1. Introduction
The advent of the current global pandemic, COVID-19, has not only highlighted the need for new active pharmaceutical ingredients (APIs), but it has also given credence to the development of novel and efficient drug delivery systems. The application of effective chemical entities coupled with advances in materials science provides a glimmer of hope for solving the most prevalent global health challenges. Moreover, the oral route is the primary and most convenient method of administering drugs; it allows self-administration and is associated with good patient compliance compared with the other options [
1,
2,
3]. Consequently, oral dosage forms of drug candidates remain the main target in research and marketing. Unfortunately, despite the advancements in delivery systems and progress in state-of-the-art of drug design and development, solubility and stability remain the Achilles heel of the APIs [
4].
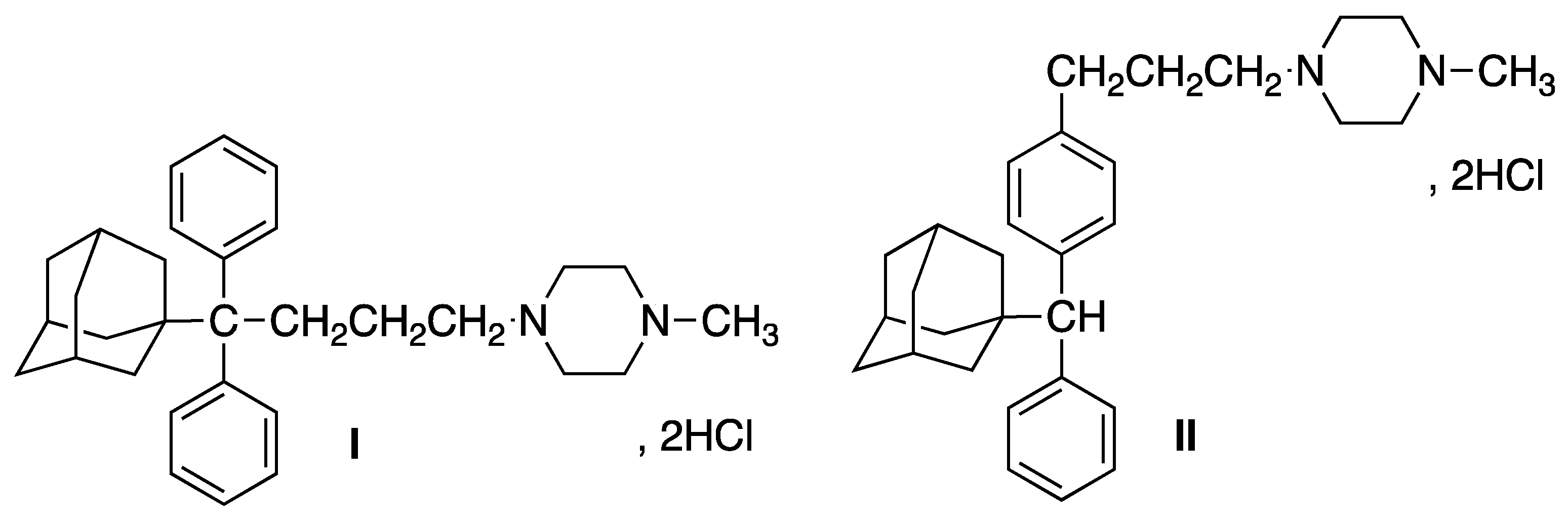
In this work, we focus on the oral modified release of two potential APIs, the isomeric 4-[4,4-diphenyl-4-[(1-adamantyl)butyl)]-1-methylpiperazine (
I) and 4-{3-[4-[
α-(1-adamantyl)phenylmethyl]phenyl]propyl}-1-methylpiperazine (
II) (
Figure 1), the synthesis and pharmacology of which we have reported [
5,
6], in the context of our ongoing effort to exploit adamantane’s role in antiproliferative activity [
7,
8,
9,
10].
The adamantane phenylalkylamine hydrochlorides
I and
II were found to exhibit a significant sigma-receptor (σR) binding affinity and antiproliferative/ anticancer activity, associated with an analgesic profile against neuropathic pain. Anavex Life Sciences Corp. Pharma has further explored these important properties by subjecting derivative
II (AV1066) to preclinical phase studies against both neuropathic and visceral pain [
11]. Sigma receptors (σRs) represent an enigmatic class of proteins [
12] involved in a plethora of biochemical pathways [
13,
14] and are accommodated by a broad structural diversity of ligands which exhibit a variety of pharmacological activities, such as anticancer [
15,
16], neuroprotective [
17,
18] and even coronavirocidal activities [
19,
20,
21].
