
Comparing Efficiency of Lysis Buffer Solutions and Sample Preparation Methods for Liquid Chromatography–Mass Spectrometry Analysis of Human Cells and Plasma

 , ,
, ,
Abstract
:1. Introduction
2. Results
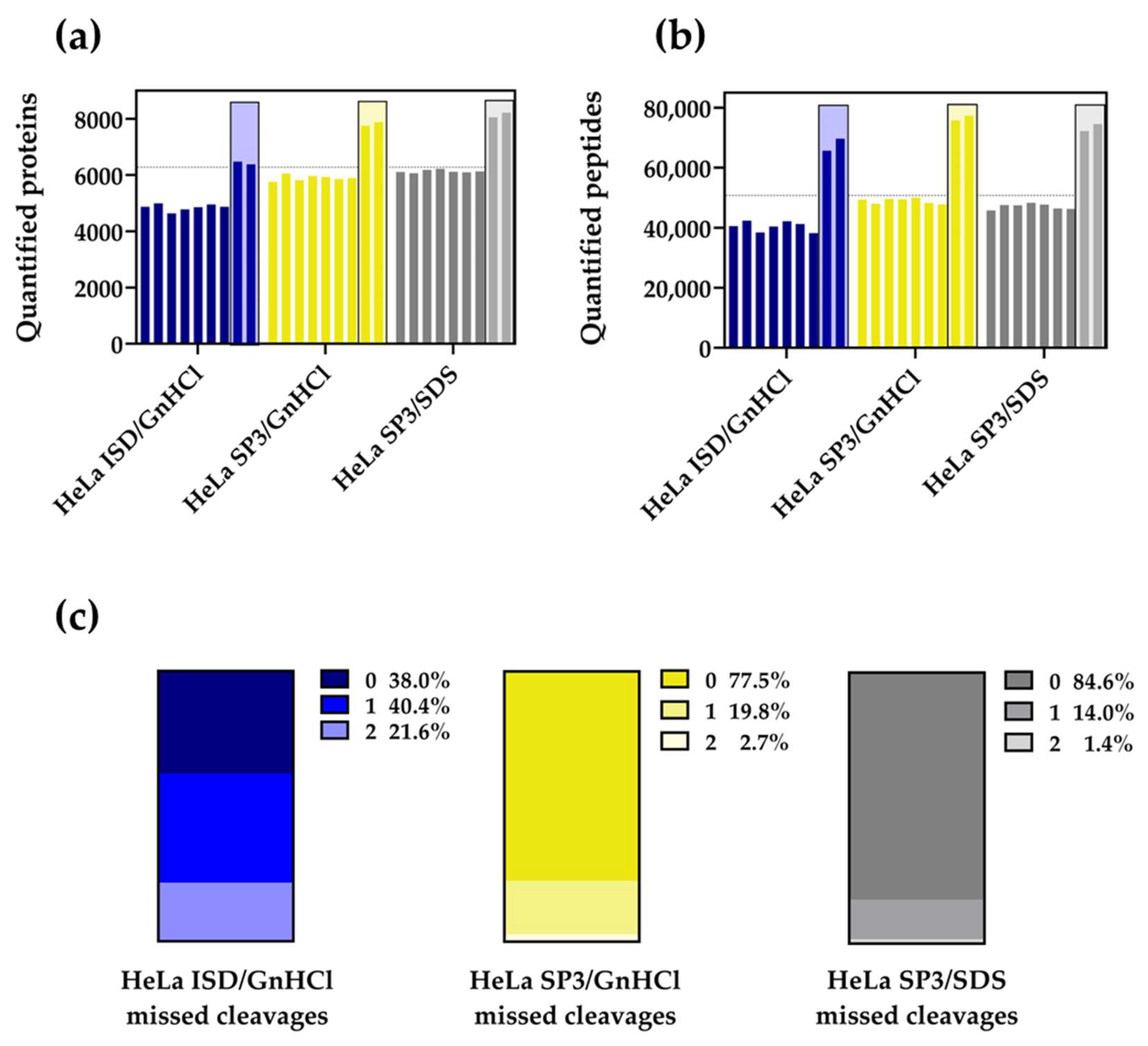
2.1. The SP3 Protocol Outperforms the ISD Strategy with HeLa Cell Extracts
2.2. The SP3 Protocol Enhances the Quantification of Membrane Proteomes
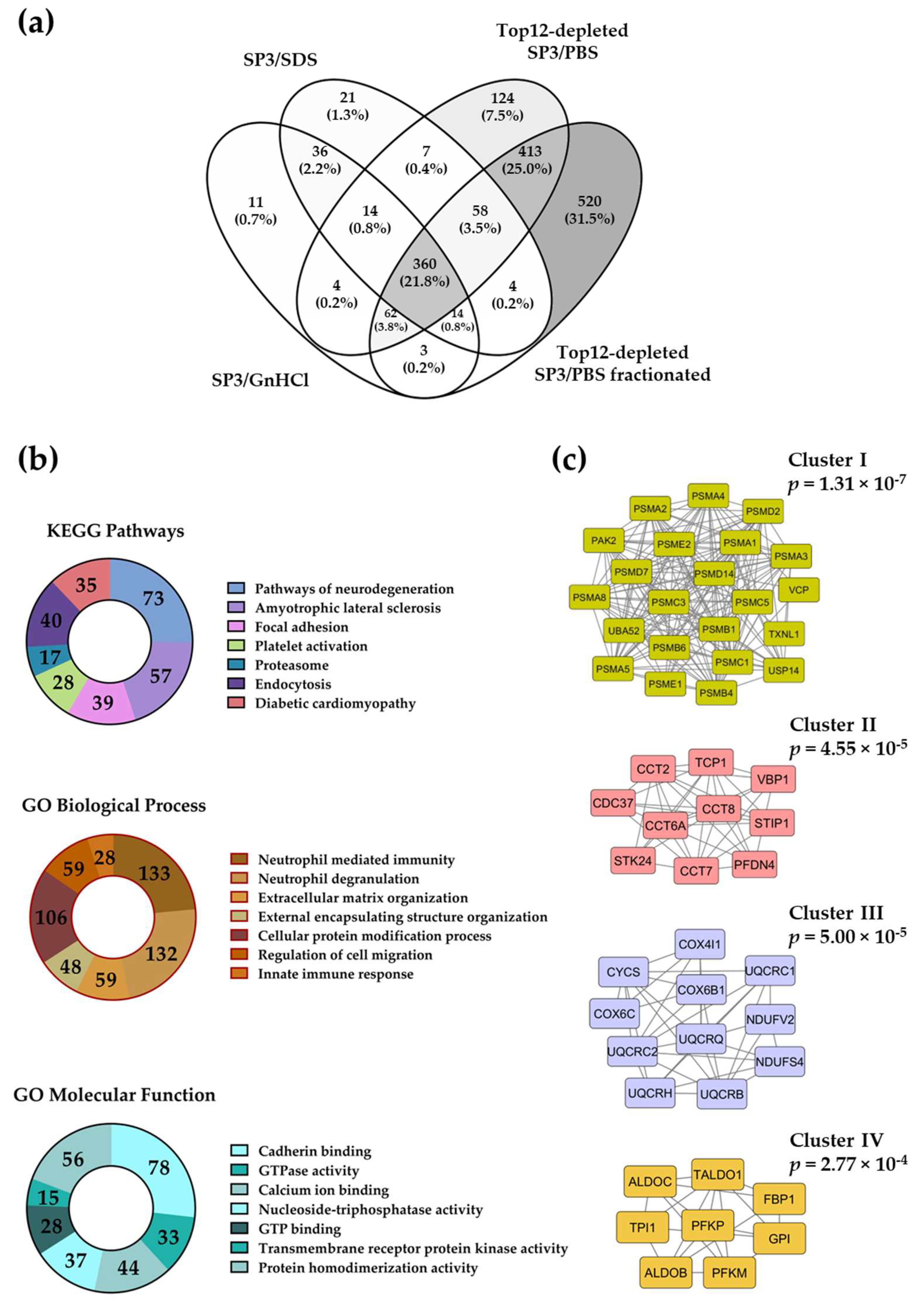
2.3. The SP3 Protocol with Depleted Plasma Samples Reaches Deeper Proteome Coverage
2.4. Evaluation of the Removal of Highly Abundant Proteins in Plasma Samples with Commercial Mini Spin Depletion Columns
3. Discussion
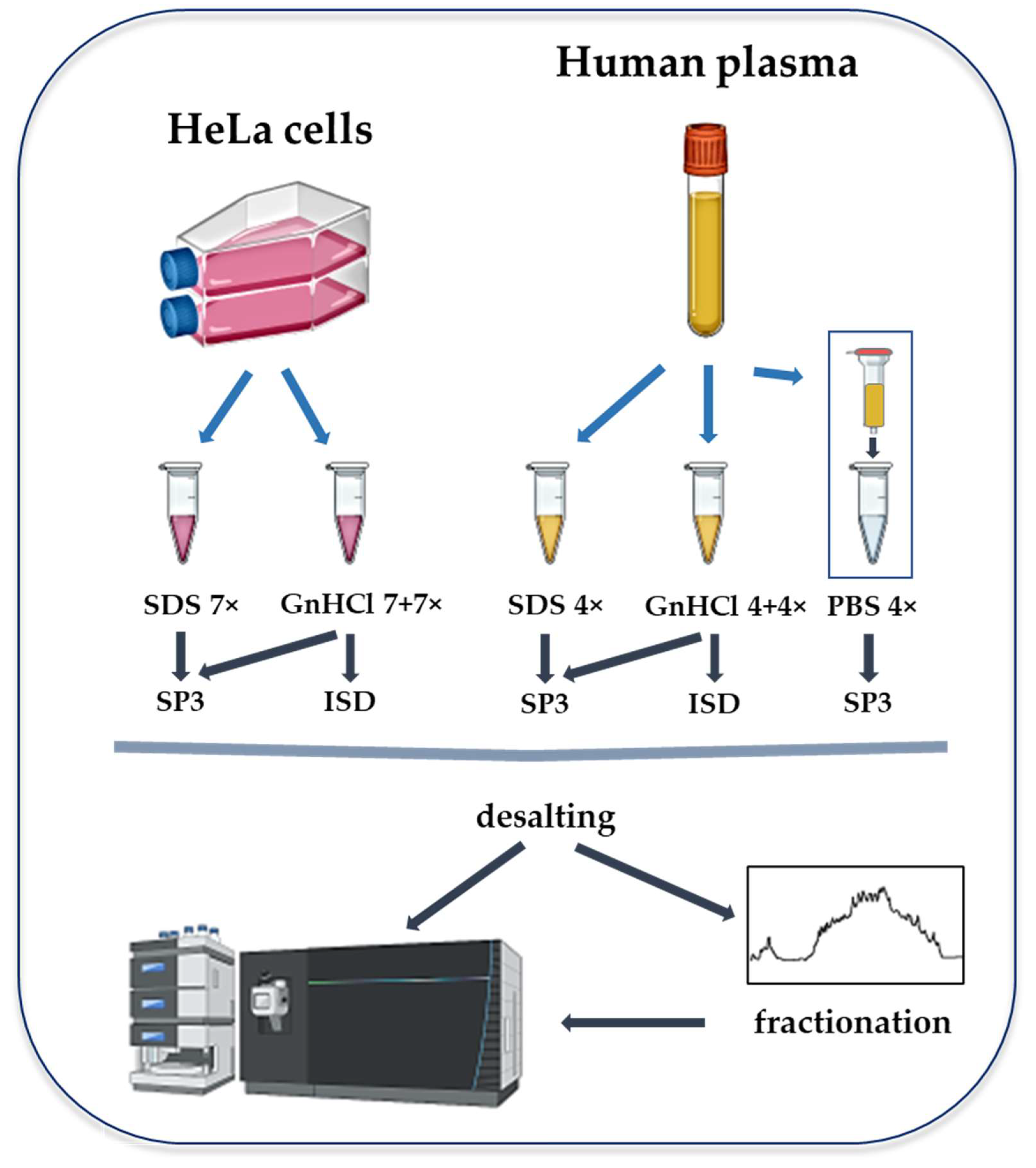
4. Materials and Methods
4.1. Cell Culture and Plasma
4.2. Protein Extraction
4.2.1. GnHCl- and SDS-Based Lysis
4.2.2. Depletion of Abundant Plasma Proteins
4.3. Protein Sample Digestion
4.3.1. ISD Protein Digestion
4.3.2. Protein Digestion with the SP3 Methodology
4.4. Peptide Cleaning
4.5. Offline High pH Reversed-Phase HPLC Fractionation
4.6. LC-MS/MS Analysis
4.7. Statistics and Data Processing
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Smith, B.J.; Martins-de-Souza, D. Biological Applications for LC-MS-Based Proteomics. Adv. Exp. Med. Biol. 2021, 1336, 17–29. [Google Scholar] [PubMed]
- Zhang, Y.; Fonslow, B.R.; Shan, B.; Baek, M.C.; Yates, J.R., 3rd. Protein analysis by shotgun/bottom-up proteomics. Chem. Rev. 2013, 113, 2343–2394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogers, J.C.; Bomgarden, R.D. Sample Preparation for Mass Spectrometry-Based Proteomics from Proteomes to Peptides. In Modern Proteomics—Sample Preparation, Analysis and Practical Applications; Mirzaei, H., Carrasco, M., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 43–62. [Google Scholar]
