
Integration of Ligand-Based and Structure-Based Methods for the Design of Small-Molecule TLR7 Antagonists

 ,
,
Abstract
:
1. Introduction
2. Results
2.1. 2D-QSAR Model
2.1.1. Development of 2D-QSAR Model
- VE3sign_D/Dt: the logarithmic coefficient sum of the last eigenvector from the distance–detour matrix [43].
- SpMin2_Bh(s): the second-smallest eigenvalue of the Burden Matrix of the H-filled molecular graph weighted by intrinsic state [44].
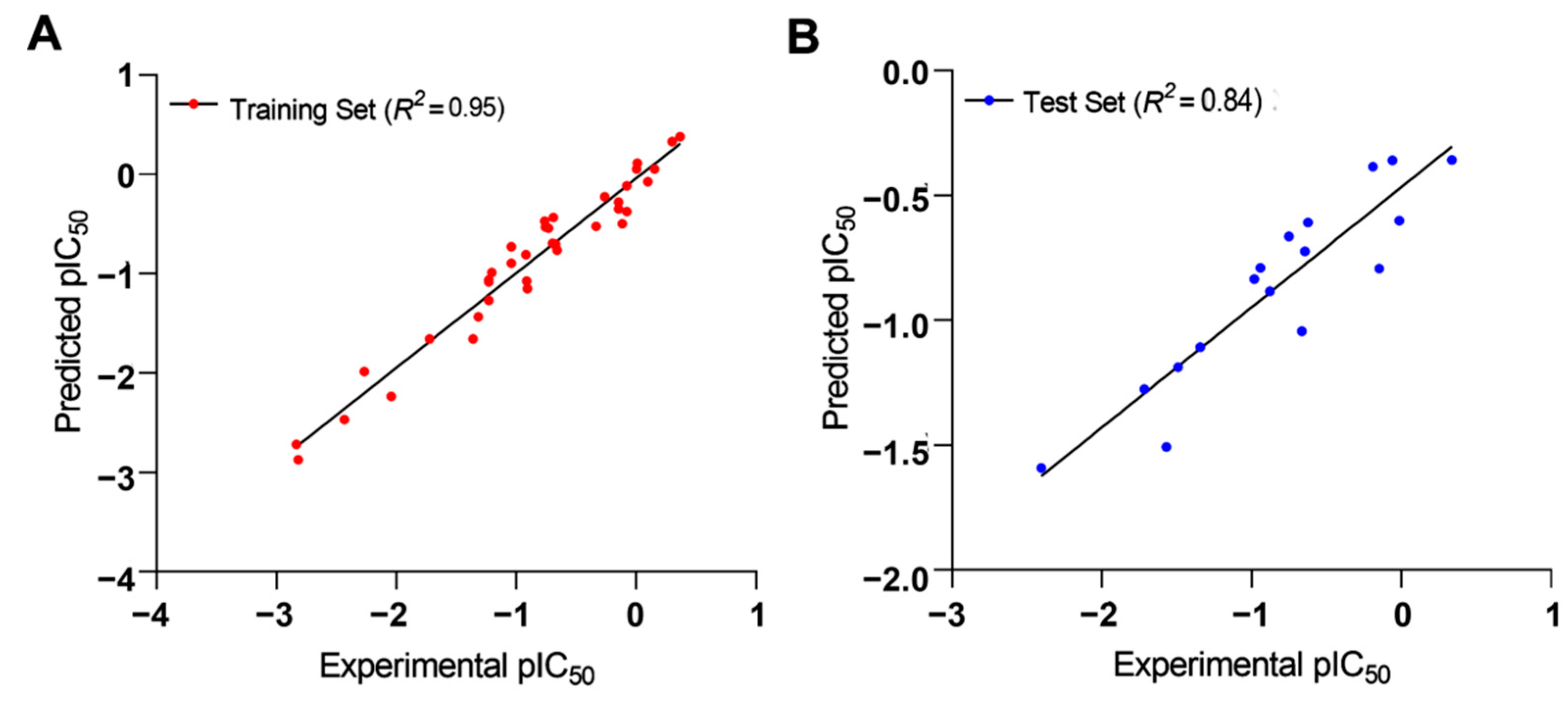
2.1.2. Validation of 2D-QSAR Model
Internal Validation
External Validation
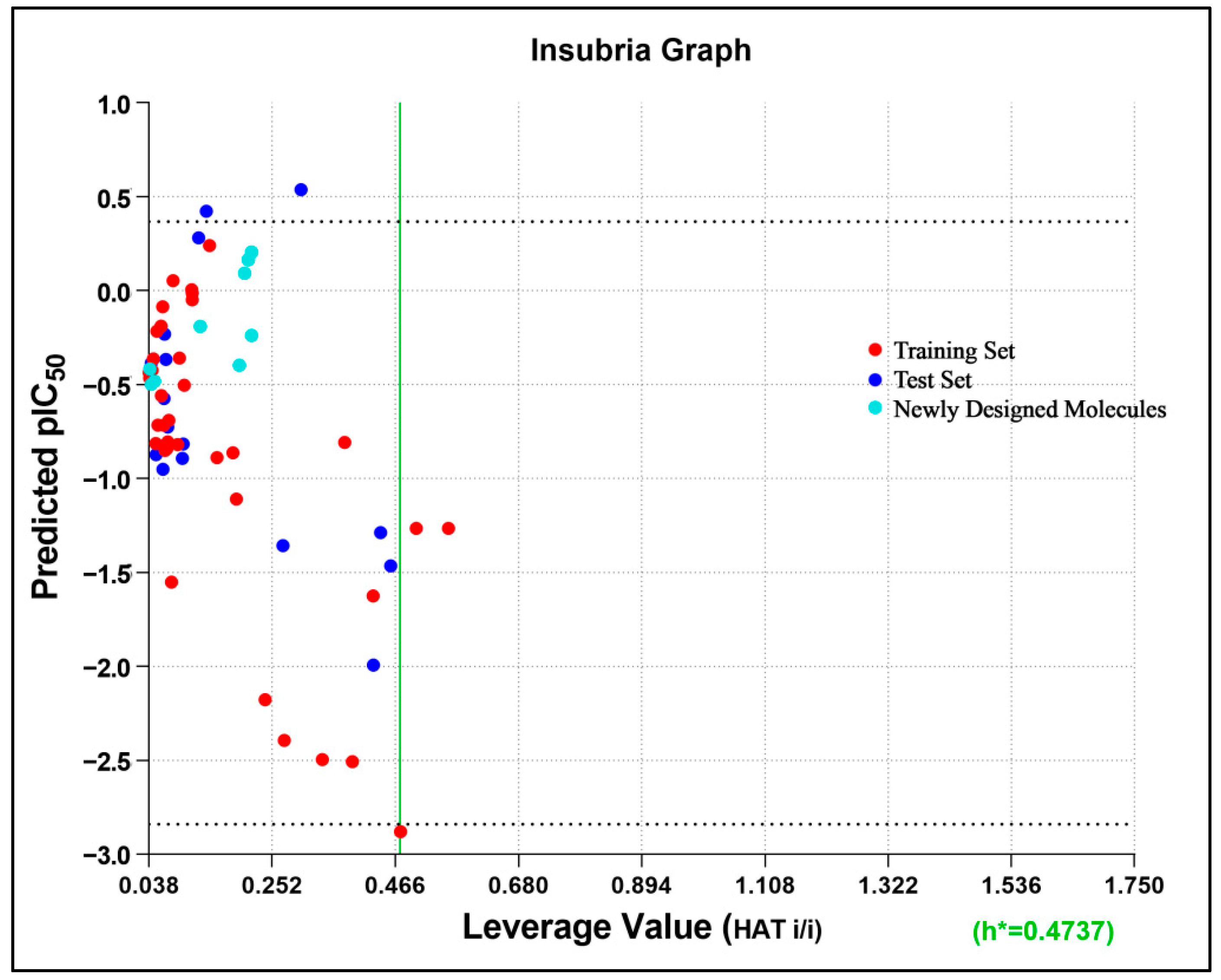
2.1.3. Applicability Domain
2.2. Pharmacophore Model
2.2.1. Development of Pharmacophore Models
2.2.2. Pharmacophore Validation
Cost Analysis Method
Test Set Analysis
Fischer Randomization
2.3. 3D-QSAR
2.3.1. Development of the 3D-QSAR Model
2.3.2. 3D-QSAR Model Validation
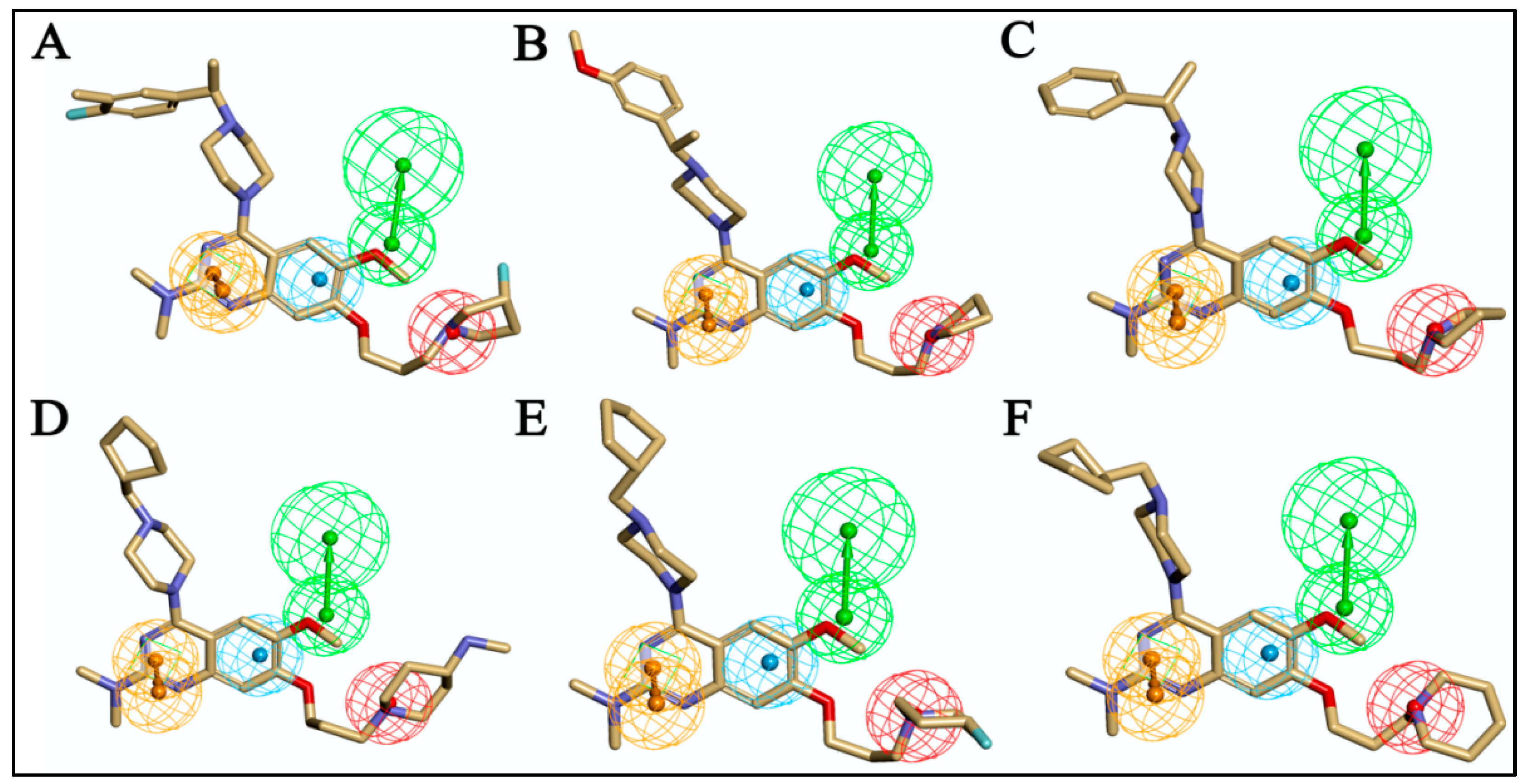
2.3.3. Analysis of 3D-QSAR Contour Maps
2.4. Design of New Compounds
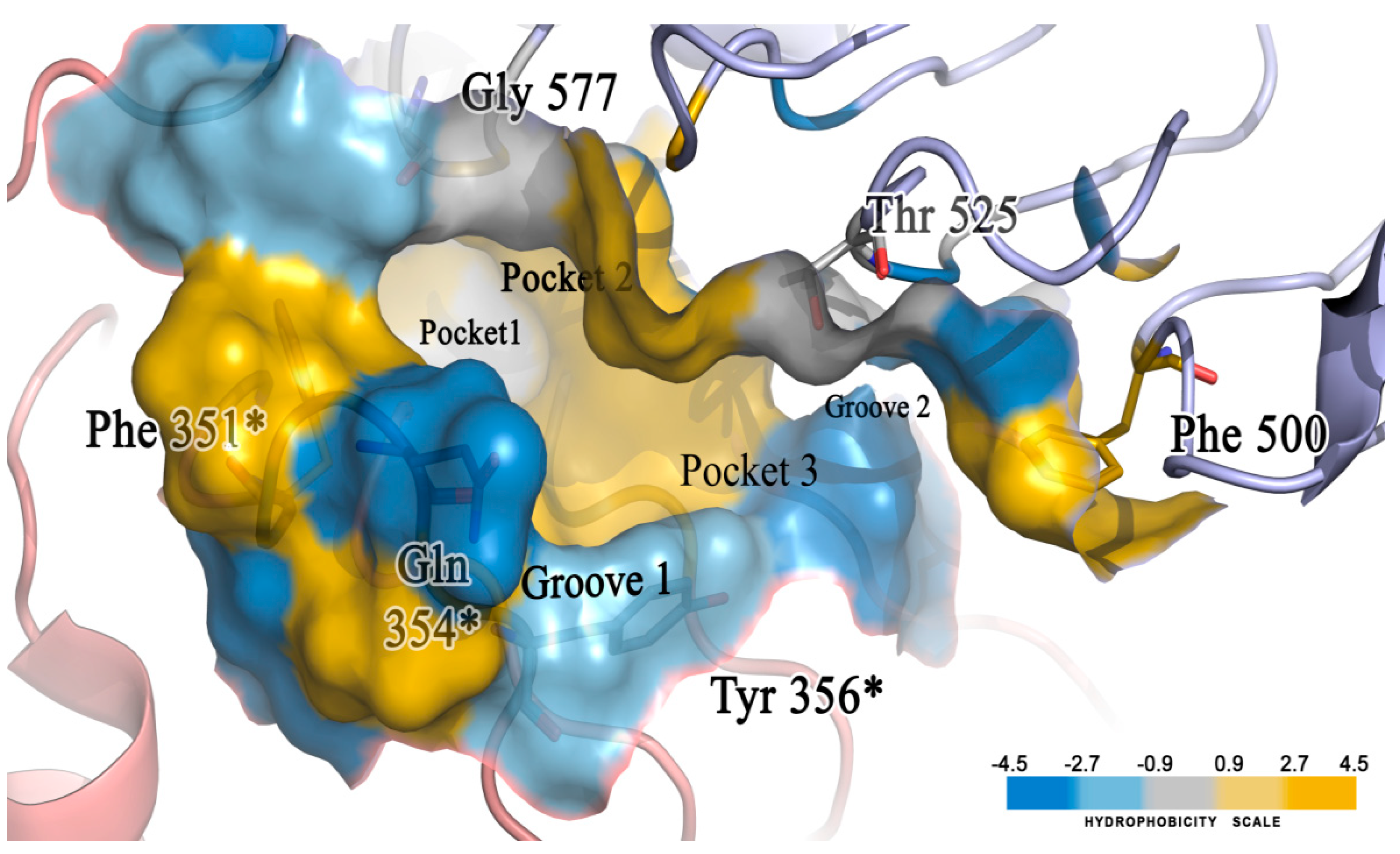
2.5. Molecular Docking of the Newly Designed Compounds
2.6. In Silico Pharmacokinetics Predictions
2.7. Toxicity Risk Assessment Screening
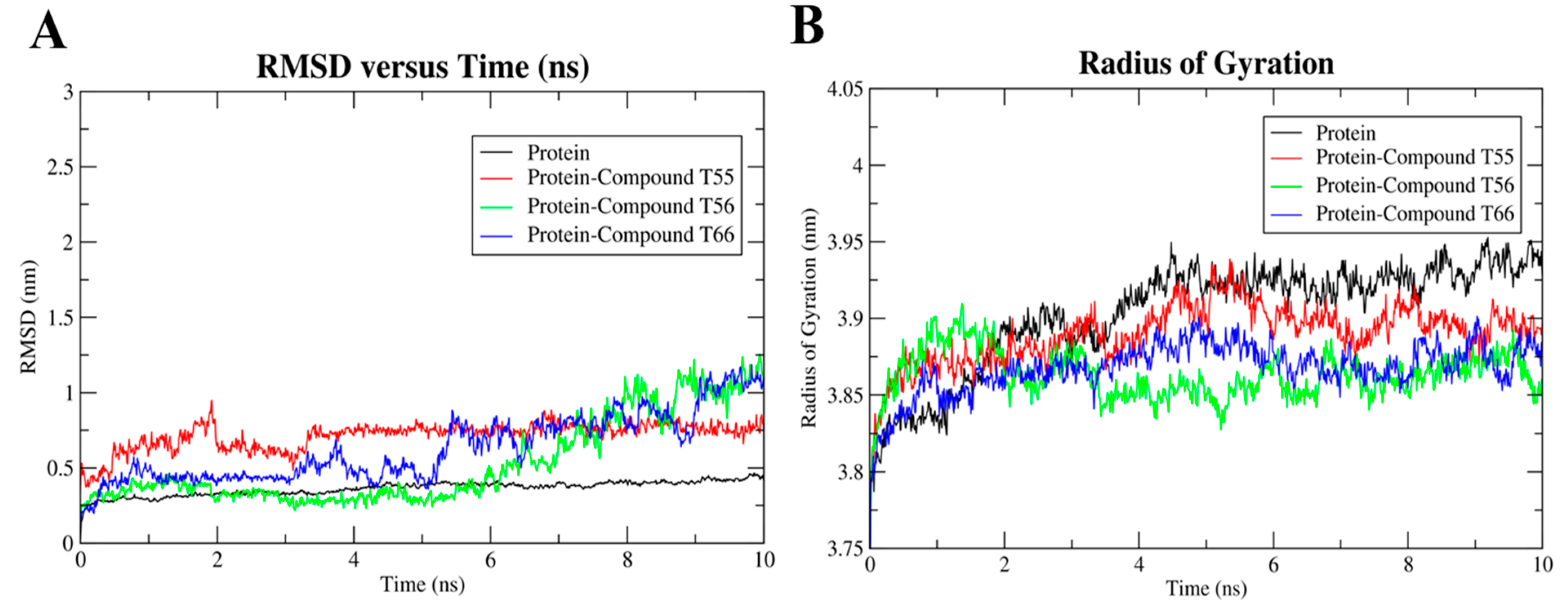
2.8. Molecular Dynamics Simulation
3. Materials and Methods
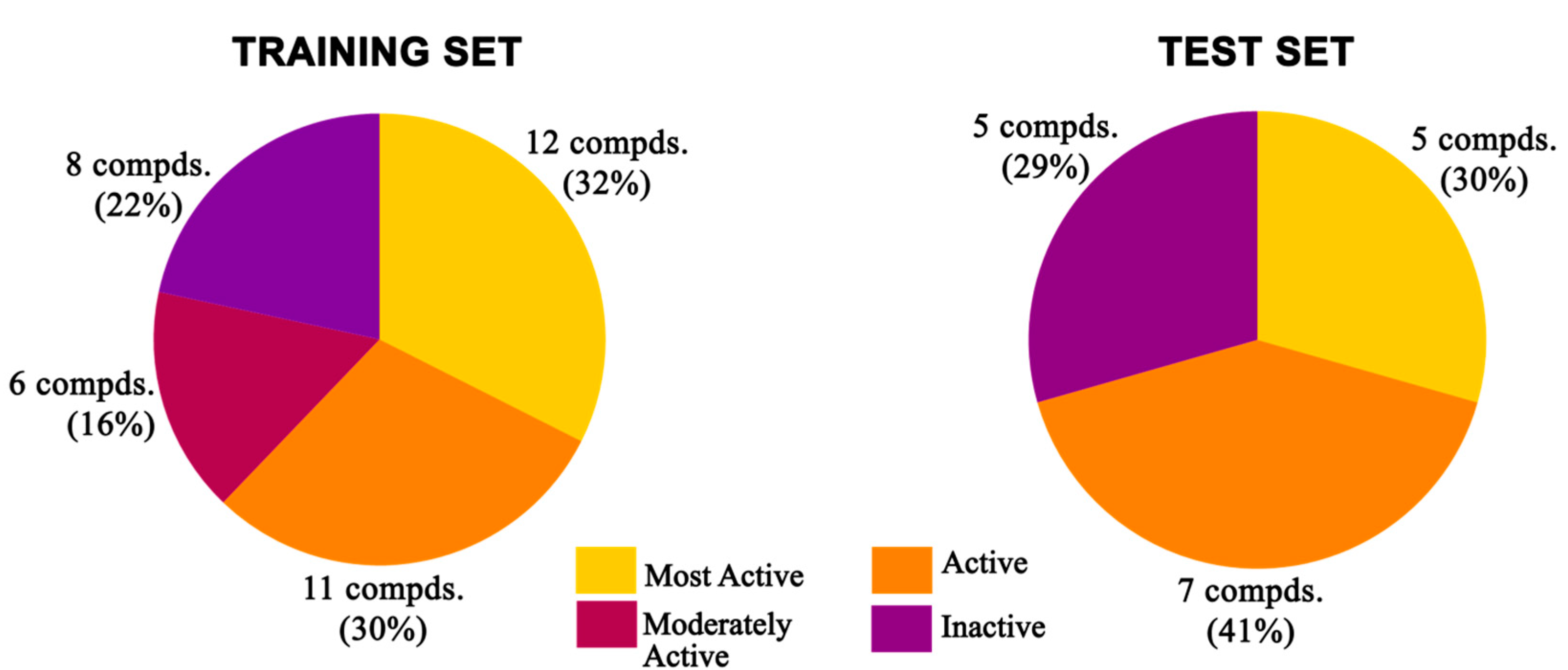
3.1. Dataset Selection
3.2. 2D-QSAR
3.2.1. 2D-QSAR Model Generation
3.2.2. 2D-QSAR Model Validation
3.2.3. Interpretation of Descriptors of the Developed 2D-QSAR Model
- VE3sign_D/Dt, the first descriptor of the 2D-QSAR model equation, is expressed as a negative coefficient. It represents the logarithmic coefficient sum of the last eigenvector from the distance–detour matrix [43] and is expressed as the following equation:where li represents the coefficient of the eigenvector associated with the largest negative eigenvalue calculated on the distance–detour matrix [78]. The distance–detour matrix is a square symmetric matrix comprising the ratio between the shortest and