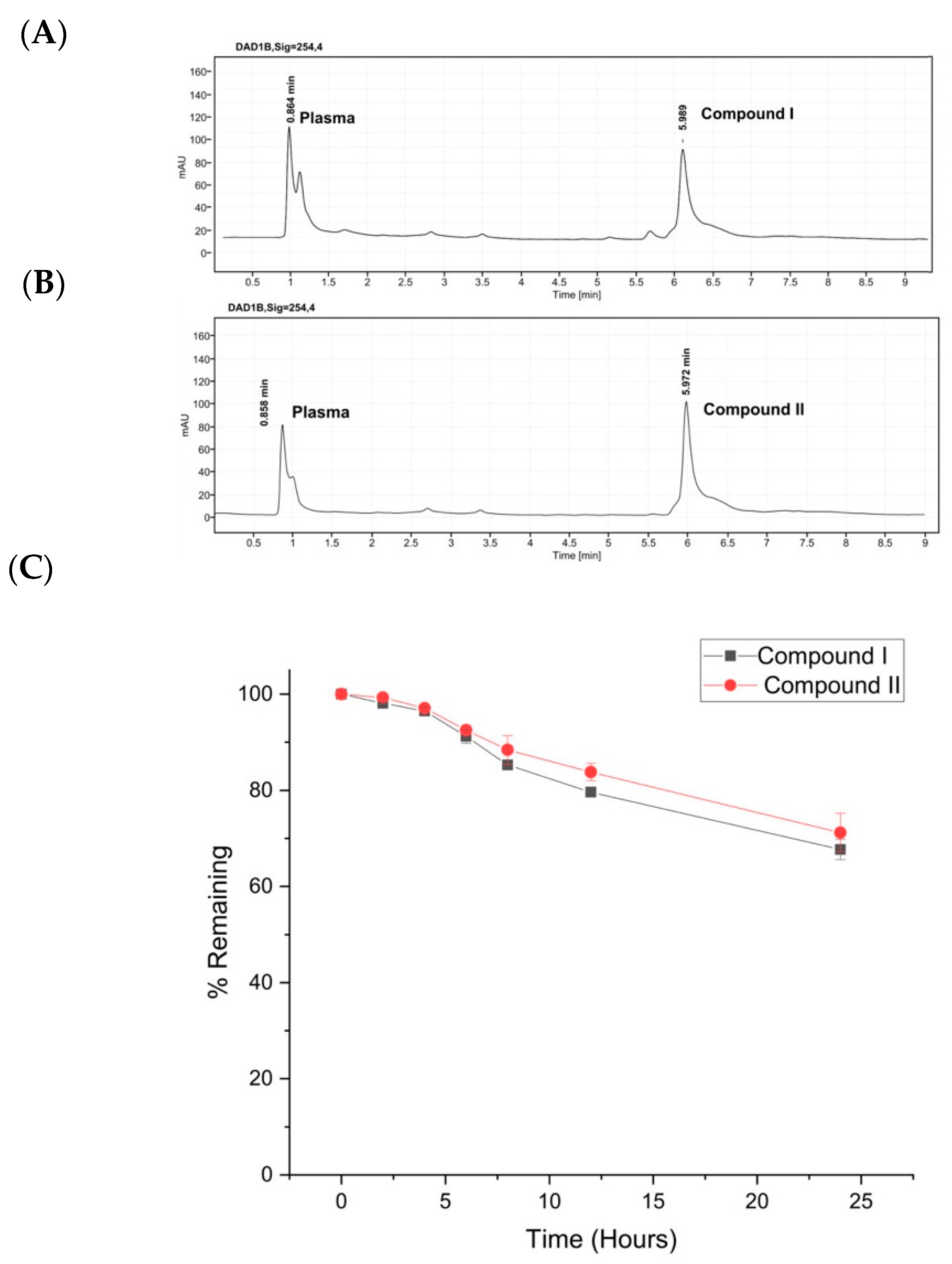
The important pharmacological properties of these two adamantane adducts prompted us to assess their enzymatic stability. Both analogues were relatively stable for 24 h of incubation in human plasma at 37 °C. Furthermore, the pharmacokinetics and toxicity characteristics of the two compounds were explored and predicted to be neither hepatotoxic nor active for all the associated toxicological pathways according to the ProTox-II platform.
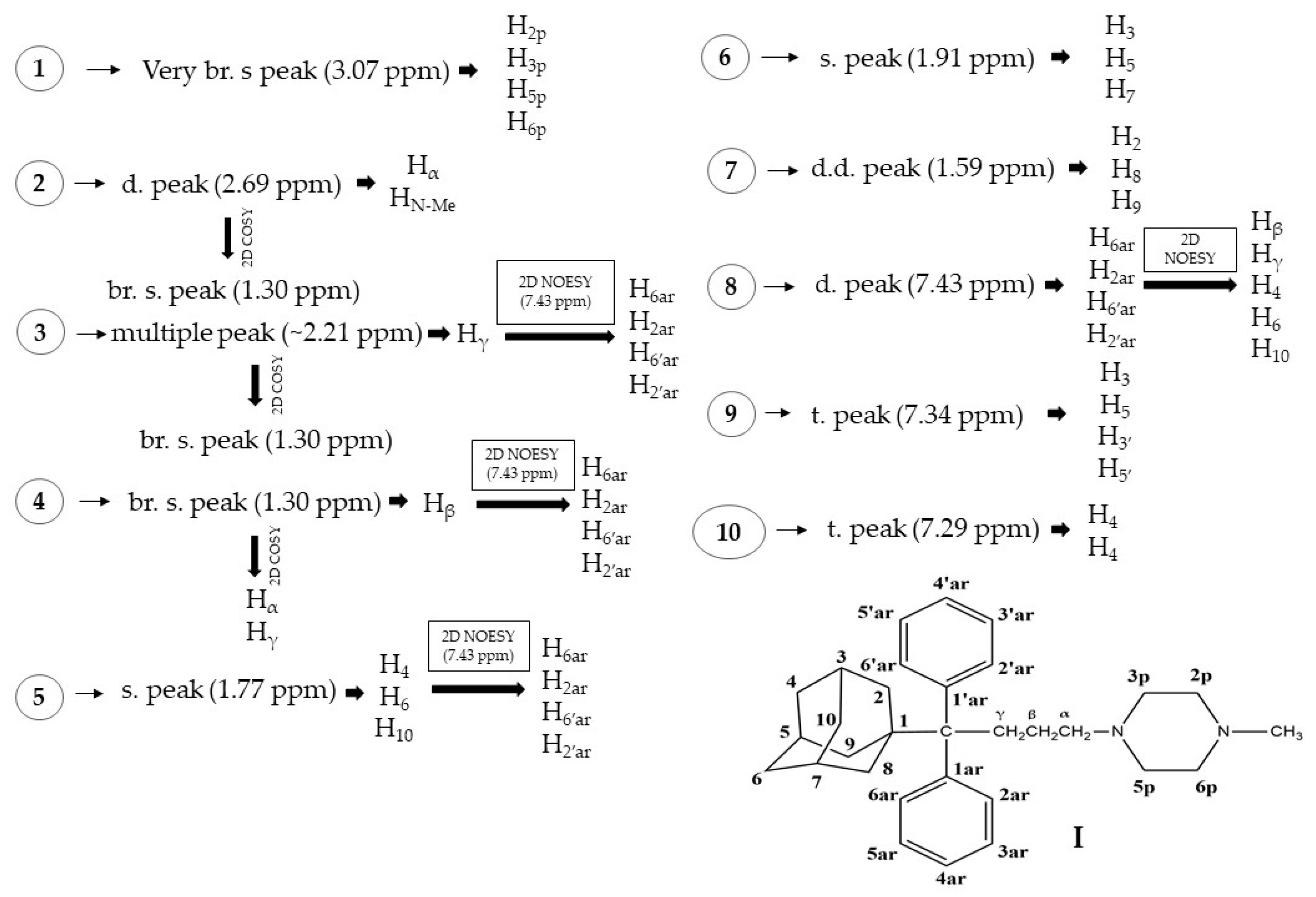
Thus, the next step in our studies was to conduct 2D NMR molecular structure elucidation experiments of compounds I and II, which were additionally confirmed by MD simulations, in an attempt to decipher whether their matrix tablets formulations could transduce them into orally administered APIs. Moreover, the human plasma stability of the corresponding adducts and their pharmacokinetic properties and toxicities were evaluated.
The in vitro release profiles of adducts I and II were monitored in the gastric and intestinal pH environment. At pH 1.2, compound I, in its hydrochloride form, is endowed with high hydrophilicity, but at neutral pH, where it is transformed to its free base, it becomes lipophilic (logP = 6.073). The hydrochloride salt of compound II shows a different profile. Specifically, at pH 1.2 it is sparingly soluble, but at neutral pH, it is equally lipophilic (logP = 6.071).
Taking into account these characteristics, we designed and prepared oral dosage forms using the appropriate matrix systems. Apart from the bioactive substances I and II, the formulations included the biopolymers hydroxypropylmethycellulose (HPMC), poly(ethylene oxide) (PEO), Eudragit, and the marine biopolymer ulvan. In order to modulate the aqueous dissolution profile of compounds I and II in simulated media along the entire gastrointestinal tract, various ratios of these formulants were used.