- Sielaff, M.; Kuharev, J.; Bohn, T.; Hahlbrock, J.; Bopp, T.; Tenzer, S.; Distler, U. Evaluation of FASP, SP3, and iST Protocols for Proteomic Sample Preparation in the Low Microgram Range. J. Proteome Res. 2017, 16, 4060–4072. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.Y.; Dann, G.P.; Shi, T.; Wang, L.; Gao, X.; Su, D.; Nicora, C.D.; Shukla, A.K.; Moore, R.J.; Liu, T.; et al. Simple sodium dodecyl sulfate-assisted sample preparation method for LC-MS-based proteomics applications. Anal. Chem. 2012, 84, 2862–2867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masuda, T.; Tomita, M.; Ishihama, Y. Phase Transfer Surfactant-Aided Trypsin Digestion for Membrane Proteome Analysis. J. Proteome Res. 2008, 7, 731–740. [Google Scholar] [CrossRef]
- Speers, A.E.; Wu, C.C. Proteomics of integral membrane proteins—Theory and application. Chem. Rev. 2007, 107, 3687–3714. [Google Scholar] [CrossRef]
- Wiśniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef]
- Wang, X.; Liang, S. Sample preparation for the analysis of membrane proteomes by mass spectrometry. Protein Cell 2012, 3, 661–668. [Google Scholar] [CrossRef] [Green Version]
- Kachuk, C.; Stephen, K.; Doucette, A. Comparison of sodium dodecyl sulfate depletion techniques for proteome analysis by mass spectrometry. J. Chromatogr. A 2015, 1418, 158–166. [Google Scholar] [CrossRef]
- Hughes, C.S.; Moggridge, S.; Müller, T.; Sorensen, P.H.; Morin, G.B.; Krijgsveld, J. Single-pot, solid-phase-enhanced sample preparation for proteomics experiments. Nat. Protoc. 2019, 14, 68–85. [Google Scholar] [CrossRef]
- Hughes, C.S.; Foehr, S.; Garfield, D.A.; Furlong, E.E.; Steinmetz, L.M.; Krijgsveld, J. Ultrasensitive proteome analysis using paramagnetic bead technology. Mol. Syst. Biol. 2014, 10, 757. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, K.R.; Schroll, M.M.; Hummon, A.B. Comparison of In-Solution, FASP, and S-Trap Based Digestion Methods for Bottom-Up Proteomic Studies. J. Proteome Res 2018, 17, 2480–2490. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Jiang, H.; Yan, Y.; Peng, B.; Chen, J.; Lin, H.; Liu, Z. Shotgun analysis of membrane proteomes by an improved SDS-assisted sample preparation method coupled with liquid chromatography-tandem mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2012, 911, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.Q.; Gilar, M.; Lee, P.J.; Bouvier, E.S.; Gebler, J.C. Enzyme-friendly, mass spectrometry-compatible surfactant for in-solution enzymatic digestion of proteins. Anal. Chem. 2003, 75, 6023–6028. [Google Scholar] [CrossRef]
- Proc, J.L.; Kuzyk, M.A.; Hardie, D.B.; Yang, J.; Smith, D.S.; Jackson, A.M.; Parker, C.E.; Borchers, C.H. A quantitative study of the effects of chaotropic agents, surfactants, and solvents on the digestion efficiency of human plasma proteins by trypsin. J. Proteome Res. 2010, 9, 5422–5437. [Google Scholar] [CrossRef] [Green Version]
- Ignjatovic, V.; Geyer, P.E.; Palaniappan, K.K.; Chaaban, J.E.; Omenn, G.S.; Baker, M.S.; Deutsch, E.W.; Schwenk, J.M. Mass Spectrometry-Based Plasma Proteomics: Considerations from Sample Collection to Achieving Translational Data. J. Proteome Res. 2019, 18, 4085–4097. [Google Scholar] [CrossRef]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.N.; Wang, Z.C.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [Green Version]
- Cao, X.F.; Sandberg, A.; Araujo, J.E.; Cvetkovski, F.; Berglund, E.; Eriksson, L.E.; Pernemalm, M. Evaluation of Spin Columns for Human Plasma Depletion to Facilitate MS-Based Proteomics Analysis of Plasma. J. Proteome Res. 2021, 20, 4610–4620. [Google Scholar] [CrossRef]
- Bekker-Jensen, D.B.; Bernhardt, O.M.; Hogrebe, A.; Martinez-Val, A.; Verbeke, L.; Gandhi, T.; Kelstrup, C.D.; Reiter, L.; Olsen, J.V. Rapid and site-specific deep phosphoproteome profiling by data-independent acquisition without the need for spectral libraries. Nat. Commun. 2020, 11, 787. [Google Scholar] [CrossRef] [Green Version]