the longest topological distances between two atoms in the constitutional molecular graph [79,80]. It is evident that this is a geometrical descriptor and does not draw any impact from the properties of the atoms. Thus, decreasing the number of detour or cyclic components (retaining at least some of them) in the molecular topology increased the value of this descriptor, which was favorable for activity. It was evident in compounds 36, 38, and 39, which had high descriptor values, where the aliphatic straight chain was abundant, and the structure followed an overall linear connection.
- SpMin2_Bh(s) bears the largest coefficient value with a positive sign. It represents the second-smallest eigenvalue of the Burden Matrix of the H-filled molecular graph weighted by intrinsic state [43,81]. It is a square symmetric matrix expressed as:where πb is the bond order (1 for single, 2 for double, 3 for triple, and 1.5 for aromatic bonds) [43,82]; ω is the intrinsic value; an electrotopological index (I) of the atom is elucidated as I = ; δv and δ are the counts of valence and sigma electrons, respectively; and N is the principal quantum number [81]. Atoms in groups such as halogens, amines, and azide hydroxyl have comparatively high intrinsic values. Also, the number of unsaturation, especially connected terminal unsaturation, can contribute to the overall increase in the component values of the matrix. These aspects can have an impact on the descriptor being a positive contributor. Because the I-state was higher for electron-withdrawing groups (=N-: 3.00, >N-: 2.00, -O-: 3.50) [81], it was reflected in compound 37 with the highest descriptor value, which had several occurrences of such atoms.
