, A.; Hamid, K.; Kam, A.; Wong, K.H.; Abdelhak, Z.; Razmovski-Naumovski, V.; Chan, K.; Li, K.M.; Groundwater, P.W.; Li, G.Q. The pentacyclic triterpenoids in herbal medicines and their pharmacological activities in diabetes and diabetic complications. Curr. Med. Chem. 2013. [Google Scholar] [CrossRef]
- Furtado, N.A.J.C.; Pirson, L.; Edelberg, H.; Miranda, L.M.; Loira-Pastoriza, C.; Preat, V.; Larondelle, Y.; André, C.M. Pentacyclic triterpene bioavailability: An overview of in vitro and in vivo studies. Molecules 2017, 4, 400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dey, B.; Mitra, A. Chemo-profiling of eucalyptus and study of its hypoglycemic potential. World J. Diabetes 2013. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Rana, S.; Singh, A. A review on phytochemistry and pharmacological profile of Eucalyptus globulus. Int. J. Pharm. Res. 2020, 11, 30–36. [Google Scholar]
- Guillén, A.; Granados, S.; Rivas, K.E.; Estrada, O.; Echeverri, L.F.; Balcázar, N. Antihyperglycemic activity of eucalyptus tereticornis in insulin-resistant cells and a nutritional model of diabetic mice. Adv. Pharmacol. Sci. 2015, 2015, 418693. [Google Scholar]
- Ceballos, S.; Guilí En, A.; Lorena, D.; Noz, M.; Castã, A.; Echeverri, L.F.; Acín, S.; Bal, N.; Bal Azar, N. Immunometabolic regulation by triterpenes of Eucalyptus tereticornis in adipose tissue cell line models. Phytomedicine 2018. [Google Scholar] [CrossRef] [PubMed]
- Acín, S.; Muñoz, D.L.; Guillen, A.; Soscue, D.; Castaño, A.; Echeverri, F.; Balcazar, N. Triterpene-enriched fractions from Eucalyptus tereticornis ameliorate metabolic alterations in a mouse model of diet-induced obesity. J. Ethnopharmacol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Seo, D.Y.; Lee, S.R.; Heo, J.W.; No, M.H.; Rhee, B.D.; Ko, K.S.; Kwak, H.B.; Han, J. Ursolic acid in health and disease. Korean J. Physiol. Pharmacol. 2018, 22, 235–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramírez-Rodríguez, A.M.; González-Ortiz, M.; Martínez-Abundis, E.; Acuña Ortega, N. Effect of ursolic acid on metabolic syndrome, insulin sensitivity, and inflammation. J. Med. Food 2017. [Google Scholar] [CrossRef] [PubMed]
- Castellano, J.M.; Guinda, A.; Delgado, T.; Rada, M.; Cayuela, J.A. Biochemical basis of the antidiabetic activity of oleanolic acid and related pentacyclic triterpenes. Diabetes 2013, 62, 1791–1799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayeleso, T.B.; Matumba, M.G.; Mukwevho, E. Oleanolic acid and its derivatives: Biological activities and therapeutic potential in chronic diseases. Molecules 2017, 22, 1915. [Google Scholar] [CrossRef] [Green Version]
- Maurya, A.; Srivastava, S.K. Determination of ursolic acid and ursolic acid lactone in the leaves of eucalyptus tereticornis by hplc. J. Braz. Chem. Soc. 2012, 23, 468–472. [Google Scholar] [CrossRef] [Green Version]
- Zatterale, F.; Longo, M.; Naderi, J.; Raciti, G.A.; Desiderio, A.; Miele, C.; Beguinot, F. Chronic adipose tissue inflammation linking obesity to insulin resistance and type 2 diabetes. Front. Physiol. 2020, 10, 1607. [Google Scholar] [CrossRef] [PubMed]
- Ebbert, J.O.; Jensen, M.D. Fat depots, free fatty acids, and dyslipidemia. Nutrients 2013, 5, 498–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marica Bakovic, N.H. Biologically active triterpenoids and their cardioprotective and anti- inflammatory effects. J. Bioanal. Biomed. 2015. [Google Scholar] [CrossRef] [Green Version]
- Mlala, S.; Oyedeji, A.O.; Gondwe, M.; Oyedeji, O.O. Ursolic acid and its derivatives as bioactive agents. Molecules 2019, 24, 2751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bano, Z.; Begum, S.; Ali, S.S.; Kiran, Z.; Siddiqui, B.S.; Ahmed, A.; Khawaja, S.; Fatima, F.; Jabeen, A. Phytochemicals from Carissa carandas with potent cytotoxic and anti-inflammatory activities. Nat. Prod. Res. 2021, 1–6. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Li, Y.; Zhao, T.; Wang, Y.; Sun, C. Ursolic acid inhibits adipogenesis in 3T3-L1 adipocytes through LKB1/AMPK pathway. PLoS ONE 2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Li, X.; Chen, J.; Chen, T.; Shi, Z.; Lei, M.; Zhang, Y.; Bai, P.; Li, Y.; Fei, X. The pentacyclic triterpene Lupeol switches M1 macrophages to M2 and ameliorates experimental inflammatory bowel disease. Int. Immunopharmacol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zheng, H.; Sui, Z.; Jing, F.; Quan, X.; Zhao, W.; Liu, G. Ursolic acid exhibits anti-inflammatory effects through blocking TLR4-MyD88 pathway mediated by autophagy. Cytokine 2019. [Google Scholar] [CrossRef] [PubMed]
- Yadav, V.R.; Prasad, S.; Sung, B.; Kannappan, R.; Aggarwal, B.B. Targeting inflammatory pathways by triterpenoids for prevention and treatment of cancer. Toxins 2010, 2, 2428–2466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-García, S.; Castañeda-Sanchez, J.I.; Jiménez-Arellanes, A.; Domínguez-López, L.; Castro-Mussot, M.E.; Hernández-Sanchéz, J.; Luna-Herrera, J. Macrophage activation by ursolic and oleanolic acids during mycobacterial infection. Molecules 2015, 20, 14348–14364. [Google Scholar] [CrossRef] [Green Version]