Matrix-based modified drug delivery systems are complex networks of either hydrophilic or hydrophobic polymers homogeneously mixed with active substances. The release rate of the active substances from matrix-type systems is strongly associated with polymer-related factors, such as the type, the combination, the proportions and the particle properties of the polymers, but also with drug-related factors, such as drug solubility, molecular weight, size, particle size and shape.
As far as drug release is concerned, solubility in aqueous media is associated with properties of the drug substances, such as the physicochemical nature of the functional ligands, the stereochemical configuration and polymorphism. When active substances are characterized by high solubility, the release rate is rapid, while substances of low solubility present a retarded release rate. In particular, in matrix-type systems, the release rate is enhanced by using a variety of polymers which contribute to the solubility potential of the active substances. Thus, the release kinetics for soluble substances are conducted by diffusion and erosion of the polymers, whilst for insoluble substances, osmosis and erosion phenomena of the polymers are dominant [
22,
23,
24]. Moreover, the utilization of pH-controlled aqueous media contributes to the creation of more stable and uniform dissolution conditions, mainly when the active substance is characterized by low solubility [
25,
26]. Polymers such as ulvan and Eudragit are pH-dependent soluble polymers, releasing the drug at a lesser rate in environments with an acidic pH (1.2).
A number of considerable aspects influence and determine drug release from matrix-type modified drug delivery systems, including the rate of aqueous medium penetration into the polymeric matrix, which results in hydration, gelation and swelling of the polymers and affects the rate of polymeric matrix erosion [
27].