- P_VSA_logP_5, the third parameter of the model, is a lipophilicity-based descriptor representing the P_VSA-like on LogP, bin 5, that is the sum of the Van der Waals surface area of atoms with logP values in the range of 0 to 0.25. This descriptor can also positively influence the activity, having a positively signed coefficient, and it is presented by both the size and hydrophobicity values of the atoms [45]. The Alvascience user manual [43] lists the individual octanol-water partition coefficient values of 115 atom-centered fragments, in which groups such as CR2X2, =CR2, =CX2, R:CR:R, R…O…R, and R-O-C=X specifically bear logP values ranging from 0 to 0.25 [83,84]. The atoms belonging to these and having larger atomic Van der Waals surface areas can be beneficially incorporated to enhance activity.where Ri is the atomic Van der Waals radius of the atom i, nAT is the number of atoms, aij are the elements of the adjacency matrix, and dij = min{max{|Ri − Rj|, bij}, (Ri + Rj). In addition, bij is the bond length between i and j (bij = rij − cij); rij is the reference bond length; and cij is 0, 0.1, 0.2, and 0.3 for single, aromatic, double, and triple bonds, respectively [43].
- Eig02_EA(dm), or the second eigenvalue from the edge adjacency matrix weighted by the dipole moment [43,46], is a negative contributor with a significant coefficient value, which emphasizes that adjacent bonds with large dipole moments are likely to decrease activity. It indicates that the substituent groups having greater charge distributions inflicted by electronegative atoms can have a negative influence if the involved bond is branched and connected to several other components in the H-depleted molecular connection map.
- CATS2D_09_AA is the number of hydrogen bond acceptors at an in-between topological distance of 9 bonds [43,47] and points out the frequency of such occurrences as a negative contributor to activity. Although the central core bears several nitrogen atoms that can be potential hydrogen bond acceptors, a topological distance of 9 bonds is not very frequent. However, as in the case of molecule 3, the symmetric pattern of the carbonyl oxygen atoms, piperazine ring, and fused pyrimidine contributed to the large value of this descriptor.