- Podder, B.; Jang, W.S.; Nam, K.W.; Lee, B.E.; Song, H.Y. Ursolic acid activates intracellular killing effect of macrophages during mycobacterium tuberculosis infection. J. Microbiol. Biotechnol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Starkov, A.A. The role of mitochondria in reactive oxygen species metabolism and signaling. Proc. Ann. N. Y. Acad. Sci. 2008, 1147, 37–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, S.; Peng, Y.; Jiang, P.; Wang, M.; Fan, M.; Wang, X.; Zhou, H.; Li, H.; Yan, Q.; Huang, T.; et al. A deafness-associated tRNAHis mutation alters the mitochondrial function, ROS production and membrane potential. Nucleic Acids Res. 2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rendra, E.; Riabov, V.; Mossel, D.M.; Sevastyanova, T.; Harmsen, M.C.; Kzhyshkowska, J. Reactive oxygen species (ROS) in macrophage activation and function in diabetes. Immunobiology 2019, 224, 242–253. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Thuong, P.T. Stimulation of glucose uptake by triterpenoids from Weigela subsessilis. Phyther. Res. 2010. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Hong, D.; Zhou, Y.; Zhang, Y.; Shen, Q.; Li, J.Y.; Li, J.-Y.; Hu, L.-H.; Li, J. Ursolic acid and its derivative inhibit protein tyrosine phosphatase 1B, enhancing insulin receptor phosphorylation and stimulating glucose uptake. Biochim. Biophys. Acta Gen. Subj. 2006. [Google Scholar] [CrossRef] [PubMed]
- Chiang, D.J.; Pritchard, M.T.; Nagy, L.E. Obesity, diabetes mellitus, and liver fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, G697–G702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, H.; Kumar, P.; Deshmukh, R.R.; Bishayee, A.; Kumar, S. Pentacyclic triterpenes: New tools to fight metabolic syndrome. Phytomedicine 2018, 50, 166–177. [Google Scholar] [CrossRef] [PubMed]
- Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Babenko, V.A.; Zorov, S.D.; Balakireva, A.V.; Juhaszova, M.; et al. Mitochondrial membrane potential. Anal. Biochem. 2018. [Google Scholar] [CrossRef]
- Garcia, D.; Shaw, R.J. AMPK: Mechanisms of cellular energy sensing and restoration of metabolic balance. Mol. Cell 2017, 66, 789–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Deng, Y.; Hu, X.; Ren, H.; Zhu, J.; Fu, S.; Xie, J.; Peng, Y. miR-128-3p regulates 3T3-L1 adipogenesis and lipolysis by targeting Pparg and Sertad2. J. Physiol. Biochem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.C.; Kim, J.S.; Kim, G.M.; Choi, S.Y. Anti-adipogenic effects of Tropaeolum majus (nasturtium) ethanol extract on 3T3-L1 cells. Food Nutr. Res. 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Y.; Luo, N.; Klein, R.L.; Garvey, W.T. Adiponectin promotes adipocyte differentiation, insulin sensitivity, and lipid accumulation. J. Lipid Res. 2005, 46, 1369–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barberá, M.J.; Schlüter, A.; Pedraza, N.; Iglesias, R.; Villarroya, F.; Giralt, M. Peroxisome proliferator-activated receptor α activates transcription of the brown fat uncoupling protein-1 gene. A link between regulation of the thermogenic and lipid oxidation pathways in the brown fat cell. J. Biol. Chem. 2001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corrales, P.; Vidal-Puig, A.; Medina-Gómez, G. PPARS and metabolic disorders associated with challenged adipose tissue plasticity. Int. J. Mol. Sci. 2018, 19, 2124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez-Pérez, A.; Sánchez-Jiménez, F.; Vilariño-García, T.; Sánchez-Margalet, V. Role of leptin in inflammation and vice versa. Int. J. Mol. Sci. 2020, 21, 5887. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balcazar, N.; Betancur, L.I.; Muñoz, D.L.; Cabrera, F.J.; Castaño, A.; Echeverri, L.F.; Acin, S. Ursolic Acid Lactone Obtained from Eucalyptus tereticornis Increases Glucose Uptake and Reduces Inflammatory Activity and Intracellular Neutral Fat: An In Vitro Study. Molecules 2021, 26, 2282. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26082282
Balcazar N, Betancur LI, Muñoz DL, Cabrera FJ, Castaño A, Echeverri LF, Acin S. Ursolic Acid Lactone Obtained from Eucalyptus tereticornis Increases Glucose Uptake and Reduces Inflammatory Activity and Intracellular Neutral Fat: An In Vitro Study. Molecules. 2021; 26(8):2282. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26082282
Chicago/Turabian StyleBalcazar, Norman, Laura I. Betancur, Diana L. Muñoz, Frankly J. Cabrera, Adriana Castaño, Luis F. Echeverri, and Sergio Acin. 2021. "Ursolic Acid Lactone Obtained from Eucalyptus tereticornis Increases Glucose Uptake and Reduces Inflammatory Activity and Intracellular Neutral Fat: An In Vitro Study" Molecules 26, no. 8: 2282. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26082282