
Optimization of Nanohybrid Biosensors Based on Electro-Crosslinked Tannic Acid Capped Nanoparticles/Enzyme


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
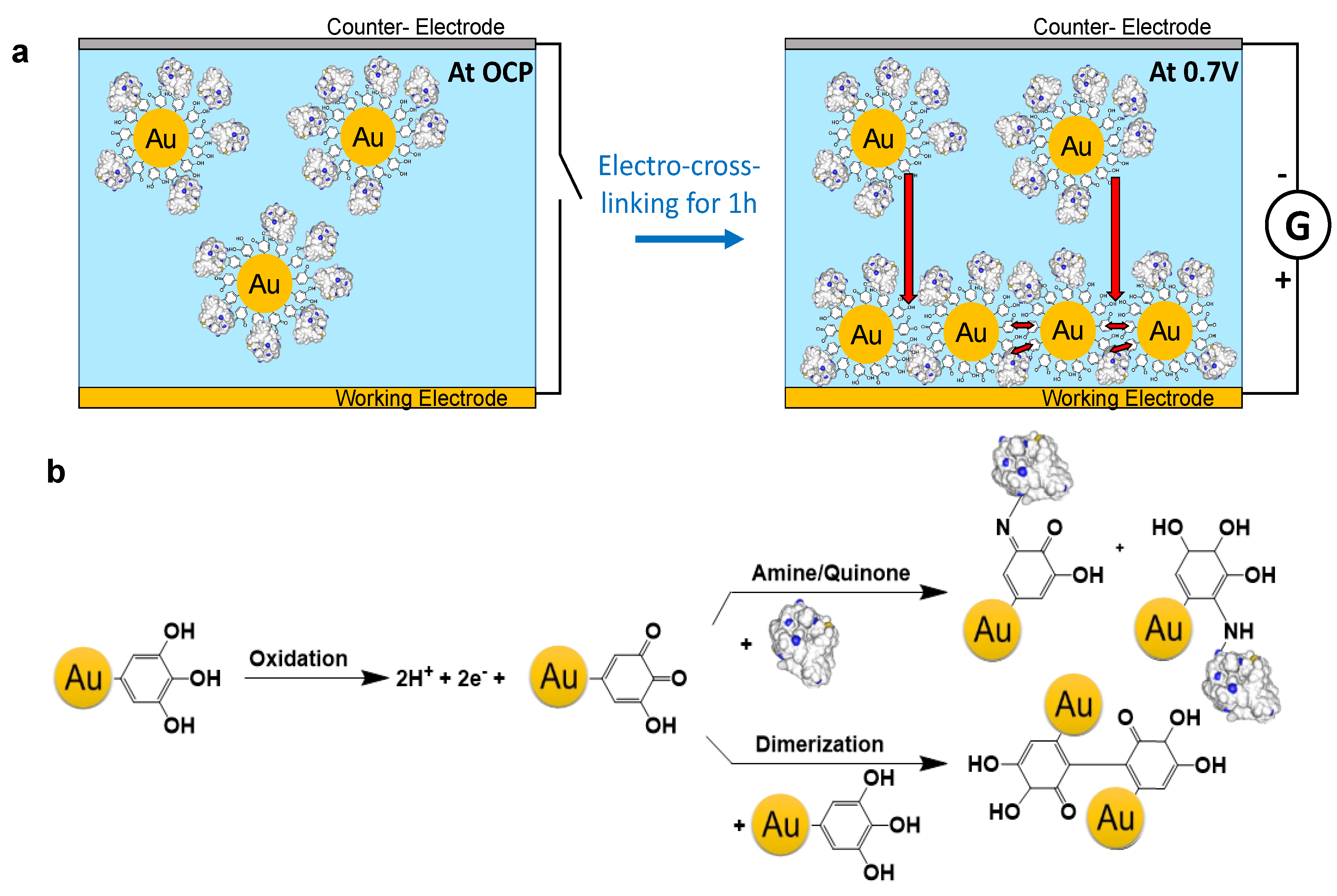
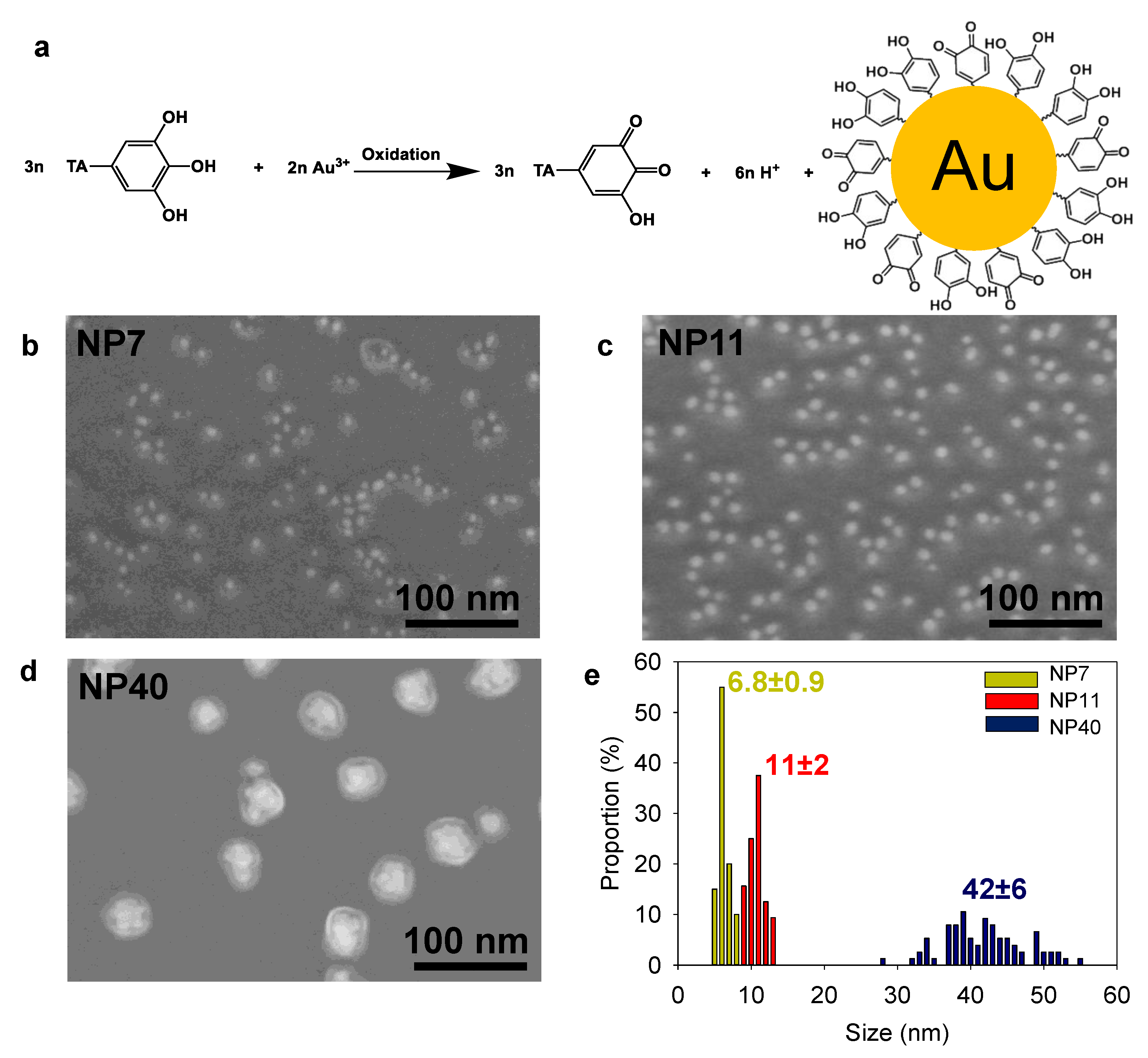
2.1. Gold Nanoparticle Synthesis and Characterization
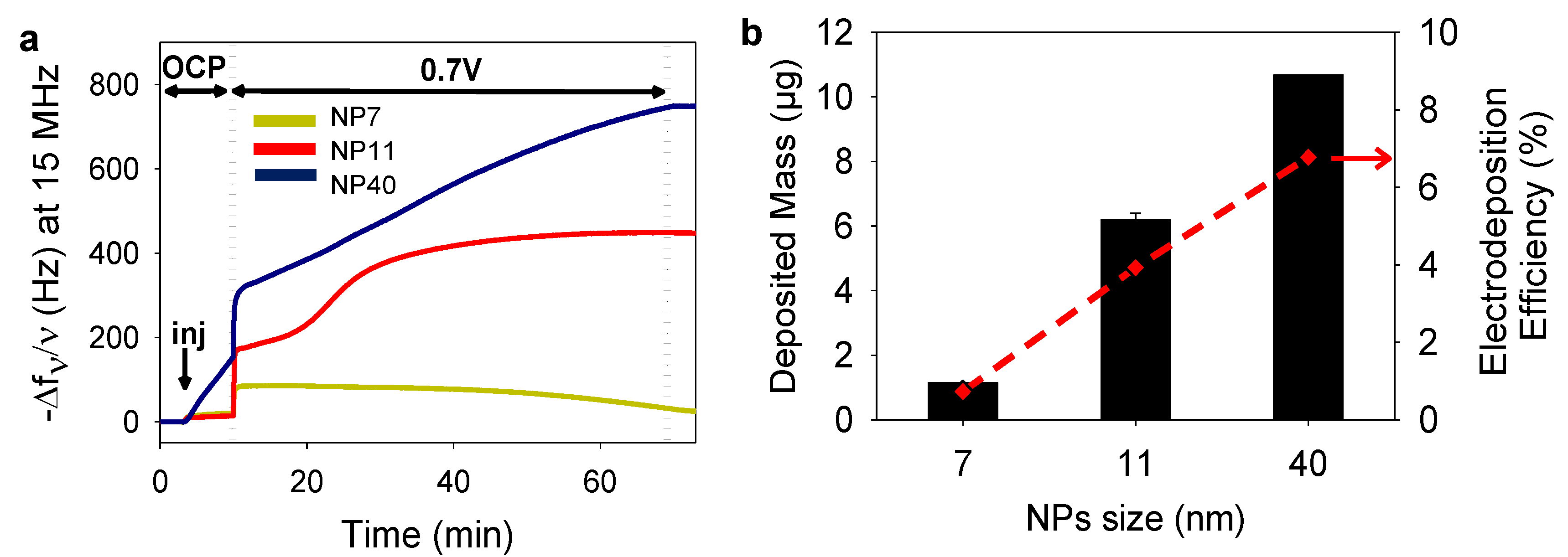
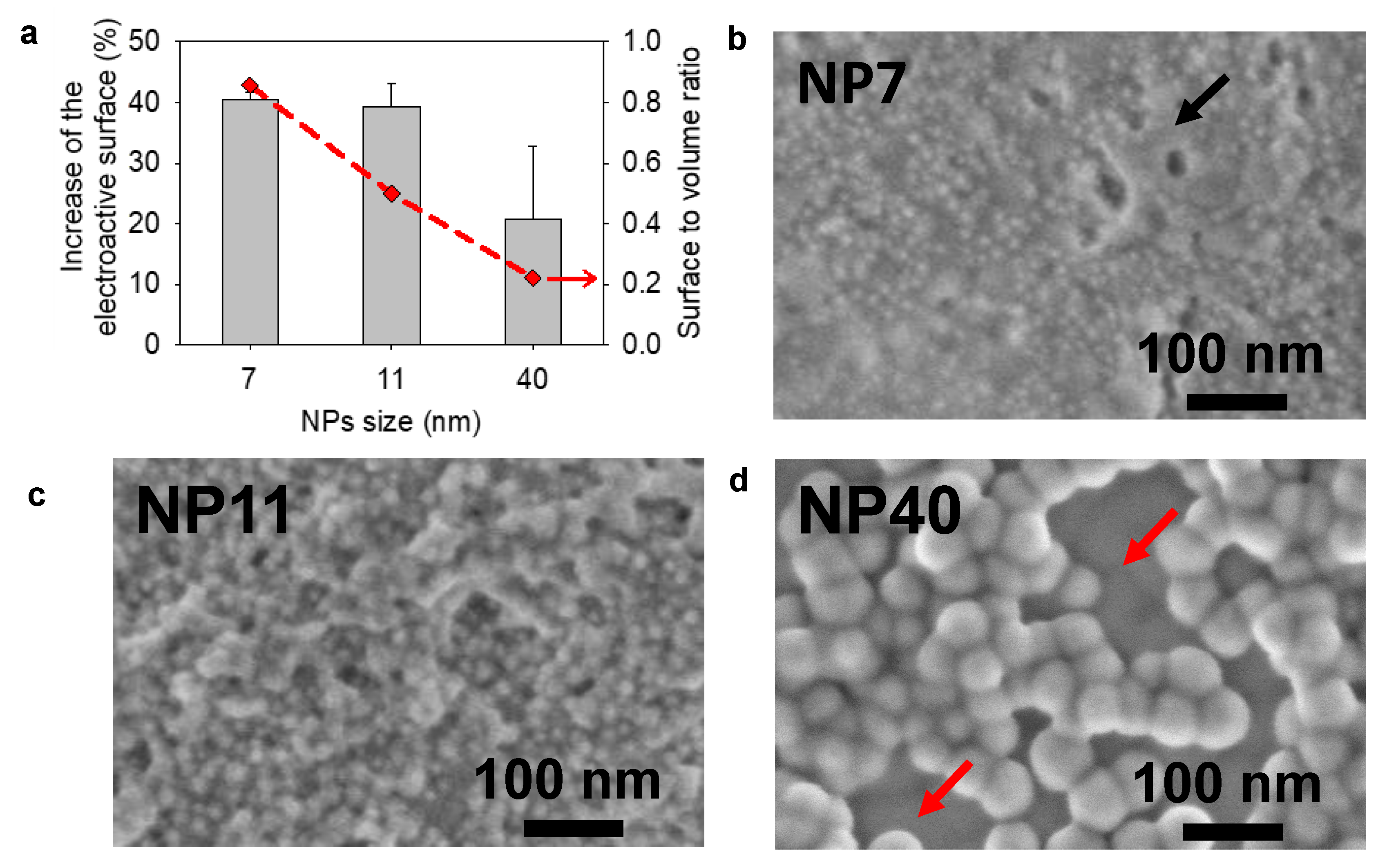
2.2. Gold Nanoparticles Electrodeposited Coatings
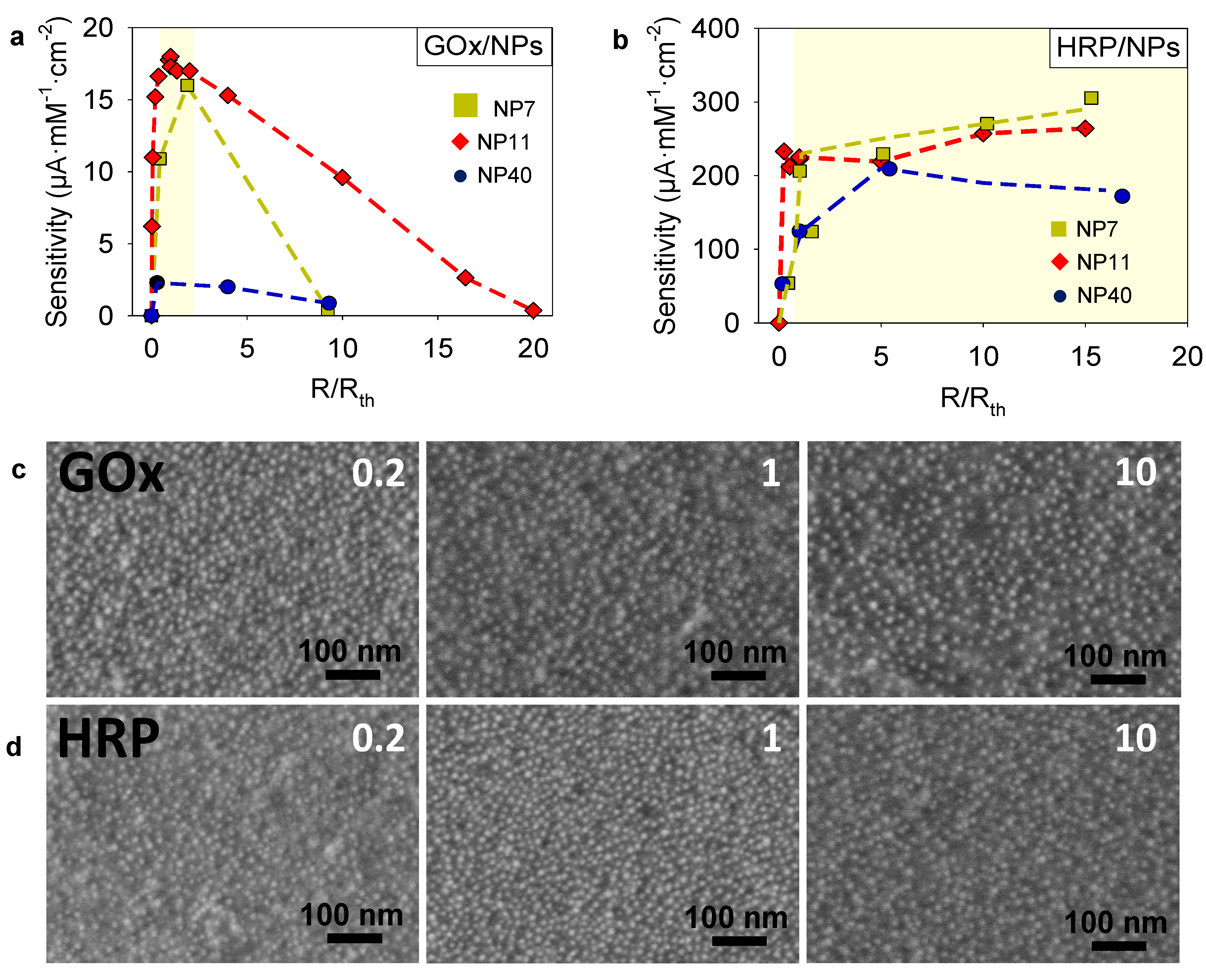
2.3. Sensitivity Optimization of Enzyme/NPs Electrodeposited Coatings
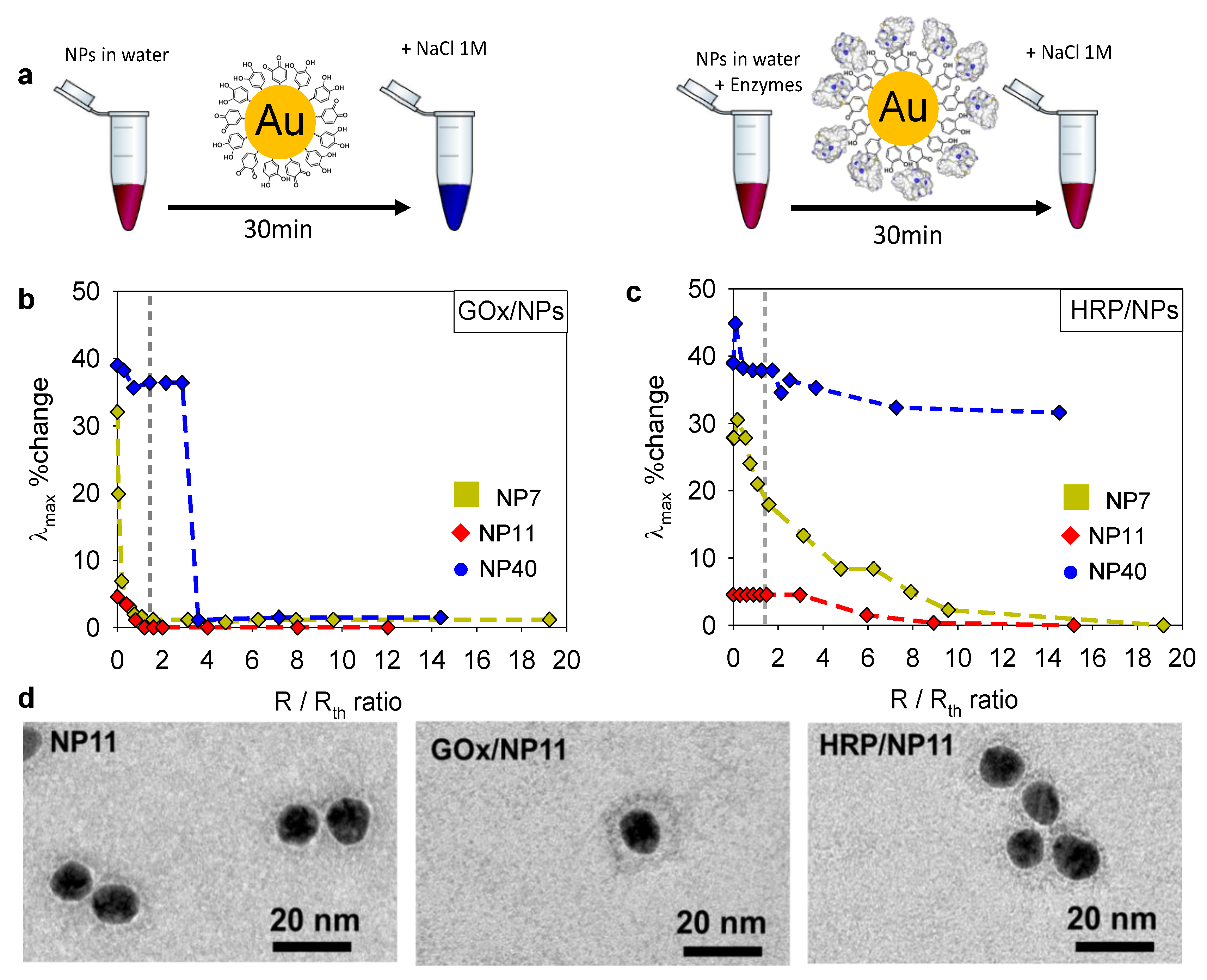
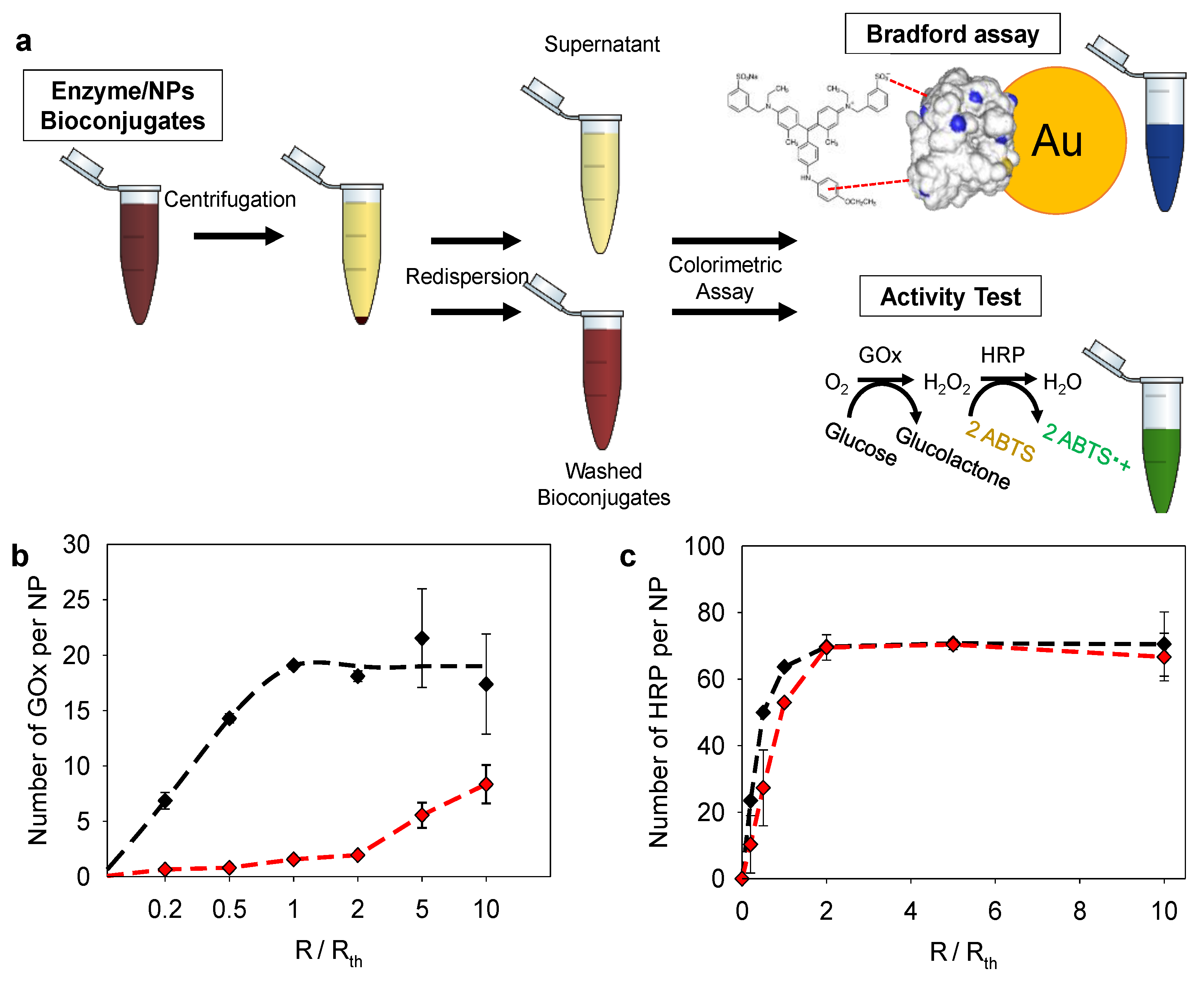
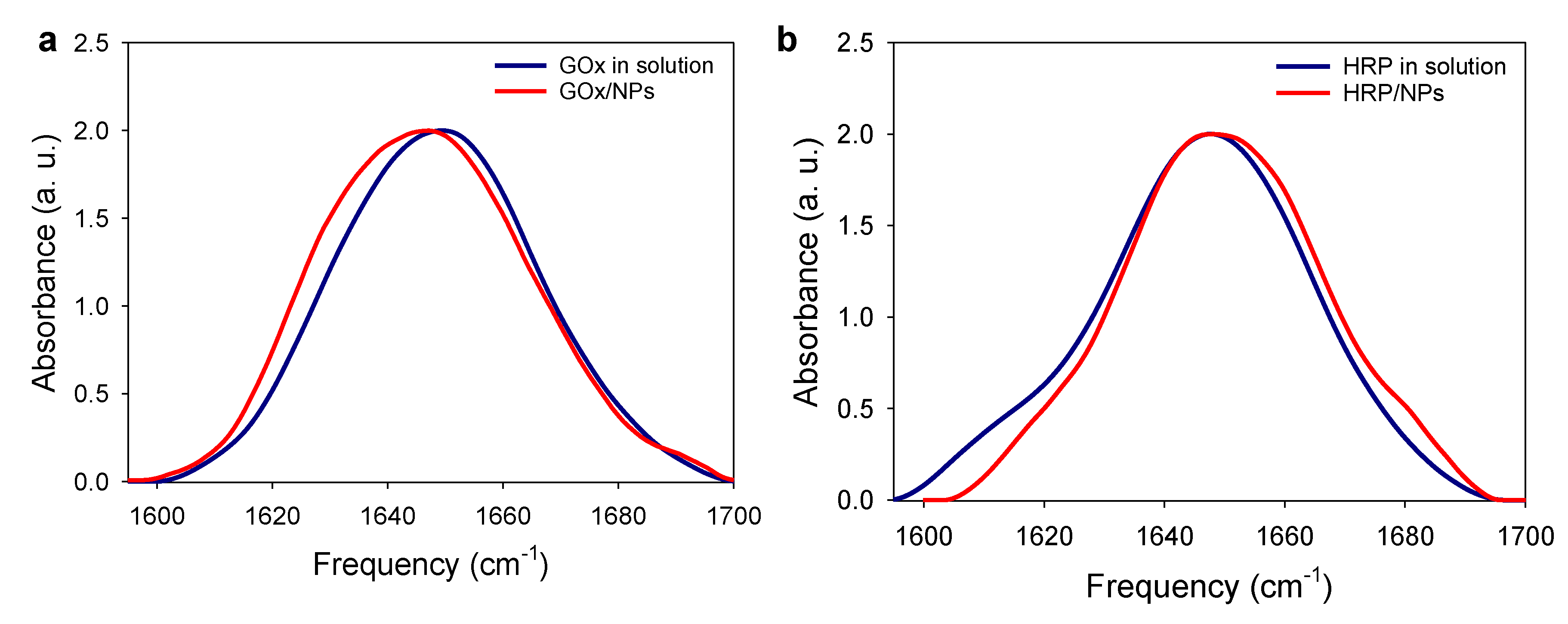
2.4. Enzyme/NPs Bioconjugate Characterization
3. Conclusions
4. Materials and Methods
4.1. Chemicals
4.2. Tannic Acid Capped Gold Nanoparticles Synthesis (NPs)
4.3. NP Characterizations
4.4. Dynamic Light Scattering Characterization
4.5. Enzyme Characterization
4.6. Enzyme/NPs Electrodeposition Solution
4.7. Electrochemical Quartz Crystal Microbalance (EC–QCM)
4.8. Determination of the Electrodeposition Efficiency
4.9. Electroactive Surface Area (EASA) Determination
4.10. Scanning Electron Microscopy (SEM)
