
Pharmaceutical Drug Metformin and MCL1 Inhibitor S63845 Exhibit Anticancer Activity in Myeloid Leukemia Cells via Redox Remodeling

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
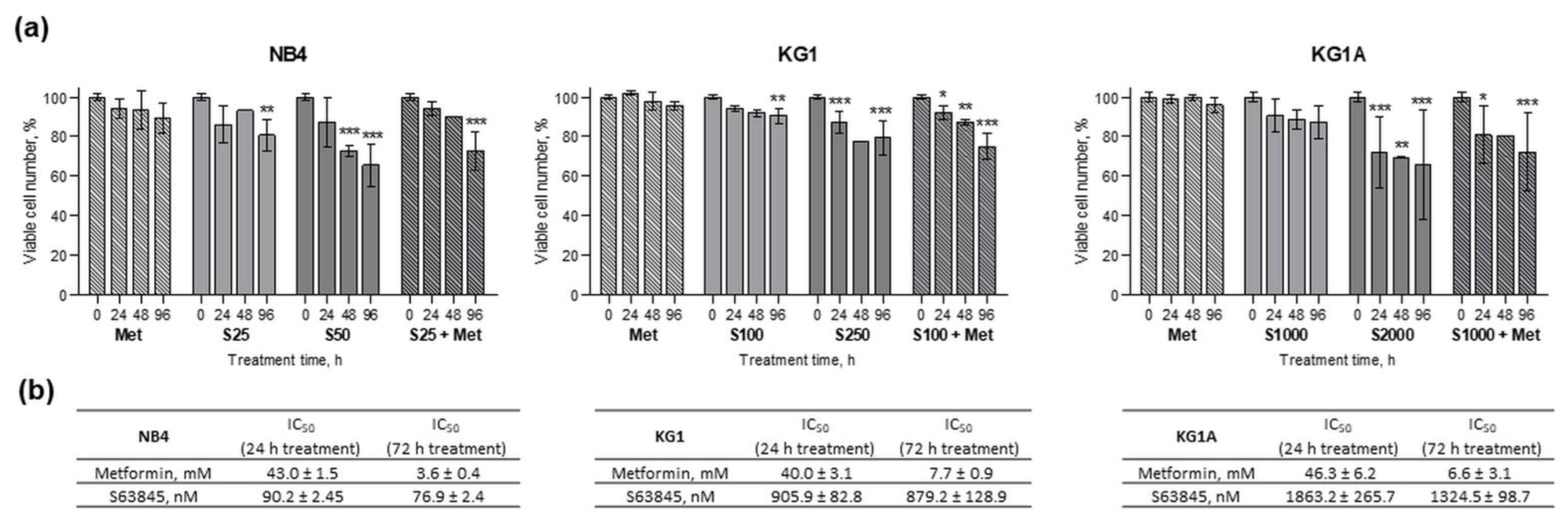
2.1. Metformin and S63845 Cytotoxicity and Effect on AML Cell Proliferation and Viability
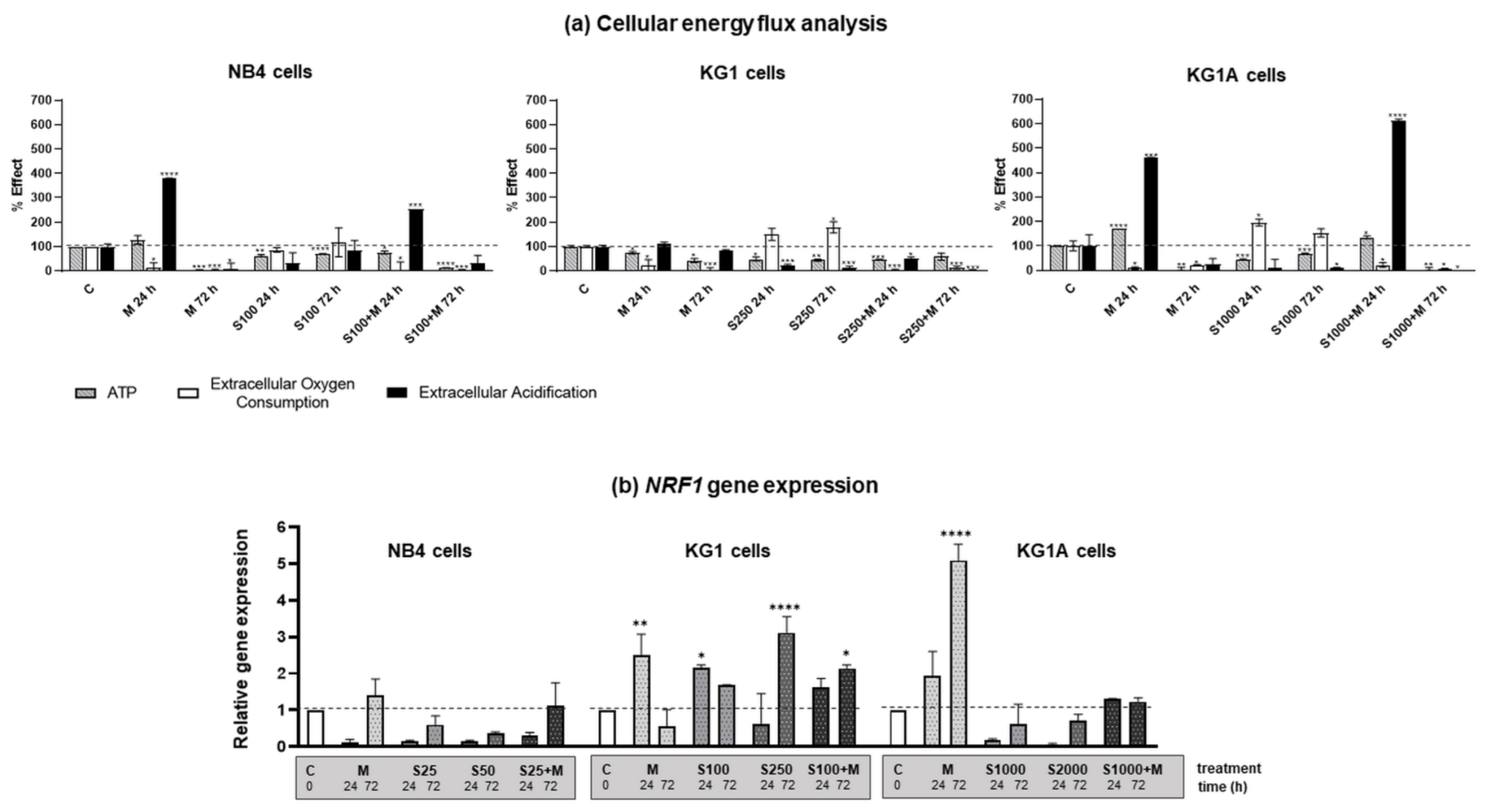
2.2. Energetic Profile Regulation by Metformin and S63845 in AML Cells
2.3. Effect of Metformin and S63845 on AML Cell Redox Modulation
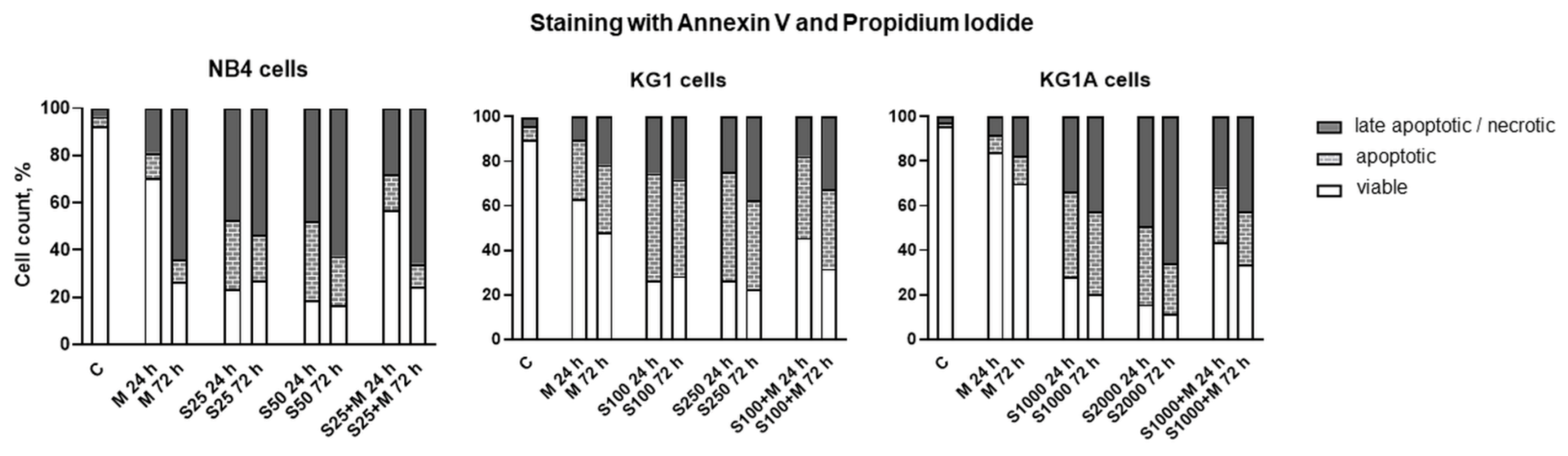
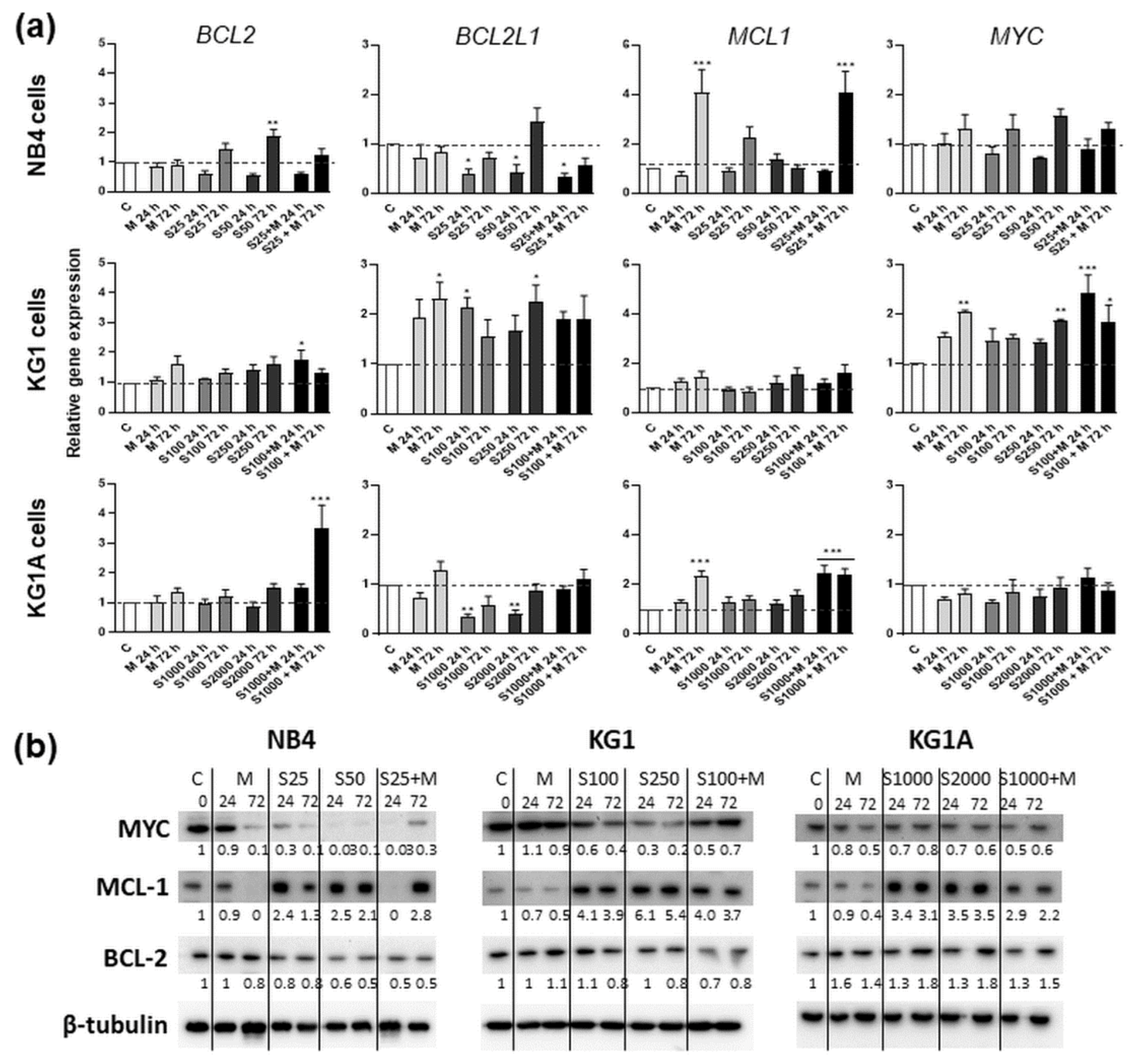
2.4. Treatment-Induced Apoptosis and Apoptosis-Related Gene and Protein Expression
3. Discussion
4. Materials and Methods
4.1. Cell Cultivation and Treatment
4.2. Apoptosis Evaluation
4.3. Cellular Energy Flux Measurement
4.4. Assessment of Mitochondrial Membrane Potential
4.5. Cellular ROS Measurement
4.6. Gene Expression Analysis by RT-qPCR
4.7. Immunoblotting
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Howlader, N.; Noone, A.M.; Krapcho, M.; Miller, D.; Brest, A.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; Lewis, D.R.; et al. (Eds.) SEER Cancer Statistics Review, 1975–2017; Based on November 2019 SEER Data Submission, Posted to the SEER Web Site; National Cancer Institute: Bethesda, MD, USA, 2020. Available online: https://seer.cancer.gov/csr/1975_2017/ (accessed on 18 January 2021).