- Franciosa, G.; Smits, J.G.A.; Minuzzo, S.; Martinez-Val, A.; Indraccolo, S.; Olsen, J.V. Proteomics of resistance to Notch1 inhibition in acute lymphoblastic leukemia reveals targetable kinase signatures. Nat. Commun. 2021, 12, 2507. [Google Scholar] [CrossRef]
- Mikulasek, K.; Konecna, H.; Potesil, D.; Holankova, R.; Havlis, J.; Zdrahal, Z. SP3 Protocol for Proteomic Plant Sample Preparation Prior LC-MS/MS. Front Plant Sci. 2021, 12, 635550. [Google Scholar] [CrossRef] [PubMed]
- Keshishian, H.; Burgess, M.W.; Specht, H.; Wallace, L.; Clauser, K.R.; Gillette, M.A.; Carr, S.A. Quantitative, multiplexed workflow for deep analysis of human blood plasma and biomarker discovery by mass spectrometry. Nat. Protoc. 2017, 12, 1683–1701. [Google Scholar] [CrossRef] [PubMed]
- Kaur, G.; Poljak, A.; Ali, S.A.; Zhong, L.; Raftery, M.J.; Sachdev, P. Extending the Depth of Human Plasma Proteome Coverage Using Simple Fractionation Techniques. J. Proteome Res. 2021, 20, 1261–1279. [Google Scholar] [CrossRef] [PubMed]
- Pernemalm, M.; Sandberg, A.; Zhu, Y.; Boekel, J.; Tamburro, D.; Schwenk, J.M.; Bjork, A.; Wahren-Herlenius, M.; Amark, H.; Ostenson, C.G.; et al. In-depth human plasma proteome analysis captures tissue proteins and transfer of protein variants across the placenta. Elife 2019, 8, e41608. [Google Scholar] [CrossRef] [PubMed]
- Blume, J.E.; Manning, W.C.; Troiano, G.; Hornburg, D.; Figa, M.; Hesterberg, L.; Platt, T.L.; Zhao, X.; Cuaresma, R.A.; Everley, P.A.; et al. Rapid, deep and precise profiling of the plasma proteome with multi-nanoparticle protein corona. Nat. Commun. 2020, 11, 3662. [Google Scholar] [CrossRef]
- Eng, J.K.; McCormack, A.L.; Yates, J.R. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 1994, 5, 976–989. [Google Scholar] [CrossRef] [Green Version]