4.11. Electrochemical Determination of the Biosensor Sensitivity
4.12. Enzymes/NPs Bioconjugate Observation with Transmission Electron Microscopy (TEM)
4.13. Flocculation Test of the Enzyme/NPs Bioconjugates
4.14. Bradford Assay Enzyme/NPs Bioconjugates
4.15. Colorimetric Enzymatic Tests for Bioconjugates
4.16. Enzyme Conformation onto NP from FTIR Experiments
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Willner, I.; Basnar, B.; Willner, B. Nanoparticle–Enzyme Hybrid Systems for Nanobiotechnology. FEBS J. 2007, 274, 302–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, D.; Liu, B.; Wei, G. Two-Dimensional Material-Based Colorimetric Biosensors: A Review. Biosensors 2021, 11, 259. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Zhang, C.; Zhang, J.; Huang, L. Development of Magnetic Single-Enzyme Nanoparticles as Electrochemical Sensor for Glucose Determination. Electrochim. Acta 2013, 111, 25–30. [Google Scholar] [CrossRef]
- Chen, J.; Jiang, Z.; Ackerman, J.D.; Yazdani, M.; Hou, S.; Nugen, S.R.; Rotello, V.M. Electrochemical Nanoparticle–Enzyme Sensors for Screening Bacterial Contamination in Drinking Water. Analyst 2015, 140, 4991–4996. [Google Scholar] [CrossRef] [Green Version]