- Hackl, H.; Astanina, K.; Wieser, R. Molecular and genetic alterations associated with therapy resistance and relapse of acute myeloid leukemia. J. Hematol. Oncol. 2017, 10, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, C.; Retzik-Stahr, C.; Singh, V.; Plomondon, R.; Anderson, V.; Rasouli, N. Should metformin remain the first-line therapy for treatment of type 2 diabetes? Ther. Adv. Endocrinol. Metab. 2021, 12, 2042018820980225. [Google Scholar] [CrossRef] [PubMed]
- Fruzzetti, F.; Perini, D.; Russo, M.; Bucci, F.; Gadducci, A. Comparison of two insulin sensitizers, metformin and myo-inositol, in women with polycystic ovary syndrome (PCOS). Gynecol. Endocrinol. 2017, 33, 39–42. [Google Scholar] [CrossRef] [PubMed]
- Schein, C.H. Repurposing approved drugs for cancer therapy. Br. Med Bull. 2021, 137, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.-Y.; Liu, Z.; Bi, M.-H.; Zhang, J.-J.; Han, Z.-Q.; Han, X.; Wang, H.-Y.; Sun, G.-P.; Liu, H. Metformin induces apoptosis via a mitochondria-mediated pathway in human breast cancer cells in vitro. Exp. Ther. Med. 2016, 11, 1700–1706. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Zhou, P.; Xu, K.; Chen, T.; Jiao, J.; Wei, H.; Yang, X.; Xu, W.; Wan, W.; Xiao, J. Metformin induces cell cycle arrest, apoptosis and autophagy through ROS/JNK signaling pathway in human osteosarcoma. Int. J. Biol. Sci. 2020, 16, 74–84. [Google Scholar] [CrossRef] [Green Version]