3.2.4. Applicability Domain
3.3. Pharmacophore Model Generation
3.3.1. Generation of Pharmacophore Hypothesis with 3D-QSAR Pharmacophore Generation (Hypogen)
3.3.2. Pharmacophore Validation
3.4. 3D-QSAR Model Generation
3.4.1. Molecular Alignment
3.4.2. 3D-QSAR Model Development and Validation
3.5. Molecular Docking
3.6. ADMET and Toxicity Prediction
3.7. MD Simulation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Takeda, K.; Kaisho, T.; Akira, S. Toll-Like Receptors. Annu. Rev. Immunol. 2003, 21, 335–376. [Google Scholar] [CrossRef]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen Recognition and Innate Immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [Green Version]
- Akira, S.; Takeda, K. Toll-like Receptor Signalling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar] [CrossRef]
- Akira, S.; Takeda, K.; Kaisho, T. Toll-like Receptors: Critical Proteins Linking Innate and Acquired Immunity. Nat. Immunol. 2001, 2, 675–680. [Google Scholar] [CrossRef]
- Gay, N.J.; Symmons, M.F.; Gangloff, M.; Bryant, C.E. Assembly and Localization of Toll-like Receptor Signalling Complexes. Nat. Rev. Immunol. 2014, 14, 546–558. [Google Scholar] [CrossRef]
- Gay, N.J.; Gangloff, M. Structure and Function of Toll Receptors and Their Ligands. Annu. Rev. Biochem. 2007, 76, 141–165. [Google Scholar] [CrossRef]
- Junt, T.; Barchet, W. Translating Nucleic Acid-Sensing Pathways into Therapies. Nat. Rev. Immunol. 2015, 15, 529–544. [Google Scholar] [CrossRef]
- He, S.; Mao, X.; Sun, H.; Shirakawa, T.; Zhang, H.; Wang, X. Potential Therapeutic Targets in the Process of Nucleic Acid Recognition: Opportunities and Challenges. Trends Pharmacol. Sci. 2015, 36, 51–64. [Google Scholar] [CrossRef]
- Lee, B.L.; Barton, G.M. Trafficking of Endosomal Toll-like Receptors. Trends Cell Biol. 2014, 24, 360–369. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.-B.; Dammer, E.B.; Ren, R.-J.; Wang, G. The Endosomal-Lysosomal System: From Acidification and Cargo Sorting to Neurodegeneration. Transl. Neurodegener. 2015, 4, 18. [Google Scholar] [CrossRef] [Green Version]
- Gilliet, M.; Cao, W.; Liu, Y.-J. Plasmacytoid Dendritic Cells: Sensing Nucleic Acids in Viral Infection and Autoimmune Diseases. Nat. Rev. Immunol. 2008, 8, 594–606. [Google Scholar] [CrossRef] [PubMed]
- Maeda, K.; Akira, S. TLR7 Structure: Cut in Z-Loop. Immunity 2016, 45, 705–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemmi, H.; Takeuchi, O.; Kawai, T.; Kaisho, T.; Sato, S.; Sanjo, H.; Matsumoto, M.; Hoshino, K.; Wagner, H.; Takeda, K.; et al. A Toll-like Receptor Recognizes Bacterial DNA. Nature 2000, 408, 740–745. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Rothenfusser, S.; Britsch, S.; Krug, A.; Jahrsdörfer, B.; Giese, T.; Endres, S.; Hartmann, G. Quantitative Expression of Toll-Like Receptor 1–10 MRNA in Cellular Subsets of Human Peripheral Blood Mononuclear Cells and Sensitivity to CpG Oligodeoxynucleotides. J. Immunol. 2002, 168, 4531–4537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lande, R.; Gregorio, J.; Facchinetti, V.; Chatterjee, B.; Wang, Y.H.; Homey, B.; Cao, W.; Wang, Y.H.; Su, B.; Nestle, F.O.; et al. Plasmacytoid Dendritic Cells Sense Self-DNA Coupled with Antimicrobial Peptide. Nature 2007, 449, 564–569. [Google Scholar] [CrossRef]
- Ganguly, D.; Chamilos, G.; Lande, R.; Gregorio, J.; Meller, S.; Facchinetti, V.; Homey, B.; Barrat, F.J.; Zal, T.; Gilliet, M. Self-RNA-Antimicrobial Peptide Complexes Activate Human Dendritic Cells through TLR7 and TLR8. J. Exp. Med. 2009, 206, 1983–1994. [Google Scholar] [CrossRef]
- Lande, R.; Ganguly, D.; Facchinetti, V.; Frasca, L.; Conrad, C.; Gregorio, J.; Meller, S.; Chamilos, G.; Sebasigari, R.; Riccieri, V.; et al. Neutrophils Activate Plasmacytoid Dendritic Cells by Releasing Self-DNA-Peptide Complexes in Systemic Lupus Erythematosus. Sci. Transl. Med. 2011, 3, 73ra19. [Google Scholar] [CrossRef] [Green Version]
- Rudnicka, W.; Burakowski, T.; Warnawin, E.; Jastzebska, M.; Bik, M.; Kontny, E.; Chorazy-Massalska, M.; Radzikowkska, A.; Buler, M.; Maldyk, P.; et al. Functional TLR9 Modulates Bone Marrow B Cells from Rheumatoid Arthritis Patients. Eur. J. Immunol. 2009, 39, 1211–1220. [Google Scholar] [CrossRef] [PubMed]
- Gottenberg, J.E.; Cagnard, N.; Lucchesi, C.; Letourneur, F.; Mistou, S.; Lazure, T.; Jacques, S.; Ba, N.; Ittah, M.; Lepajolec, C.; et al. Activation of IFN Pathways and Plasmacytoid Dendritic Cell Recruitment in Target Organs of Primary Sjögren’s Syndrome. Proc. Natl. Acad. Sci. USA 2006, 103, 2770–2775. [Google Scholar] [CrossRef] [Green Version]
- Diana, J.; Simoni, Y.; Furio, L.; Beaudoin, L.; Agerberth, B.; Barrat, F.; Lehuen, A. Crosstalk between Neutrophils, B-1a Cells and Plasmacytoid Dendritic Cells Initiates Autoimmune Diabetes. Nat. Med. 2013, 19, 65–73. [Google Scholar] [CrossRef]
- Ghosh, A.R.; Bhattacharya, R.; Bhattacharya, S.; Nargis, T.; Rahaman, O.; Duttagupta, P.; Raychaudhuri, D.; Liu, C.S.C.; Roy, S.; Ghosh, P.; et al. Adipose Recruitment and Activation of Plasmacytoid Dendritic Cells Fuel Metaflammation. Diabetes 2016, 65, 3440–3452. [Google Scholar] [CrossRef] [Green Version]
- Patinote, C.; Karroum, N.B.; Moarbess, G.; Cirnat, N.; Kassab, I.; Bonnet, P.-A.; Deleuze-Masquéfa, C. Agonist and Antagonist Ligands of Toll-like Receptors 7 and 8: Ingenious Tools for Therapeutic Purposes. Eur. J. Med. Chem. 2020, 193, 112238. [Google Scholar] [CrossRef] [PubMed]
- Federico, S.; Pozzetti, L.; Papa, A.; Carullo, G.; Gemma, S.; Butini, S.; Campiani, G.; Relitti, N. Modulation of the Innate Immune Response by Targeting Toll-like Receptors: A Perspective on Their Agonists and Antagonists. J. Med. Chem. 2020, 63, 13466–13513. [Google Scholar] [CrossRef] [PubMed]
- Hennessy, E.J.; Parker, A.E.; O’Neill, L.A.J. Targeting Toll-like Receptors: Emerging Therapeutics? Nat. Rev. Drug Discov. 2010, 9, 293–307. [Google Scholar] [CrossRef]
- Marshak-Rothstein, A. Toll-like Receptors in Systemic Autoimmune Disease. Nat. Rev. Immunol. 2006, 6, 823–835. [Google Scholar] [CrossRef]
- El-Zayat, S.R.; Sibaii, H.; Mannaa, F.A. Toll-like Receptors Activation, Signaling, and Targeting: An Overview. Bull. Natl. Res. Cent. 2019, 43, 187. [Google Scholar] [CrossRef] [Green Version]
- Anwar, M.A.; Shah, M.; Kim, J.; Choi, S. Recent Clinical Trends in Toll-like Receptor Targeting Therapeutics. Med. Res. Rev. 2019, 39, 1053–1090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, W.; Xiong, Y.; Li, Q.; Yang, H. Inhibition of Toll-Like Receptor Signaling as a Promising Therapy for Inflammatory Diseases: A Journey from Molecular to Nano Therapeutics. Front. Physiol. 2017, 8, 508. [Google Scholar] [CrossRef]
- Paul, B.; Rahaman, O.; Roy, S.; Pal, S.; Satish, S.; Mukherjee, A.; Ghosh, A.R.; Raychaudhuri, D.; Bhattacharya, R.; Goon, S.; et al. Activity-Guided Development of Potent and Selective Toll-like Receptor 9 Antagonists. Eur. J. Med. Chem. 2018, 159, 187–205. [Google Scholar] [CrossRef]
- Talukdar, A.; Ganguly, D.; Paul, B.; Mukherjee, A.; Roy, S.; Roy, S.; Ghosh, A.R.; Bhattacharya, R.; Rahaman, O.; Kundu, B. Blocking Toll-like Receptor 9 Signaling with Small Molecule Antagonist. U.S. Patent US20190092758, 28 March 2019. [Google Scholar]
- Lamphier, M.; Zheng, W.; Latz, E.; Spyvee, M.; Hansen, H.; Rose, J.; Genest, M.; Yang, H.; Shaffer, C.; Zhao, Y.; et al. Novel Small Molecule Inhibitors of TLR7 and TLR9: Mechanism of Action and Efficacy in Vivo. Mol. Pharmacol. 2014, 85, 429–440. [Google Scholar] [CrossRef] [Green Version]
- Zheng, W.; Spyvee, M.; Gusovsky, F.; Ishizaka, S.T.; Gazzinelli, R.; Golenbock, D.T. Use of Benzoxazole Compounds in the Treatment of Malaria. WO Patent WO2010036908, 1 April 2010. [Google Scholar]
- Roy, S.; Mukherjee, A.; Paul, B.; Rahaman, O.; Roy, S.; Maithri, G.; Ramya, B.; Pal, S.; Ganguly, D.; Talukdar, A. Design and Development of Benzoxazole Derivatives with Toll-like Receptor 9 Antagonism. Eur. J. Med. Chem. 2017, 134, 334–347. [Google Scholar] [CrossRef] [PubMed]
- Shukla, N.M.; Mutz, C.A.; Malladi, S.S.; Warshakoon, H.J.; Balakrishna, R.; David, S.A. Toll-Like Receptor (TLR)-7 and -8 Modulatory Activities of Dimeric Imidazoquinolines. J. Med. Chem. 2012, 55, 1106–1116. [Google Scholar] [CrossRef] [Green Version]
- Švajger, U.; Horvat, Ž.; Knez, D.; Rožman, P.; Turk, S.; Gobec, S. New Antagonists of Toll-like Receptor 7 Discovered through 3D Ligand-Based Virtual Screening. Med. Chem. Res. 2015, 24, 362–371. [Google Scholar] [CrossRef]
- Mukherjee, A.; Raychaudhuri, D.; Sinha, B.P.; Kundu, B.; Mitra, M.; Paul, B.; Bandopadhyay, P.; Ganguly, D.; Talukdar, A. A Chemical Switch for Transforming a Purine Agonist for Toll-like Receptor 7 to a Clinically Relevant Antagonist. J. Med. Chem. 2020, 63, 4776–4789. [Google Scholar] [CrossRef] [PubMed]
- Kundu, B.; Raychaudhuri, D.; Mukherjee, A.; Sinha, B.P.; Sarkar, D.; Bandopadhyay, P.; Pal, S.; Das, N.; Dey, D.; Ramarao, K.; et al. Systematic Optimization of Potent and Orally Bioavailable Purine Scaffold as a Dual Inhibitor of Toll-Like Receptors 7 and 9. J. Med. Chem. 2021, 64, 9279–9301. [Google Scholar] [CrossRef]
- Talukdar, A.; Ganguly, D.; Mukherjee, A.; Paul, B.; Rahaman, O.; Kundu, B.; Roy, S.; Raychaudhuri, D. Purine Based Compounds as Toll-Like Receptor 9 Antagonist. U.S. Patent US20200347062, 5 November 2020. [Google Scholar]
- Talukdar, A.; Ganguly, D.; Roy, S.; Das, N.; Sarkar, D. Structural Evolution and Translational Potential for Agonists and Antagonists of Endosomal Toll-like Receptors. J. Med. Chem. 2021, 64, 8010–8041. [Google Scholar] [CrossRef]
- Talukdar, A.; Mukherjee, A.; Ganguly, D. Small Molecule Modulators of Endo-Lysosomal Toll-like Receptors. In Protein–Protein Interaction Regulators; The Royal Society of Chemistry: London, UK, 2021; pp. 339–372. ISBN 978-1-78801-187-7. [Google Scholar]
- Pal, S.; Paul, B.; Bandopadhyay, P.; Preethy, N.; Sarkar, D.; Rahaman, O.; Goon, S.; Roy, S.; Ganguly, D.; Talukdar, A. Synthesis and Characterization of New Potent TLR7 Antagonists Based on Analysis of the Binding Mode Using Biomolecular Simulations. Eur. J. Med. Chem. 2021, 210, 112978. [Google Scholar] [CrossRef]
- Sushko, I.; Novotarskyi, S.; Körner, R.; Pandey, A.K.; Rupp, M.; Teetz, W.; Brandmaier, S.; Abdelaziz, A.; Prokopenko, V.V.; Tanchuk, V.Y.; et al. Online Chemical Modeling Environment (OCHEM): Web Platform for Data Storage, Model Development and Publishing of Chemical Information. J. Comput. Aided. Mol. Des. 2011, 25, 533–554. [Google Scholar] [CrossRef] [Green Version]
- Andrea Mauri AlvaDesc: A Tool to Calculate and Analyze Molecular Descriptors and Fingerprints. In Ecotoxicological QSARs; Springer: New York, NY, USA, 2020; pp. 801–820. ISBN 978-1-0716-0150-1.