- Lundqvist, M.; Sethson, I.; Jonsson, B.H. Protein Adsorption onto Silica Nanoparticles: Conformational Changes Depend on the Particles’ Curvature and the Protein Stability. Langmuir 2004, 20, 10639–10647. [Google Scholar] [CrossRef]
- Gagner, J.E.; Lopez, M.D.; Dordick, J.S.; Siegel, R.W. Effect of Gold Nanoparticle Morphology on Adsorbed Protein Structure and Function. Biomaterials 2011, 32, 7241–7252. [Google Scholar] [CrossRef]
- Gagner, J.E.; Qian, X.; Lopez, M.M.; Dordick, J.S.; Siegel, R.W. Effect of Gold Nanoparticle Structure on the Conformation and Function of Adsorbed Proteins. Biomaterials 2012, 33, 8503–8516. [Google Scholar] [CrossRef]
- Tellechea, E.; Wilson, K.J.; Bravo, E.; Hamad-Schifferli, K. Engineering the Interface between Glucose Oxidase and Nanoparticles. Langmuir 2012, 28, 5190–5200. [Google Scholar] [CrossRef]
- Wu, H.; Liu, Y.; Li, M.; Chong, Y.; Zeng, M.; Lo, Y.M.; Yin, J.J. Size-Dependent Tuning of Horseradish Peroxidase Bioreactivity by Gold Nanoparticles. Nanoscale 2015, 7, 4505–4513. [Google Scholar] [CrossRef]