- Yuan, F.; Cheng, C.; Xiao, F.; Liu, H.; Cao, S.; Zhou, G. Inhibition of mTORC1/P70S6K pathway by Metformin synergistically sensitizes Acute Myeloid Leukemia to Ara-C. Life Sci. 2020, 243, 117276. [Google Scholar] [CrossRef]
- Vitkevičienė, A.; Janulis, V.; Žučenka, A.; Borutinskaitė, V.; Kaupinis, A.; Valius, M.; Griškevičius, L.; Navakauskienė, R. Oxidative phosphorylation inhibition induces anticancerous changes in therapy-resistant–acute myeloid leukemia patient cells. Mol. Carcinog. 2019, 58, 2008–2016. [Google Scholar] [CrossRef]
- Fontaine, E. Metformin-Induced Mitochondrial Complex I Inhibition: Facts, Uncertainties, and Consequences. Front. Endocrinol. 2018, 9, 753. [Google Scholar] [CrossRef]
- Carter, J.L.; Hege, K.; Kalpage, H.A.; Edwards, H.; Hüttemann, M.; Taub, J.W.; Ge, Y. Targeting mitochondrial respiration for the treatment of acute myeloid leukemia. Biochem. Pharmacol. 2020, 182, 114253. [Google Scholar] [CrossRef]
- Maji, S.; Panda, S.; Samal, S.K.; Shriwas, O.; Rath, R.; Pellecchia, M.; Emdad, L.; Das, S.K.; Fisher, P.B.; Dash, R. Bcl-2 Antiapoptotic Family Proteins and Chemoresistance in Cancer. Adv. Cancer Res. 2018, 137, 37–75. [Google Scholar] [CrossRef]
- Ewald, L.; Dittmann, J.; Vogler, M.; Fulda, S. Side-by-side comparison of BH3-mimetics identifies MCL-1 as a key therapeutic target in AML. Cell Death Dis. 2019, 10, 917. [Google Scholar] [CrossRef]