- Burden, F.R. Molecular Identification Number for Substructure Searches. J. Chem. Inf. Comput. Sci. 1989, 29, 225–227. [Google Scholar] [CrossRef]
- Labute, P. A Widely Applicable Set of Descriptors. J. Mol. Graph. Model. 2000, 18, 464–477. [Google Scholar] [CrossRef]
- Mauri, A.; Consonni, V.; Pavan, M.; Todeschini, R.; Chemometrics, M. DRAGON Software: An Easy Approach to Molecular Descriptor Calculations. MATCH Commun. Math. Comput. Chem. 2006, 56, 237–248. [Google Scholar]
- Carhart, R.E.; Smith, D.H.; Venkataraghavan, R. Atom Pairs as Molecular Features in Structure-Activity Studies: Definition and Applications. J. Chem. Inf. Comput. Sci. 1985, 25, 64–73. [Google Scholar] [CrossRef]
- Gramatica, P. On the Development and Validation of QSAR Models. Methods Mol. Biol. 2013, 930, 499–526. [Google Scholar] [CrossRef]
- Pratim Roy, P.; Paul, S.; Mitra, I.; Roy, K. On Two Novel Parameters for Validation of Predictive QSAR Models. Molecules 2009, 14, 1660–1701. [Google Scholar] [CrossRef]
- Wehrens, R.; Putter, H.; Buydens, L.M.C. The Bootstrap: A Tutorial. Chemom. Intell. Lab. Syst. 2000, 54, 35–52. [Google Scholar] [CrossRef]
- Eriksson, L.; Jaworska, J.; Worth, A.P.; Cronin, M.T.D.; McDowell, R.M.; Gramatica, P. Methods for Reliability and Uncertainty Assessment and for Applicability Evaluations of Classification- and Regression-Based QSARs. Environ. Health Perspect. 2003, 111, 1361–1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, L.M.; Fang, H.; Tong, W.; Wu, J.; Perkins, R.; Blair, R.M.; Branham, W.S.; Dial, S.L.; Moland, C.L.; Sheehan, D.M. QSAR Models Using a Large Diverse Set of Estrogens. J. Chem. Inf. Comput. Sci. 2001, 41, 186–195. [Google Scholar] [CrossRef]
- Tropsha, A.; Gramatica, P.; Gombar, V.K. The Importance of Being Earnest: Validation Is the Absolute Essential for Successful Application and Interpretation of QSPR Models. QSAR Comb. Sci. 2003, 22, 69–77. [Google Scholar] [CrossRef]
- Schüürmann, G.; Ebert, R.-U.; Chen, J.; Wang, B.; Kühne, R. External Validation and Prediction Employing the Predictive Squared Correlation Coefficient—Test Set Activity Mean vs Training Set Activity Mean. J. Chem. Inf. Model. 2008, 48, 2140–2145. [Google Scholar] [CrossRef]
- Consonni, V.; Ballabio, D.; Todeschini, R. Comments on the Definition of the Q2 Parameter for QSAR Validation. J. Chem. Inf. Model. 2009, 49, 1669–1678. [Google Scholar] [CrossRef] [PubMed]
- Chirico, N.; Gramatica, P. Real External Predictivity of QSAR Models: How to Evaluate It? Comparison of Different Validation Criteria and Proposal of Using the Concordance Correlation Coefficient. J. Chem. Inf. Model. 2011, 51, 2320–2335. [Google Scholar] [CrossRef]
- Roy, K.; Kar, S.; Ambure, P. On a Simple Approach for Determining Applicability Domain of QSAR Models. Chemom. Intell. Lab. Syst. 2015, 145, 22–29. [Google Scholar] [CrossRef]
- Jaworska, J.; Nikolova-Jeliazkova, N.; Aldenberg, T. QSAR Applicabilty Domain Estimation by Projection of the Training Set Descriptor Space: A Review. Altern. Lab. Anim. 2005, 33, 445–459. [Google Scholar] [CrossRef]
- John, S.; Thangapandian, S.; Arooj, M.; Hong, J.C.; Kim, K.D.; Lee, K.W. Development, Evaluation and Application of 3D QSAR Pharmacophore Model in the Discovery of Potential Human Renin Inhibitors. BMC Bioinform. 2011, 12, S4. [Google Scholar] [CrossRef] [Green Version]
- Islam, M.A.; Pillay, T.S. Structural Requirements for Potential HIV-Integrase Inhibitors Identified Using Pharmacophore-Based Virtual Screening and Molecular Dynamics Studies. Mol. Biosyst. 2016, 12, 982–993. [Google Scholar] [CrossRef] [Green Version]
- Schuster, D.; Laggner, C.; Steindl, T.M.; Palusczak, A.; Hartmann, R.W.; Langer, T. Pharmacophore Modeling and in Silico Screening for New P450 19 (Aromatase) Inhibitors. J. Chem. Inf. Model. 2006, 46, 1301–1311. [Google Scholar] [CrossRef] [PubMed]
- Gramatica, P.; Cassani, S.; Roy, P.P.; Kovarich, S.; Yap, C.W.; Papa, E. QSAR Modeling Is Not “Push a Button and Find a Correlation”: A Case Study of Toxicity of (Benzo-)Triazoles on Algae. Mol. Inform. 2012, 31, 817–835. [Google Scholar] [CrossRef]
- Brandmaier, S.; Peijnenburg, W.; Durjava, M.; Kolar, B.; Gramatica, P.; Papa, E.; Bhhatarai, B.; Kovarich, S.; Cassani, S.; Roy, P.P.; et al. The QSPR-THESAURUS: The Online Platform of the CADASTER Project. Altern. Lab. Anim. 2014, 42, 13–24. [Google Scholar] [CrossRef]
- Watanabe, M.; Kasai, M.; Tomizawa, H.; Aoki, M.; Eiho, K.; Isobe, Y.; Asano, S. Dihydropyrrolo[2,3-d]Pyrimidines: Selective Toll-Like Receptor 9 Antagonists from Scaffold Morphing Efforts. ACS Med. Chem. Lett. 2014, 5, 1235–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponnan, P.; Gupta, S.; Chopra, M.; Tandon, R.; Baghel, A.S.; Gupta, G.; Prasad, A.K.; Rastogi, R.C.; Bose, M.; Raj, H.G. 2D-QSAR, Docking Studies, and In Silico ADMET Prediction of Polyphenolic Acetates as Substrates for Protein Acetyltransferase Function of Glutamine Synthetase of Mycobacterium Tuberculosis. ISRN Struct. Biol. 2013, 2013, 373516. [Google Scholar] [CrossRef] [Green Version]
- Gaur, R.; Cheema, H.S.; Kumar, Y.; Singh, S.P.; Yadav, D.K.; Darokar, M.P.; Khan, F.; Bhakuni, R.S. In Vitro Antimalarial Activity and Molecular Modeling Studies of Novel Artemisinin Derivatives. RSC Adv. 2015, 5, 47959–47974. [Google Scholar] [CrossRef]
- Palm, K.; Stenberg, P.; Luthman, K.; Artursson, P. Polar Molecular Surface Properties Predict the Intestinal Absorption of Drugs in Humans. Pharm. Res. 1997, 14, 568–571. [Google Scholar] [CrossRef]
- Egan, W.J.; Merz, K.M.; Baldwin, J.J. Prediction of Drug Absorption Using Multivariate Statistics. J. Med. Chem. 2000, 43, 3867–3877. [Google Scholar] [CrossRef]
- Debnath, U.; Verma, S.; Singh, P.; Rawat, K.; Gupta, S.K.; Tripathi, R.K.; Siddiqui, H.H.; Katti, S.B.; Prabhakar, Y.S. Synthesis, Biological Evaluation and Molecular Modeling Studies of New 2,3-Diheteroaryl Thiazolidin-4-Ones as NNRTIs. Chem. Biol. Drug Des. 2015, 86, 1285–1291. [Google Scholar] [CrossRef]
- Elmchichi, L.; Belhassan, A.; Lakhlifi, T.; Bouachrine, M. 3D-QSAR Study of the Chalcone Derivatives as Anticancer Agents. J. Chem. 2020, 2020, 5268985. [Google Scholar] [CrossRef]
- Gramatica, P. Principles of QSAR Modeling: Comments and Suggestions from Personal Experience. Int. J. Quant. Struct. Relatsh. 2020, 5, 61–97. [Google Scholar] [CrossRef]
- Gramatica, P.; Chirico, N.; Papa, E.; Cassani, S.; Kovarich, S. QSARINS: A New Software for the Development, Analysis, and Validation of QSAR MLR Models. J. Comput. Chem. 2013, 34, 2121–2132. [Google Scholar] [CrossRef]