3. Materials and Methods
Compounds I (MW: 515.61 g/mol) and II (MW: 515.61 g/mol) were provided by colleagues in the Pharmaceutical Chemistry Department. Hydroxypropylmethycellulose (HPMC K100M) and poly(ethylene oxide) (PEO) (MW: 7 × 106 g/mol) were obtained from Sigma-Aldrich (Steinheim, Germany). Eudragit L100-55 was purchased from Rohm GmbH Pharma Polymers (Darmstadt, Germany), and ulvan (Cat. No. YU11689) was bought from Carbosynth® Ltd. (Berkshire, UK). Alginic acid sodium salt (low viscosity) was supplied by Alfa Aesar GmbH & Co KG (Karlsruhe, Germany), and magnesium stearate was purchased from Riedel-De Haen (Hannover, Germany). Deuterium oxide 99.90% D (D214F) was purchased from Eurisotop via Fluorochem (Hadfield, UK). All chemicals were of reagent grade and used in this study without further purification.
3.1. Formulation of Compounds I and II into Modified Release Tablets
The formulations of compounds
I and
II were prepared using a variety of polymeric excipients into matrix-type tablet forms (vide
Table 5 and
Table 6). In specific, the APIs (compounds
I and
II) and excipients (HPMC, PEO, Eudragit L100-55, ulvan and sodium alginate) were mixed homogenously in a laboratory scale powder blender at 32 rpm for a time duration of 10 min. Subsequently, the lubricant (magnesium stearate) was added to the mixture, and the blending was continued for another 5 min. Finally, the powder mixture was precisely weighed (200 mg), loaded on a 10 mm diameter matrix and directly compressed using a hydraulic press (Maassen type, MP 150).
3.2. Post Compression Parameters
Thickness Test: The thickness of the tablets was measured using a vernier caliper.
Hardness Test: The hardness of the tablets was determined using an Erweka hardness tester (Erweka, type TBH28). The force applied was equal to breaking the tablet in a diametric compression. The surface hardness of each tablet is expressed in N.