- Aubin-Tam, M.E.; Zhou, H.; Hamad-Schifferli, K. Structure of Cytochrome c at the Interface with Magnetic CoFe2O4nanoparticles. Soft Matter 2008, 4, 554–559. [Google Scholar] [CrossRef]
- Tadepalli, S.; Wang, Z.; Slocik, J.; Naik, R.R.; Singamaneni, S. Effect of Size and Curvature on the Enzyme Activity of Bionanoconjugates. Nanoscale 2017, 9, 15666–15672. [Google Scholar] [CrossRef] [PubMed]
- Breger, J.C.; Oh, E.; Susumu, K.; Klein, W.P.; Walper, S.A.; Ancona, M.G.; Medintz, I.L. Nanoparticle Size Influences Localized Enzymatic Enhancement - A Case Study with Phosphotriesterase. Bioconjug. Chem. 2019, 30, 2060–2074. [Google Scholar] [CrossRef] [PubMed]
- Marichal, L.; Degrouard, J.; Gatin, A.; Raffray, N.; Aude, J.C.; Boulard, Y.; Combet, S.; Cousin, F.; Hourdez, S.; Mary, J.; et al. From Protein Corona to Colloidal Self-Assembly: The Importance of Protein Size in Protein-Nanoparticle Interactions. Langmuir 2020, 36, 8218–8230. [Google Scholar] [CrossRef] [PubMed]
- De Roe, C.; Courtoy, P.J.; Baudhuin, P. A Model of Protein-Colloidal Gold Interactions. J. Histochem. & Cytochem. 1987, 35, 1191–1198. [Google Scholar]