- Leardi, R.; Boggia, R.; Terrile, M. Genetic Algorithms as a Strategy for Feature Selection. J. Chemom. 1992, 6, 267–281. [Google Scholar] [CrossRef]
- Environment Directorate, Organization for Economic Co-operation and Development. Guidance Document on the Validation of (Quantitative) Structure-Activity Relationship [(Q)SAR] Models; OECD Series on Testing and Assessment, No. 69; OECD Publishing: Paris, France, 2007. [Google Scholar] [CrossRef]
- Stone, M. Cross-Validatory Choice and Assessment of Statistical Predictions. J. R. Stat. Soc. Ser. B 1974, 36, 111–133. [Google Scholar] [CrossRef]
- Roy, K.; Mitra, I. On Various Metrics Used for Validation of Predictive QSAR Models with Applications in Virtual Screening and Focused Library Design. Comb. Chem. High Throughput Screen. 2011, 14, 450–474. [Google Scholar] [CrossRef]
- Consonni, V.; Ballabio, D.; Todeschini, R. Evaluation of Model Predictive Ability by External Validation Techniques. J. Chemom. 2010, 24, 194–201. [Google Scholar] [CrossRef]
- Balaban, A.T.; Ciubotariu, D.; Medeleanu, M. Topological Indices and Real Number Vertex Invariants Based on Graph Eigenvalues or Eigenvectors. J. Chem. Inf. Comput. Sci. 1991, 31, 517–523. [Google Scholar] [CrossRef]
- Randić, M. On Characterization of Cyclic Structures. J. Chem. Inf. Comput. Sci. 1997, 37, 1063–1071. [Google Scholar] [CrossRef]
- Balaban, A. Topological Indices Based on Topological Distances in Molecular Graph. Pure Appl. Chem. 1988, 55, 199–206. [Google Scholar] [CrossRef]
- Kier, L.B.; Hall, L.H. An Electrotopological-State Index for Atoms in Molecules. Pharm. Res. 1990, 7, 801–807. [Google Scholar] [CrossRef]
- Ivanciuc, O. QSAR Comparative Study of Wiener Descriptors for Weighted Molecular Graphs. J. Chem. Inf. Comput. Sci. 2000, 40, 1412–1422. [Google Scholar] [CrossRef]
- Viswanadhan, V.N.; Ghose, A.K.; Revankar, G.R.; Robins, R.K. Atomic Physicochemical Parameters for Three Dimensional Structure Directed Quantitative Structure-Activity Relationships. 4. Additional Parameters for Hydrophobic and Dispersive Interactions and Their Application for an Automated Superposition of Certain. J. Chem. Inf. Comput. Sci. 1989, 29, 163–172. [Google Scholar] [CrossRef]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. Prediction of Hydrophobic (Lipophilic) Properties of Small Organic Molecules Using Fragmental Methods: An Analysis of ALOGP and CLOGP Methods. J. Phys. Chem. A 1998, 102, 3762–3772. [Google Scholar] [CrossRef]
- Atkinson, A.C. Plots, Transformations, and Regression: An Introduction to Graphical Methods of Diagnostic Regression Analysis; Oxford Statistical Science Series; Clarendon Press: Oxford, UK, 1985; ISBN 0198533594. [Google Scholar]
- Gramatica, P. Principles of QSAR Models Validation: Internal and External. QSAR Comb. Sci. 2007, 26, 694–701. [Google Scholar] [CrossRef]
- BIOVIA. Dassault Systèmes, Discovery Studio, V18.1; Dassault Systèmes: San Diego, CA, USA, 2018. [Google Scholar]
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and Ligand Preparation: Parameters, Protocols, and Influence on Virtual Screening Enrichments. J. Comput. Aided. Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Kandakatla, N.; Ramakrishnan, G. Ligand Based Pharmacophore Modeling and Virtual Screening Studies to Design Novel HDAC2 Inhibitors. Adv. Bioinform. 2014, 2014, 812148. [Google Scholar] [CrossRef] [Green Version]
- Yang, B.; Yang, Y.S.; Yang, N.; Li, G.; Zhu, H.L. Design, Biological Evaluation and 3D QSAR Studies of Novel Dioxin-Containing Pyrazoline Derivatives with Thiourea Skeleton as Selective HER-2 Inhibitors. Sci. Rep. 2016, 6, 27571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shahin, R.; Swellmeen, L.; Shaheen, O.; Aboalhaija, N.; Habash, M. Identification of Novel Inhibitors for Pim-1 Kinase Using Pharmacophore Modeling Based on a Novel Method for Selecting Pharmacophore Generation Subsets. J. Comput. Aided. Mol. Des. 2016, 30, 39–68. [Google Scholar] [CrossRef]
- Che, J.; Wang, Z.; Sheng, H.; Huang, F.; Dong, X.; Hu, Y.; Xie, X.; Hu, Y. Ligand-Based Pharmacophore Model for the Discovery of Novel CXCR2 Antagonists as Anti-Cancer Metastatic Agents. R. Soc. Open Sci. 2018, 5, 180176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.H.; Yi, G.S. Prediction of Novel Anoctamin1 (ANO1) Inhibitors Using 3D-QSAR Pharmacophore Modeling and Molecular Docking. Int. J. Mol. Sci. 2018, 19, 3204. [Google Scholar] [CrossRef] [Green Version]
- Pal, S.; Kumar, V.; Kundu, B.; Bhattacharya, D.; Preethy, N.; Reddy, M.P.; Talukdar, A. Ligand-Based Pharmacophore Modeling, Virtual Screening and Molecular Docking Studies for Discovery of Potential Topoisomerase I Inhibitors. Comput. Struct. Biotechnol. J. 2019, 17, 291–310. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Choi, J.; Lee, K.; No, K.T. Comparison of Three-Dimensional Ligand-Based Pharmacophores among 11 Phosphodiesterases (PDE 1 to PDE 11) Pharmacophores. Bull. Korean Chem. Soc. 2017, 38, 1033–1037. [Google Scholar] [CrossRef]
- Zou, X.J.; Lai, L.H.; Jin, G.Y.; Zhang, Z.X. Synthesis, Fungicidal Activity, and 3D-QSAR of Pyridazinone-Substituted 1,3,4-Oxadiazoles and 1,3,4-Thiadiazoles. J. Agric. Food Chem. 2002, 50, 3757–3760. [Google Scholar] [CrossRef]
- Ren, J.X.; Zhang, R.T.; Zhang, H. Identifying Novel ATX Inhibitors via Combinatory Virtual Screening Using Crystallography-Derived Pharmacophore Modelling, Docking Study, and QSAR Analysis. Molecules 2020, 25, 1107. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; Yang, H.; Wu, K.; Ji, Y.; Ju, L.; Lu, X. Novel Pyrazoline Derivatives as Bi-Inhibitor of COX-2 and B-Raf in Treating Cervical Carcinoma. Bioorg. Med. Chem. 2014, 22, 4109–4118. [Google Scholar] [CrossRef]
- Halgren, T.A. Maximally Diagonal Force Constants in Dependent Angle-Bending Coordinates. II. Implications for the Design of Empirical Force Fields. J. Am. Chem. Soc. 1990, 112, 4710–4723. [Google Scholar] [CrossRef]
- Podlogar, B.L.; Ferguson, D.M. QSAR and CoMFA: A Perspective on the Practical Application to Drug Discovery. Drug Des. Discov. 2000, 17, 4–12. [Google Scholar] [PubMed]
- Zhang, Z.; Ohto, U.; Shibata, T.; Krayukhina, E.; Taoka, M.; Yamauchi, Y.; Tanji, H.; Isobe, T.; Uchiyama, S.; Miyake, K.; et al. Structural Analysis Reveals That Toll-like Receptor 7 Is a Dual Receptor for Guanosine and Single-Stranded RNA. Immunity 2016, 45, 737–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, A.; Merz, K.M. Prediction of Aqueous Solubility of a Diverse Set of Compounds Using Quantitative Structure−Property Relationships. J. Med. Chem. 2003, 46, 3572–3580. [Google Scholar] [CrossRef]