- Kall, L.; Storey, J.D.; MacCoss, M.J.; Noble, W.S. Assigning significance to peptides identified by tandem mass spectrometry using decoy databases. J. Proteome Res. 2008, 7, 29–34. [Google Scholar] [CrossRef]
- Tyanova, S.; Cox, J. Perseus: A Bioinformatics Platform for Integrative Analysis of Proteomics Data in Cancer Research. Methods Mol. Biol. 2018, 1711, 133–148. [Google Scholar]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Nepusz, T.; Yu, H.; Paccanaro, A. Detecting overlapping protein complexes in protein-protein interaction networks. Nat. Methods 2012, 9, 471–472. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ISD/GnHCl | SP3/GnHCl | SP3/SDS | Top12-Depleted SP3/PBS | Top12-Depleted SP3/PBS & Fractionated | |
|---|---|---|---|---|---|
| Quantifiedproteins [1] | 361 (35) | 397 (32) | 411 (35) | 833 (76) | 1397 (48) [2] |
| Quantifiedpeptides [1] | 3822 (309) | 3726 (172) | 3117 (134) | 5860 (624) | 8431 (617) [2] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neset, L.; Takayidza, G.; Berven, F.S.; Hernandez-Valladares, M. Comparing Efficiency of Lysis Buffer Solutions and Sample Preparation Methods for Liquid Chromatography–Mass Spectrometry Analysis of Human Cells and Plasma. Molecules 2022, 27, 3390. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27113390
Neset L, Takayidza G, Berven FS, Hernandez-Valladares M. Comparing Efficiency of Lysis Buffer Solutions and Sample Preparation Methods for Liquid Chromatography–Mass Spectrometry Analysis of Human Cells and Plasma. Molecules. 2022; 27(11):3390. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27113390
Chicago/Turabian StyleNeset, Lasse, Gracious Takayidza, Frode S. Berven, and Maria Hernandez-Valladares. 2022. "Comparing Efficiency of Lysis Buffer Solutions and Sample Preparation Methods for Liquid Chromatography–Mass Spectrometry Analysis of Human Cells and Plasma" Molecules 27, no. 11: 3390. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27113390