- Suzuki, M.; Murata, K.; Nakamura, N.; Ohno, H. The Effect of Particle Size on the Direct Electron Transfer Reactions of Metalloproteins Using Au Nanoparticle-Modified Electrodes. Electrochemistry 2012, 80, 337–339. [Google Scholar] [CrossRef] [Green Version]
- Lata, J.P.; Gao, L.; Mukai, C.; Cohen, R.; Nelson, J.L.; Anguish, L.; Coonrod, S.; Travis, A.J. Effects of Nanoparticle Size on Multilayer Formation and Kinetics of Tethered Enzymes. Bioconjug. Chem. 2015, 26, 1931–1938. [Google Scholar] [CrossRef]
- Park, H.J.; McConnell, J.T.; Boddohi, S.; Kipper, M.J.; Johnson, P.A. Synthesis and Characterization of Enzyme–Magnetic Nanoparticle Complexes: Effect of Size on Activity and Recovery. Colloids Surfaces B Biointerfaces 2011, 83, 198–203. [Google Scholar] [CrossRef]
- Talbert, J.N.; Goddard, J.M. Influence of Nanoparticle Diameter on Conjugated Enzyme Activity. Food Bioprod. Process. 2013, 91, 693–699. [Google Scholar] [CrossRef]
- Vertegel, A.A.; Siegel, R.W.; Dordick, J.S. Silica Nanoparticle Size Influences the Structure and Enzymatic Activity of Adsorbed Lysozyme. Langmuir 2004, 20, 6800–6807. [Google Scholar] [CrossRef]