3.3. In Vitro Dissolution Studies
The dissolution tests were performed in a tablet dissolution test apparatus USP type II (Pharmatest, Hainerp, Germany). The experiments were carried out in 2 different aqueous media: at pH 1.2, Vmax = 450 mL for 2 h, and at pH 6.8, Vmax = 900 mL for 6 more hours, in order to simulate, in vitro, the pH range along the gastrointestinal tract. The temperature of the dissolution medium was maintained at 37 ± 0.5 °C. The tablet was placed in the bottom of a vessel, equipped with paddles, under sink conditions, and the apparatus was operated at 50 rpm. Samples were withdrawn at predetermined time intervals, and the withdrawn volume of the medium was replenished. The withdrawn samples were filtered and analyzed using UV-Vis spectrophotometry (LLG-uniSPEC 2 Spectrophotometer) at λmax = 214 nm for pH 1.2 and at λmax = 210 nm for pH 6.8 for compound I and at λmax = 217 nm for pH 1.2 and pH 6.8 for compound II. The % dissolution curve versus time was determined according to the calibration curve of the corresponding API.
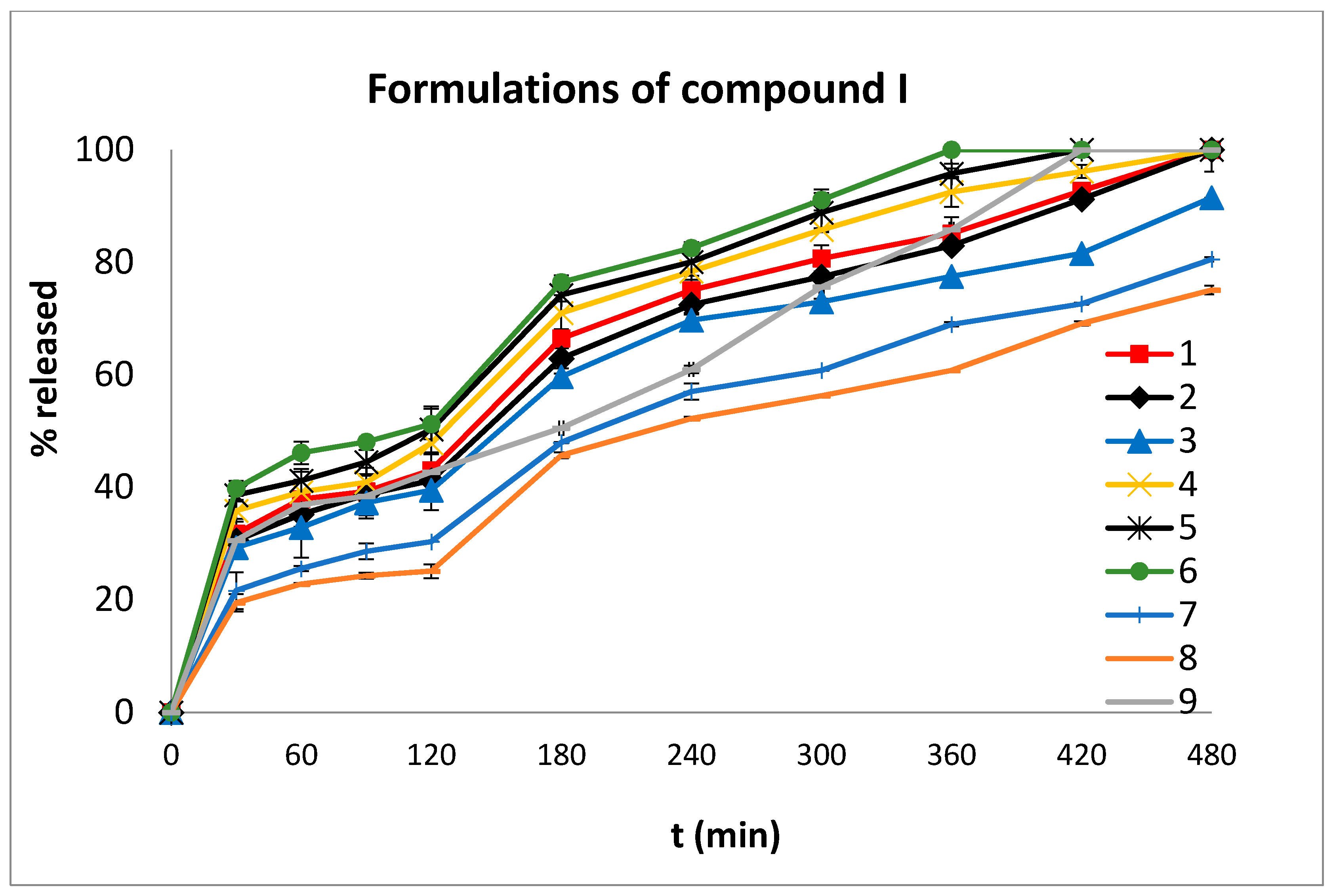
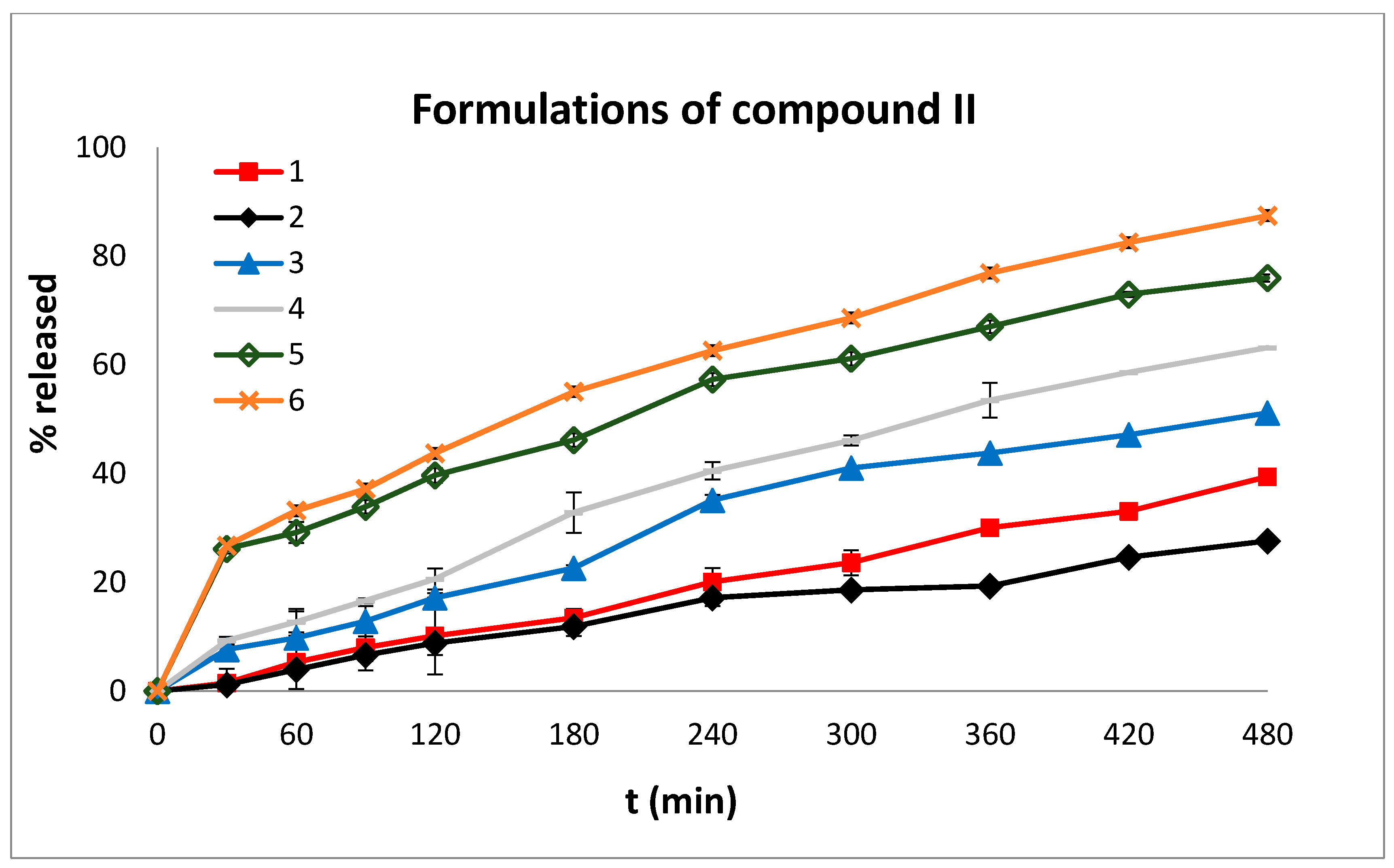
In order to compare the dissolution profiles, graphs of % drug release versus time were constructed (
Figure 5 and
Figure 6) and the
D.E. (%) value was estimated. According to Khan [
40],
D.E. (%) is a useful parameter for the evaluation of dissolution in vitro and is calculated from the following equation:
where
y is the percentage of dissolved product and
D.E. (%) is the area under the dissolution curve between time points
t1 and
t2. They are expressed as a percentage of the curve at maximum dissolution
y100 over the same time period. Dissolution efficiency, which considers the dissolution profile as a whole, was employed to interrelate dissolution with the other variables used in this study.