- Glaser, S.P.; Lee, E.F.; Trounson, E.; Bouillet, P.; Wei, A.; Fairlie, W.D.; Izon, D.J.; Zuber, J.; Rappaport, A.R.; Herold, M.J.; et al. Anti-apoptotic Mcl-1 is essential for the development and sustained growth of acute myeloid leukemia. Genes Dev. 2012, 26, 120–125. [Google Scholar] [CrossRef] [Green Version]
- Kotschy, A.; Szlavik, Z.; Murray, J.; Davidson, J.; Maragno, A.L.; Le Toumelin-Braizat, G.; Chanrion, M.; Kelly, G.L.; Gong, J.-N.; Moujalled, D.M.; et al. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature 2016, 538, 477–482. [Google Scholar] [CrossRef]
- Hormi, M.; Birsen, R.; Belhadj, M.; Huynh, T.; Aguilar, L.C.; Grignano, E.; Haddaoui, L.; Guillonneau, F.; Mayeux, P.; Hunault, M.; et al. Pairing MCL-1 inhibition with venetoclax improves therapeutic efficiency of BH3-mimetics in AML. Eur. J. Haematol. 2020, 105, 588–596. [Google Scholar] [CrossRef]
- Yuan, J.; Zhang, S.; Zhang, Y. Nrf1 is paved as a new strategic avenue to prevent and treat cancer, neurodegenerative and other diseases. Toxicol. Appl. Pharmacol. 2018, 360, 273–283. [Google Scholar] [CrossRef]
- Jia, J.-J.; Geng, W.-S.; Wang, Z.-Q.; Chen, L.; Zeng, X.-S. The role of thioredoxin system in cancer: Strategy for cancer therapy. Cancer Chemother. Pharmacol. 2019, 84, 453–470. [Google Scholar] [CrossRef]