- German, N.; Ramanaviciene, A.; Voronovic, J.; Ramanavicius, A. Glucose Biosensor Based on Graphite Electrodes Modified with Glucose Oxidase and Colloidal Gold Nanoparticles. Microchim. Acta 2010, 168, 221–229. [Google Scholar] [CrossRef]
- Cans, A.-S.; Dean, S.L.; Reyes, F.E.; Keating, C.D. Synthesis and Characterization of Enzyme-Au Bioconjugates: HRP and Fluorescein-Labeled HRP. NanoBiotechnology 2007, 3, 12–22. [Google Scholar] [CrossRef]
- Wang, Y.; Jonkute, R.; Lindmark, H.; Keighron, J.D.; Cans, A.-S. Molecular Crowding and a Minimal Footprint at a Gold Nanoparticle Support Stabilize Glucose Oxidase and Boost Its Activity. Langmuir 2019, 36, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Savin, R.; Benzaamia, N.-O.; Njel, C.; Pronkin, S.N.; Blanck, C.; Schmutz, M.; Boulmedais, F. Nanohybrid Biosensor Based on Mussel-Inspired Electro-Cross-Linking of Tannic Acid Capped Gold Nanoparticles and Enzymes. Mater. Adv. 2022, 3, 2222–2233. [Google Scholar] [CrossRef]
- Sivaraman, S.K.; Kumar, S.; Santhanam, V. Room-Temperature Synthesis of Gold Nanoparticles, Size-Control by Slow Addition. Gold Bull. 2010, 43, 275–286. [Google Scholar] [CrossRef] [Green Version]