Furthermore, t
20%, t
50%, and t
90%, as well as the mean dissolution time (
MDT) values, were estimated. The t
20%, t
50%, and t
90% values refer to the time where the 20%, 50%, and 90% of the active substance is released.
MDT is the value used to characterize the drug release rate from a dosage form, and the following is used to derive an estimate of
MDT from experimental dissolution data [
38]:
where
W∞ is the maximum amount of the drug substance that is dissolved, and
ABC is the area between the drug dissolution curve and its asymptote.
The in vitro release data were fitted to the Korsmeyer–Peppas equation:
where
Mt and
M∞ denote the absolute cumulative amount of drug released at time
t and infinite time, respectively,
k represents the release rate constant and
n is the diffusion coefficient. This equation is only valid for the first 60% of the fractional release [
37]. In the case of cylindrical tablets,
n ≤ 0.45 corresponds to a Fickian diffusion release (case I diffusional), 0.45 <
n < 0.89 corresponds to an anomalous transport, and
n = 0.89 corresponds to zero-order (case II) release kinetics.
3.4. Stability of Compounds I and II in Human Plasma
In order to examine the stability of the different compounds in human plasma, we followed the assay described by Di et al. [
41]. A stock solution of 1 mg/mL of compounds
I and
II were prepared by dissolving the appropriate amounts in DMSO. The final working solutions of 50 μM were prepared by further diluting the stock solutions with H