- Pecinová, A.; Brázdová, A.; Drahota, Z.; Houštěk, J.; Mráček, T. Mitochondrial targets of metformin—Are they physiologically relevant? BioFactors 2019, 45, 703–711. [Google Scholar] [CrossRef]
- Karimian, A.; Ahmadi, Y.; Yousefi, B. Multiple functions of p21 in cell cycle, apoptosis and transcriptional regulation after DNA damage. DNA Repair 2016, 42, 63–71. [Google Scholar] [CrossRef]
- Snezhkina, A.V.; Kudryavtseva, A.V.; Kardymon, O.L.; Savvateeva, M.V.; Melnikova, N.V.; Krasnov, G.S.; Dmitriev, A.A. ROS Generation and Antioxidant Defense Systems in Normal and Malignant Cells. Oxidative Med. Cell. Longev. 2019, 2019, 1–17. [Google Scholar] [CrossRef]
- Nicholls, D.G. Mitochondrial membrane potential and aging. Aging Cell 2003, 3, 35–40. [Google Scholar] [CrossRef]
- Khan, N.H.; Sexton, K.J.; Grimm, M.J.; Segal, B.H.; E Vigil, C. NADPH Oxidase Regulates Cytarabine-Induced Apoptotic Death in Acute Myeloid Leukemia Cells. Blood 2011, 118, 4258. [Google Scholar] [CrossRef]
- Carter, B.Z.; Mak, P.Y.; Tao, W.; Warmoes, M.; Lorenzi, P.L.; Mak, D.; Ruvolo, V.; Tan, L.; Cidado, J.; Drew, L.; et al. Targeting MCL-1 dysregulates cell metabolism and leukemia-stroma interactions and resensitizes acute myeloid leukemia to BCL-2 inhibition. Haematologica 2020. [Google Scholar] [CrossRef]
- Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Babenko, V.A.; Zorov, S.D.; Balakireva, A.V.; Juhaszova, M.; et al. Mitochondrial membrane potential. Anal. Biochem. 2018, 552, 50–59. [Google Scholar] [CrossRef]