- Egan, W.J.; Lauri, G. Prediction of Intestinal Permeability. Adv. Drug Deliv. Rev. 2002, 54, 273–289. [Google Scholar] [CrossRef]
- Susnow, R.G.; Dixon, S.L. Use of Robust Classification Techniques for the Prediction of Human Cytochrome P450 2D6 Inhibition. J. Chem. Inf. Comput. Sci. 2003, 43, 1308–1315. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindah, E. Gromacs: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; Van Der Spoel, D.; et al. GROMACS 4.5: A High-Throughput and Highly Parallel Open Source Molecular Simulation Toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef]
- Showalter, S.A.; Brüschweiler, R. Validation of Molecular Dynamics Simulations of Biomolecules Using NMR Spin Relaxation as Benchmarks: Application to the AMBER99SB Force Field. J. Chem. Theory Comput. 2007, 3, 961–975. [Google Scholar] [CrossRef]
- Sousa Da Silva, A.W.; Vranken, W.F. ACPYPE—AnteChamber PYthon Parser InterfacE. BMC Res. Notes 2012, 5, 367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, B.; Lin, S.J.; Ren, J.Y.; Liu, T.; Wang, Y.M.; Li, C.M.; Xu, W.W.; He, Y.W.; Zheng, W.H.; Zhao, J.; et al. Molecular Docking and Molecular Dynamics (MD) Simulation of Human Anti-Complement Factor h (CFH) Antibody Ab42 and CFH Polypeptide. Int. J. Mol. Sci. 2019, 20, 2568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sommer, M.E.; Elgeti, M.; Hildebrand, P.W.; Szczepek, M.; Hofmann, K.P.; Scheerer, P. Structure-Based Biophysical Analysis of the Interaction of Rhodopsin with G Protein and Arrestin. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 2015; Volume 556, pp. 563–608. ISBN 0076-6879. [Google Scholar]
- Nath, O.; Singh, A.; Singh, I.K. In-Silico Drug Discovery Approach Targeting Receptor Tyrosine Kinase-like Orphan Receptor 1 for Cancer Treatment. Sci. Rep. 2017, 7, 1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A Linear Constraint Solver for Molecular Simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Yuan, X.H.; Wang, Y.C.; Jin, W.J.; Zhao, B.B.; Chen, C.F.; Yang, J.; Wang, J.F.; Guo, Y.Y.; Liu, J.J.; Zhang, D.; et al. Structure-Based High-Throughput Epitope Analysis of Hexon Proteins in B and C Species Human Adenoviruses (HAdVs). PLoS ONE 2012, 7, e32938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersen, H.G. Accuracy and Efficiency of the Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 3668–3679. [Google Scholar] [CrossRef]

























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hypo. No. | Total Cost | Cost Difference | RMSD | Correlation | Max. Fit | Features |
|---|---|---|---|---|---|---|
| 1 | 141.90 | 89.77 | 0.87 | 0.94 | 5.40 | HBA, HYA, PI, RA |
| 2 | 146.16 | 85.50 | 0.99 | 0.92 | 5.29 | HBA, HBA, HYA, PI |
| 3 | 146.47 | 85.19 | 1.02 | 0.92 | 5.77 | HBA, HYA, PI, RA |
| 4 | 148.27 | 83.40 | 1.03 | 0.92 | 6.10 | HBA, HBA, HYA, PI, PI |
| 5 | 148.27 | 83.40 | 1.03 | 0.92 | 5.01 | HBA, HBA, HYA, PI |
| 6 | 149.04 | 82.63 | 0.99 | 0.92 | 4.00 | HYA, PI, PI, RA |
| 7 | 149.04 | 82.62 | 1.05 | 0.91 | 4.93 | HBA, HYA, PI, RA |
| 8 | 149.11 | 82.55 | 1.07 | 0.91 | 5.34 | HBA, HYA, PI, RA |
| 9 | 149.13 | 82.53 | 0.97 | 0.93 | 3.78 | HYA, PI, PI, RA |
| 10 | 149.48 | 82.19 | 1.03 | 0.92 | 5.55 | HBA, HBA, HYA, PI, PI |
| Comp No. | IC50 (μM) | Errors a | Fit Value b | Activity Scale c | ||
|---|---|---|---|---|---|---|
| Experimental | Estimated | Experimental | Estimated | |||
| 32 | 0.43 | 0.63 | +1.47 | 5.06 | ++++ | ++++ |
| 33 | 0.5 | 0.75 | +1.5 | 4.98 | ++++ | ++++ |
| 14 | 0.7 | 1.7 | +2.43 | 4.64 | ++++ | ++++ |
| 38 | 0.8 | 0.73 | −1.1 | 5.00 | ++++ | ++++ |
| 36 | 0.98 | 0.91 | −1.08 | 4.92 | ++++ | ++++ |
| 35 | 0.99 | 0.73 | −1.36 | 4.99 | ++++ | ++++ |
| 37 | 1.14 | 0.84 | −1.36 | 4.95 | ++++ | ++++ |
| 19 | 1.2 | 3.1 | +2.58 | 4.37 | ++++ | +++ |
| 39 | 1.4 | 3.1 | +2.21 | 4.88 | ++++ | +++ |
| 13 | 1.4 | 1 | −1.4 | 4.37 | ++++ | ++++ |
| 34 | 1.55 | 0.99 | −1.57 | 4.88 | ++++ | ++++ |
| 15 | 4.4 | 8.3 | +1.89 | 3.95 | +++ | +++ |
| 53 | 4.57 | 5.8 | +1.27 | 4.11 | +++ | +++ |
| 48 | 4.71 | 5.4 | +1.15 | 4.12 | +++ | +++ |
| 24 | 4.9 | 5.6 | +1.14 | 4.13 | +++ | +++ |
| 50 | 4.99 | 5.7 | +1.14 | 4.10 | +++ | +++ |
| 17 | 5.4 | 7.3 | +1.35 | 4.01 | +++ | +++ |
| 22 | 5.7 | 8.1 | +1.42 | 3.96 | +++ | +++ |
| 12 | 5.8 | 2.7 | −2.15 | 4.44 | +++ | +++ |
| 52 | 8.09 | 7.8 | −1.04 | 3.98 | +++ | +++ |
| HCQ | 8.2 | 17 | +2.07 | 3.62 | +++ | ++ |
| 49 | 8.3 | 11 | +1.33 | 3.83 | +++ | ++ |
| 21 | 8.7 | 4.1 | −2.12 | 4.25 | +++ | +++ |
| 18 | 9.6 | 6.9 | −1.39 | 4.03 | +++ | +++ |
| 16 | 11 | 8.6 | −1.28 | 3.94 | ++ | +++ |
| 26 | 16 | 8.1 | −1.98 | 3.97 | ++ | +++ |
| 27 | 17 | 7.6 | −2.24 | 3.99 | ++ | +++ |
| 25 | 17 | 7.5 | −2.27 | 3.97 | ++ | +++ |
| 7 | 20.7 | 37 | +1.79 | 3.30 | + | + |
| 5 | 22 | 9.1 | −2.42 | 3.91 | + | +++ |
| 4 | 31 | 31 | +1 | 3.36 | + | + |
| 45 | 37 | 160 | +4.32 | 2.67 | + | + |
| 1 | 53 | 37 | −1.43 | 3.30 | + | + |
| 41 | 185 | 190 | +1.03 | 2.60 | + | + |
| 42 | 253 | 530 | +2.09 | 2.15 | + | + |
| 43 | 272 | 160 | −1.7 | 2.66 | + | + |
| 44 | 684 | 180 | −3.8 | 2.62 | + | + |
| Comp No. | IC50 (μM) | Errors a | Activity Scale b | ||
|---|---|---|---|---|---|
| Experimental | Estimated | Experimental | Estimated | ||
| 31 | 0.46 | 0.92 | +2.01 | ++++ | ++++ |
| 28 | 1.03 | 10.47 | +10.17 | ++++ | ++ |
| 23 | 1.2 | 4.27 | +3.55 | ++++ | +++ |
| 9 | 1.3 | 8.69 | +6.68 | ++++ | +++ |
| 2 | 1.4 | 8.69 | +6.21 | ++++ | +++ |
| 29 | 1.83 | 5.89 | +3.22 | ++++ | +++ |
| 30 | 2.16 | 7.59 | +3.63 | +++ | +++ |
| 20 | 4.2 | 6.17 | +1.47 | +++ | +++ |
| 6 | 4.6 | 8.71 | +1.89 | +++ | +++ |
| 11 | 5.6 | 6.61 | +1.18 | +++ | +++ |
| 51 | 7.55 | 5.62 | −1.34 | +++ | +++ |
| 10 | 11 | 39.81 | +3.62 | ++ | + |
| 8 | 17 | 38.90 | +2.29 | ++ | + |
| 40 | 23 | 157.04 | +6.83 | + | + |
| 3 | 52 | 36.31 | −1.43 | + | + |
| 47 | 110 | 190.55 | +1.73 | + | + |
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Comp. | R1 | R2 | R3 | 2D-QSAR | 3D-QSAR | Pharmacophore | ||
| pIC50 (µM) | IC50 (µM) | pIC50 (µM) | IC50 (µM) | IC50 (µM) | ||||
| T55 |  |  |  | −0.40 | 2.50 | −0.09 | 1.24 | 1.55 |
| T56 |  |  |  | −0.19 | 1.55 | −0.08 | 1.20 | 1.97 |
| T57 |  |  |  | −0.24 | 1.73 | 0.01 | 0.98 | 1.51 |
| T58 |  |  |  | −0.48 | 3.04 | −0.03 | 1.07 | 1.73 |
| T59 |  |  |  | 0.20 | 0.63 | −0.20 | 1.58 | 1.78 |
| T60 |  |  |  | 0.20 | 0.63 | −0.10 | 1.25 | 1.50 |
| T61 |  |  |  | 0.16 | 0.69 | 0.03 | 0.94 | 1.65 |
| T62 |  |  |  | 0.16 | 0.69 | −0.08 | 1.21 | 1.42 |
| T63 |  |  |  | −0.42 | 2.62 | 0.00 | 1.01 | 1.62 |