2O/MeCN (1:1). Human plasma samples were prepared by incubating 10 μL of each compound separately from the prepared stocks with 90 μL human plasma for 2, 4, 6, 8, 12, and 24 h at 37 °C. To terminate the reactions and precipitate the plasma proteins, 300 μL of ice-cold acetonitrile were added to each sample. Samples were then vortex-mixed and centrifuged at 12,000×
g for 10 min. The supernatants were collected, syringe filtered and transferred to vials for RP-HPLC analysis. Each sample was studied in triplicate, and the concentration of each analogue was calculated by standard curves.
3.5. NMR Experiments
The 1D and 2D COSY and NOESY NMR experiments were conducted in a Bruker Avance 600 spectrometer (600 MHz) at 25 °C in D2O (99.5%) and water (0.5%). Standard pulse sequences were used and stored in the library of the spectrometer.
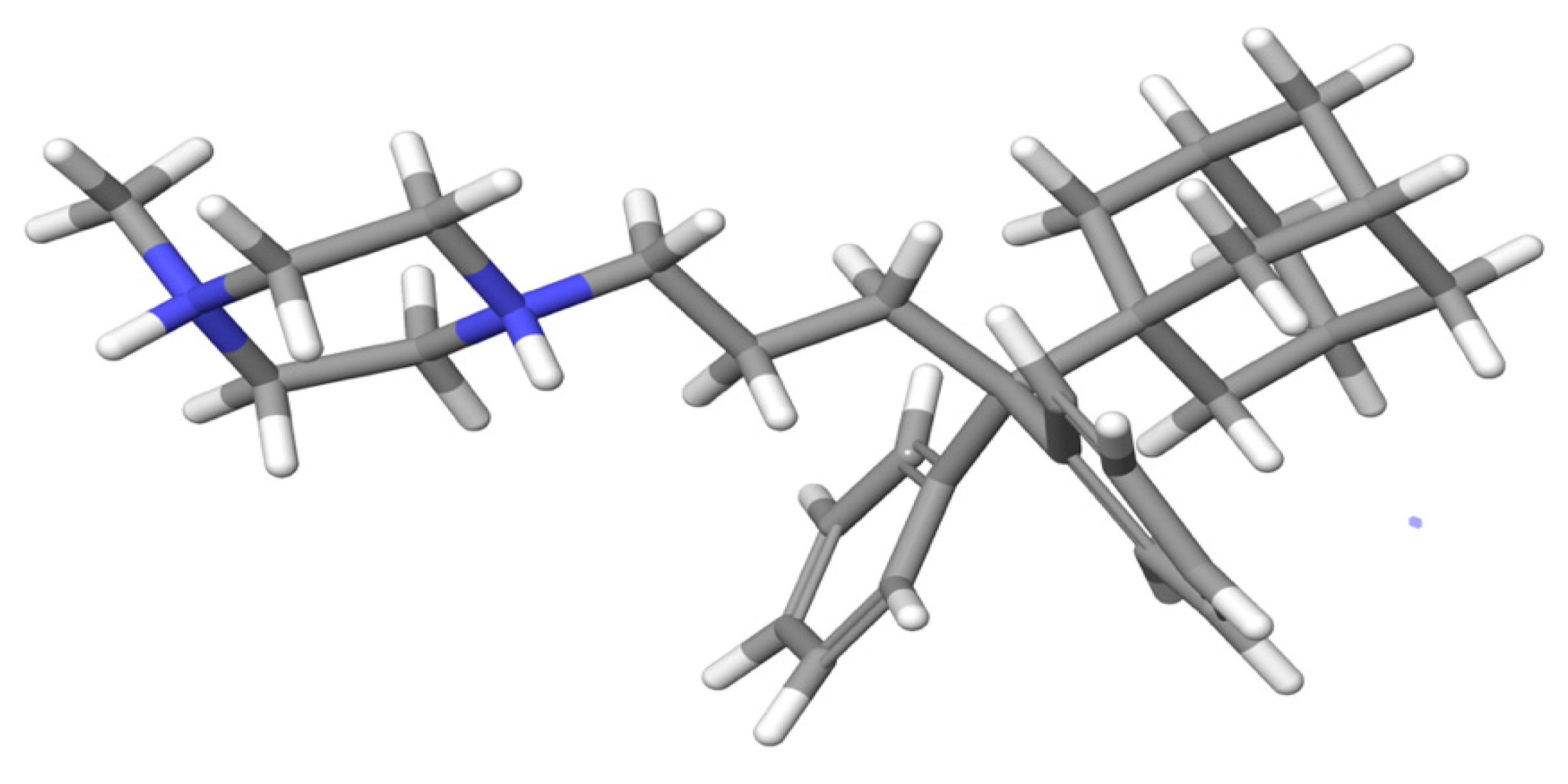
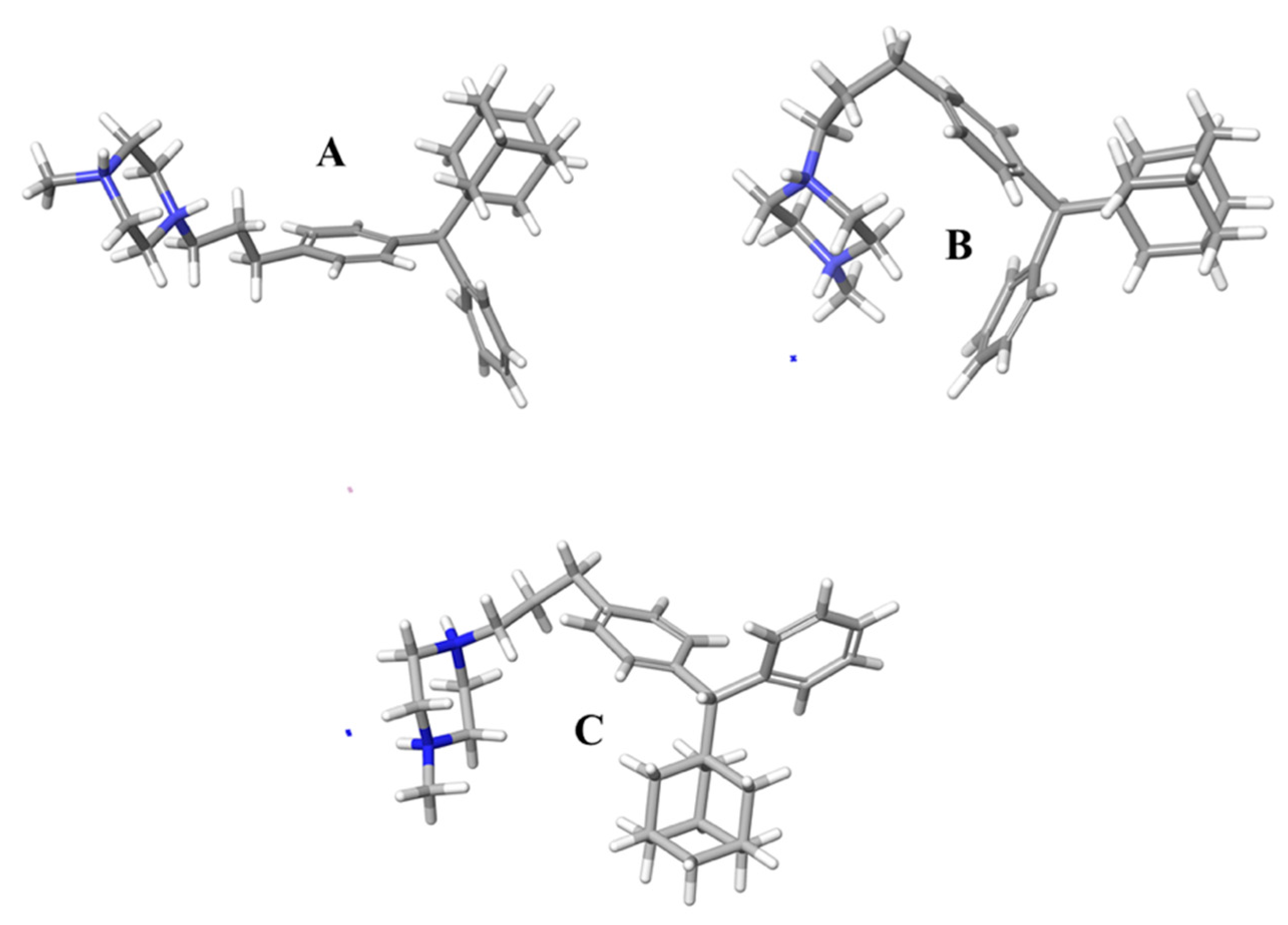
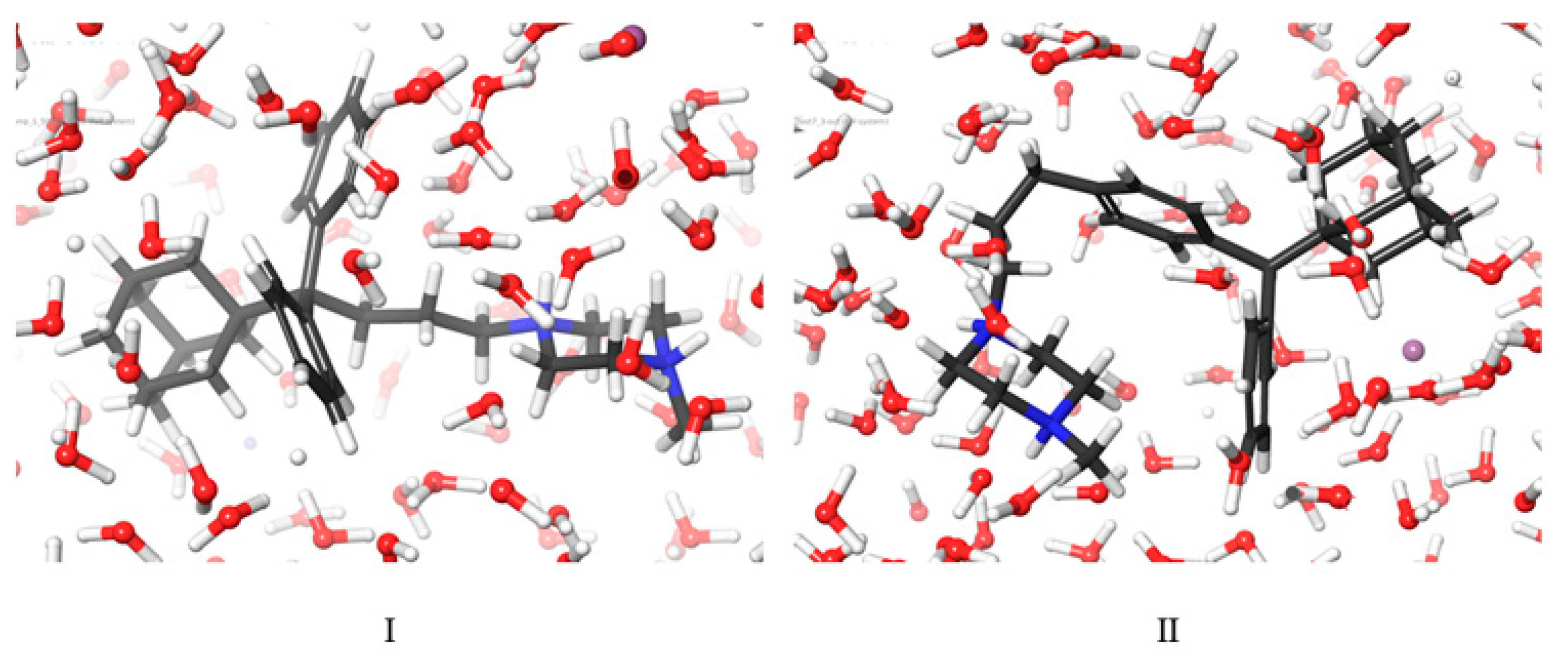
3.6. Molecular Dynamics Simulations Method
The structures of compounds I and II were designed with the aid of the 2D Sketcher in Maestro software. After energy minimization, compounds I and II underwent molecular dynamics (MD) simulations in order to determine their conformational properties in water.
The MD study system was built with SPC/E modeled water around the structure and neutralized with Na
+ and Cl
- ions until the experimental salt concentration of 0.15 M NaCl was reached. The OPLS2005 force field was used to simulate the system, and long-range electrostatics were treated using the particle mesh Ewald (PME) method and a grid spacing of 0.8 Å. Van der Waals and short-range electrostatic interactions were smoothly truncated at 9.0 Å [