- Savickiene, J.; Treigyte, G.; Valiuliene, G.; Stirblyte, I.; Navakauskiene, R. Epigenetic and molecular mechanisms underlying the antileukemic activity of the histone deacetylase inhibitor belinostat in human acute promyelocytic leukemia cells. Anti-Cancer Drugs 2014, 25, 938–949. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valiulienė, G.; Vitkevičienė, A.; Skliutė, G.; Borutinskaitė, V.; Navakauskienė, R. Pharmaceutical Drug Metformin and MCL1 Inhibitor S63845 Exhibit Anticancer Activity in Myeloid Leukemia Cells via Redox Remodeling. Molecules 2021, 26, 2303. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26082303
Valiulienė G, Vitkevičienė A, Skliutė G, Borutinskaitė V, Navakauskienė R. Pharmaceutical Drug Metformin and MCL1 Inhibitor S63845 Exhibit Anticancer Activity in Myeloid Leukemia Cells via Redox Remodeling. Molecules. 2021; 26(8):2303. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26082303
Chicago/Turabian StyleValiulienė, Giedrė, Aida Vitkevičienė, Giedrė Skliutė, Veronika Borutinskaitė, and Rūta Navakauskienė. 2021. "Pharmaceutical Drug Metformin and MCL1 Inhibitor S63845 Exhibit Anticancer Activity in Myeloid Leukemia Cells via Redox Remodeling" Molecules 26, no. 8: 2303. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26082303