| T64 |  |  |  | 0.09 | 0.81 | −0.19 | 1.53 | 1.39 |
| T65 |  |  |  | −0.42 | 2.62 | −0.06 | 1.15 | 2.10 |
| T66 |  |  |  | −0.50 | 3.15 | 0.07 | 0.85 | 1.82 |
| Comp. | Antagonist Structure | Interacting Residue | Hydrogen Bond Formed | H-Bond Distance (Å) | Docking Score (kcal/mol) |
|---|---|---|---|---|---|
| T55 |  | Lys432*, Gln324, Gly577, Tyr356*, Ser523 | B:Lys432:HZ2–F38:T55 H44: T55O:Gln354:B T55:H60O:Gly577:A | 2.02 | 123.23 |
| T56 |  | Gln354*, Val355*, Gly577, Lys432* | B:Lys432:HZ2F38: T56 T56:H42O:Gln354:B T56:H42O:Val355:B T56:H58O:Gly577:A | 1.77 | 124.08 |
| T57 |  | Gln354*, Val355*, Gly577, Phe500 | T57:H43–O:Gln354:B T57:H43–O:Val355:B T57:H59–O:Gly577:A | 1.85 | 114.97 |
| T58 |  | Gln354*, Val355*, Gly577 | T58:H41–O:Gln354:B T58:H41–O:Val355:B T58:H57–O:Gly577:A | 1.81 | 110.54 |
| T59 |  | Thr525, Gln354*, Thr579, Tyr356* | T59:H42–OE1:Gln354:B T59:H42–O:Gln354:B T59:H58–OG1:Thr579:A T59:H77–OG1:Thr525:A | 1.81 | 115.88 |
| T60 |  | Gln354*, Thr525, Gly577 | T60:H42–O:Gln354:B T60:H63–OG1:Thr525:A T60:H89–O:Gly577:A | 2.53 | 133.30 |
| T61 |  | Gln354*, Thr525, Gly577, Tyr356* | T61:H41–O:Gln354:B T61:H62–OG1:Thr525:A T61:H87–O:Gly577:A | 1.74 | 111.78 |
| T62 |  | Gln354*, Gly577, Thr525, Asn255*, Tyr356* | T62:H41–B:Gln354:O T62:H57–A:Gly577:O T62:H62–A:Thr525:OG1 | 2.41 | 88.21 |
| T63 |  | Gln354*, Thr525, Gly577, Tyr356* | T63:H40–B:Gln354:O T63:H61–A:Thr525:OG1 T63:H85–A:Gly577:O | 2.16 | 108.50 |
| T64 |  | Gln354*, Thr525, Gly577, Tyr356* | T64:H40–O:Gln354:B T64:H56–O:Gly577:A T64:H61–OG1:Thr525:A | 2.98 | 110.24 |
| T65 |  | Gln354*, Thr525, Tyr356*, Thr406* | T65:H40–O:Gln354:B T65:H61–OG1:Thr525:A | 1.82 | 112.45 |
| T66 |  | Gln354*, Gly577, Thr525, Tyr356* | T66:H40–O:Gln354:B T66:H50–O:GLY577:A T66:H63–A:Thr525:OG1 | 2.47 | 116.22 |
| Comp. | Absorption Level | AlogP98 | PSA | BBB | BBB Level | Solubility | Solubility Level | Hepato- Toxicity | CYP2D6 | CYP2D6 Probability |
|---|---|---|---|---|---|---|---|---|---|---|
| 14 | 0 | 5.091 | 53.79 | 0.57 | 1.00 | −5.71 | 2 | False | −1.98 | False |
| T55 | 1 | 5.861 | 53.79 | 0.81 | 0.00 | −6.00 | 1 | False | −0.78 | False |
| T56 | 0 | 5.634 | 53.79 | 0.74 | 0.00 | −5.94 | 2 | False | −0.81 | False |
| T57 | 0 | 5.303 | 62.72 | 0.49 | 1.00 | −5.49 | 2 | False | 0.52 | True |
| T58 | 0 | 5.32 | 53.79 | 0.64 | 1.00 | −5.79 | 2 | False | 1.32 | True |
| T59 | 1 | 6.256 | 53.79 | - | 4.00 | −6.31 | 1 | False | −3.40 | False |
| T60 | 0 | 4.205 | 66.60 | 0.09 | 1.00 | −4.89 | 2 | False | −1.58 | False |
| T61 | 0 | 4.063 | 74.61 | −0.08 | 2.00 | −4.45 | 2 | False | −2.75 | False |
| T62 | 0 | 5.003 | 53.79 | 0.54 | 1.00 | −5.44 | 2 | False | −2.28 | False |
| T63 | 0 | 3.711 | 80.33 | −0.28 | 2.00 | −5.18 | 2 | False | −2.64 | False |
| T64 | 0 | 5.548 | 53.79 | 0.71 | 0.00 | −6.01 | 1 | False | −1.26 | False |
| T65 | 0 | 4.941 | 53.79 | 0.52 | 1.00 | −5.51 | 2 | False | −2.91 | False |
| T66 | 0 | 5.469 | 53.79 | 0.69 | 1.00 | −6.03 | 1 | False | −0.70 | False |
| Molecule | Molecular Weight (g/mol) | LogP | H-Bond Donors | H-Bond Acceptors | Number of Rotatable Bond | Polar Surface Area |
|---|---|---|---|---|---|---|
| 14 | 496.69 | 5.09 | 0 | 8 | 10 | 57.2 |
| T55 | 568.70 | 5.86 | 0 | 8 | 10 | 57.2 |
| T56 | 536.68 | 5.63 | 0 | 8 | 10 | 57.2 |
| T57 | 548.72 | 5.30 | 0 | 9 | 11 | 66.43 |
| T58 | 518.69 | 5.32 | 0 | 8 | 10 | 57.2 |
| T59 | 538.77 | 6.26 | 0 | 8 | 11 | 57.2 |
| T60 | 539.76 | 4.21 | 1 | 9 | 11 | 69.23 |
| T61 | 526.71 | 4.06 | 1 | 9 | 10 | 77.43 |
| T62 | 528.71 | 5.00 | 0 | 8 | 10 | 57.2 |
| T63 | 511.70 | 3.71 | 1 | 9 | 10 | 83.22 |
| T64 | 510.72 | 5.55 | 0 | 8 | 10 | 57.2 |
| T65 | 514.68 | 4.94 | 0 | 8 | 10 | 57.2 |
| T66 | 510.72 | 5.47 | 0 | 8 | 10 | 57.2 |
| Comp. | FDA Carcinogenicity | FDA Carcinogenicity | AMES Mutagenicity | Rat oral LD50 (mg/kg) | Skin Irritation | Probability of Biodegradability | ||
|---|---|---|---|---|---|---|---|---|
| Male Mouse | Female Mouse | Male Rat | Female Rat | |||||
| 14 | Non- carcinogen | Non- carcinogen | Non- carcinogen | Non- carcinogen | Non- mutagen | 81.63 | None | Non- degradable |
| T55 | Non- carcinogen | Non- carcinogen | Non- carcinogen | Non- carcinogen | Non- mutagen | 9.70 | None | Non- degradable |
| T56 | Non- carcinogen | Non- carcinogen | Non- carcinogen | Non- carcinogen | Non- mutagen | 53.89 | None | Non- degradable |
| T57 | Non- carcinogen | Non- carcinogen | Non- carcinogen | Non- carcinogen | Non- mutagen | 171.96 | None | Non- degradable |
| T58 | Non- carcinogen | Non- carcinogen | Non- carcinogen | Non- carcinogen | Non- mutagen | 73.78 | None | Non- degradable |
| T59 | Non- carcinogen | Non- carcinogen | Non- carcinogen | Non- carcinogen | Non- mutagen | 65.11 | Mild | Non- degradable |
| T60 | Non- carcinogen | Non- carcinogen | Non- carcinogen | Non- carcinogen | Non- mutagen | 36.50 | Mild | Non- degradable |
| T61 | Non- carcinogen | Non- carcinogen | Non- carcinogen | Non- carcinogen | Non- mutagen | 79.04 | Mild | Non- degradable |
| T62 | Non- carcinogen | Non- carcinogen | Non- carcinogen | Non- carcinogen | Non- mutagen | 17.71 | Mild | Non- degradable |
| T63 | Non- carcinogen | Non- carcinogen | Non- carcinogen | Non- carcinogen | Non- mutagen | 44.17 | Mild | Non- degradable |
| T64 | Non- carcinogen | Non- carcinogen | Non- carcinogen | Non- carcinogen | Non- mutagen | 101.52 | None | Non- degradable |
| T65 | Non- carcinogen | Non- carcinogen | Non- carcinogen | Non- carcinogen | Non- mutagen | 13.12 | Mild | Non- degradable |
| T66 | Non- carcinogen | Non- carcinogen | Non- carcinogen | Non- carcinogen | Non- mutagen | 42.90 | None | Non- degradable |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pal, S.; Ghosh Dastidar, U.; Ghosh, T.; Ganguly, D.; Talukdar, A. Integration of Ligand-Based and Structure-Based Methods for the Design of Small-Molecule TLR7 Antagonists. Molecules 2022, 27, 4026. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27134026
Pal S, Ghosh Dastidar U, Ghosh T, Ganguly D, Talukdar A. Integration of Ligand-Based and Structure-Based Methods for the Design of Small-Molecule TLR7 Antagonists. Molecules. 2022; 27(13):4026. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27134026
Chicago/Turabian StylePal, Sourav, Uddipta Ghosh Dastidar, Trisha Ghosh, Dipyaman Ganguly, and Arindam Talukdar. 2022. "Integration of Ligand-Based and Structure-Based Methods for the Design of Small-Molecule TLR7 Antagonists" Molecules 27, no. 13: 4026. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27134026