42,
43]. A Nosé–Hoover thermostat kept the temperature constant [
44], while the pressure was controlled with the Martyna–Tobias–Klein method [
42]. Periodic boundary conditions were applied, and the dimensions of the simulation box were (30.0 Å × 30.0 Å × 30.0 Å). The volume of the box was minimized by reorienting the solutes in the box. The equations of motion were integrated using the multilevel RESPA integrator [
45] with an outer time step of 2 fs for bound interactions and unbound interactions within a cutoff of 9 Å. Each system was equilibrated using the standard Desmond protocol [
45]. The system was first relaxed with a Brownian dynamics simulation in the NVT ensemble at T = 298 K, with constraints on the solute heavy atoms. MD simulations were performed using the standard Desmond relaxation protocol, which involves gradual heating and reduction of harmonic constraints first for the solvent atoms and later for the solute heavy atoms. Before the start of the production phase, the system was relaxed in the NPT ensemble without any constraints for 1.0 ns. The production phase of the MD simulation was set to 500 ns, which provides a reasonable sample size to discover the predominant conformation during the simulation time. The MD simulations were run on a workstation using the GPU implementation of the MD simulation code.
3.7. Methods to Predict Pharmacokinetic and Toxicity of These Compounds
The pharmacokinetic properties and toxicities were determined using the online programs SwissADME, pro-TOX and pkCSM [
36,
37,
46].


 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}