- Wuithschick, M.; Birnbaum, A.; Witte, S.; Sztucki, M.; Vainio, U.; Pinna, N.; Rademann, K.; Emmerling, F.; Kraehnert, R.; Polte, J. Turkevich in New Robes: Key Questions Answered for the Most Common Gold Nanoparticle Synthesis. ACS Nano 2015, 9, 7052–7071. [Google Scholar] [CrossRef]
- Karasyova, O.N.; Ivanova, L.I.; Lakshtanov, L.Z.; Lövgren, L.; Sjöberg, S. Complexation of Gold(III)-Chloride at the Surface of Hematite. Aquat. Geochemistry 1998, 4, 215–231. [Google Scholar] [CrossRef]
- Ciganda, R.; Irigoyen, J.; Gregurec, D.; Hernández, R.; Moya, S.; Wang, C.; Ruiz, J.; Astruc, D. Liquid-Liquid Interfacial Electron Transfer from Ferrocene to Gold(III): An Ultrasimple and Ultrafast Gold Nanoparticle Synthesis in Water under Ambient Conditions. Inorg. Chem. 2016, 55, 6361–6363. [Google Scholar] [CrossRef]
- Sauerbrey, G. Verwendung von Schwingquarzen Zur Wägung Dünner Schichten Und Zur Mikrowägung. Zeitschrift für Phys. 1959, 155, 206–222. [Google Scholar] [CrossRef]
- Scientific, B. Specification Data from QSense QSX 301 and QEM401. Available online: https://www.biolinscientific.com/qsense/instruments/qsense-analyzer#specifications (accessed on 20 April 2022).
- Chandrasekharan, N.; Kamat, P.V. Assembling Gold Nanoparticles as Nanostructured Films Using an Electrophoretic Approach. Nano Lett. 2000, 1, 67–70. [Google Scholar] [CrossRef]
- Ammam, M. Electrochemical and Electrophoretic Deposition of Enzymes: Principles, Differences and Application in Miniaturized Biosensor and Biofuel Cell Electrodes. Biosens. Bioelectron. 2014, 58, 121–131. [Google Scholar] [CrossRef]
- Cordelair, J.; Greil, P. Discrete Element Modeling of Solid Formation during Electrophoretic Deposition. J. Mater. Sci. 2004, 39, 1017–1021. [Google Scholar] [CrossRef]
- Bergman, J.; Wang, Y.; Wigström, J.; Cans, A.S. Counting the Number of Enzymes Immobilized onto a Nanoparticle-Coated Electrode. Anal. Bioanal. Chem. 2018, 410, 1775–1783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagerman, A.E. Fifty Years of Polyphenol-Protein Complexes. In Recent Advances in Polyphenol Research; Cheynier, V., Sarni-Manchado, P., Quideau, S., Eds.; Wiley-Blackwell: Hoboken, NJ, USA, 2012; Volume 3, pp. 71–91. [Google Scholar]
- Yang, J.; Stuart, M.A.C.; Kamperman, M. Jack of All Trades: Versatile Catechol Crosslinking Mechanisms. Chem. Soc. Rev. 2014, 43, 8271–8298. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Xu, W.; Wu, P.; Zhang, H.; Cai, C.; Zhao, B. New Insights into the Effects of Thermal Treatment on the Catalytic Activity and Conformational Structure of Glucose Oxidase Studied by Electrochemistry, IR Spectroscopy, and Theoretical Calculation. J. Phys. Chem. B 2010, 114, 12754–12764. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Xu, Y.; Xia, C.; Li, S. Effect of Cyclodextrin on the Activity and Secondary Structure of Horseradish Peroxidase. Protein Pept. Lett. 2005, 11, 509–513. [Google Scholar] [CrossRef] [PubMed]
- Zhong, K.; Hu, X.; Zhao, G.; Chen, F.; Liao, X. Inactivation and Conformational Change of Horseradish Peroxidase Induced by Pulsed Electric Field. Food Chem. 2005, 92, 473–479. [Google Scholar] [CrossRef]
- Liu, X.; Atwater, M.; Wang, J.; Huo, Q. Extinction Coefficient of Gold Nanoparticles with Different Sizes and Different Capping Ligands. Colloids Surfaces B Biointerfaces 2007, 53, 3–7. [Google Scholar] [CrossRef]
- El-Maiss, J.; Cuccarese, M.; Lupattelli, P.; Chiummiento, L.; Funicello, M.; Schaaf, P.; Boulmedais, F. Mussel-Inspired Electro-Cross-Linking of Enzymes for the Development of Biosensors. ACS Appl. Mater. Inter. 2018, 10, 18574–18584. [Google Scholar] [CrossRef] [Green Version]
- Niethammer, P.; Grabher, C.; Look, A.T.; Mitchison, T.J. A Tissue-Scale Gradient of Hydrogen Peroxide Mediates Rapid Wound Detection in Zebrafish. Nature 2009, 459, 996–999. [Google Scholar] [CrossRef]
- Dunnill, C.; Patton, T.; Brennan, J.; Barrett, J.; Dryden, M.; Cooke, J.; Leaper, D.; Georgopoulos, N.T. Reactive Oxygen Species (ROS) and Wound Healing: The Functional Role of ROS and Emerging ROS-Modulating Technologies for Augmentation of the Healing Process. Int. Wound J. 2017, 14, 89–96. [Google Scholar] [CrossRef]
- Singh, K.; McArdle, T.; Sullivan, P.R.; Blanford, C.F. Sources of Activity Loss in the Fuel Cell Enzyme Bilirubin Oxidase. Energy Environ. Sci. 2013, 6, 2460. [Google Scholar] [CrossRef] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Savin, R.; Blanck, C.; Benzaamia, N.-O.; Boulmedais, F. Optimization of Nanohybrid Biosensors Based on Electro-Crosslinked Tannic Acid Capped Nanoparticles/Enzyme. Molecules 2022, 27, 3309. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27103309
Savin R, Blanck C, Benzaamia N-O, Boulmedais F. Optimization of Nanohybrid Biosensors Based on Electro-Crosslinked Tannic Acid Capped Nanoparticles/Enzyme. Molecules. 2022; 27(10):3309. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27103309
Chicago/Turabian StyleSavin, Rémy, Christian Blanck, Nour-Ouda Benzaamia, and Fouzia Boulmedais. 2022. "Optimization of Nanohybrid Biosensors Based on Electro-Crosslinked Tannic Acid Capped Nanoparticles/Enzyme" Molecules 27, no. 10: 3309. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27103309