
Design, Synthesis, Docking, DFT, MD Simulation Studies of a New Nicotinamide-Based Derivative: In Vitro Anticancer and VEGFR-2 Inhibitory Effects

 , ,
, ,  , ,
, ,  and
and 
Abstract
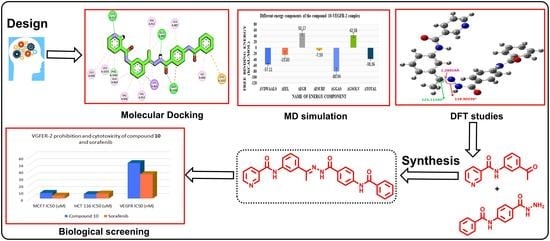
:
1. Introduction
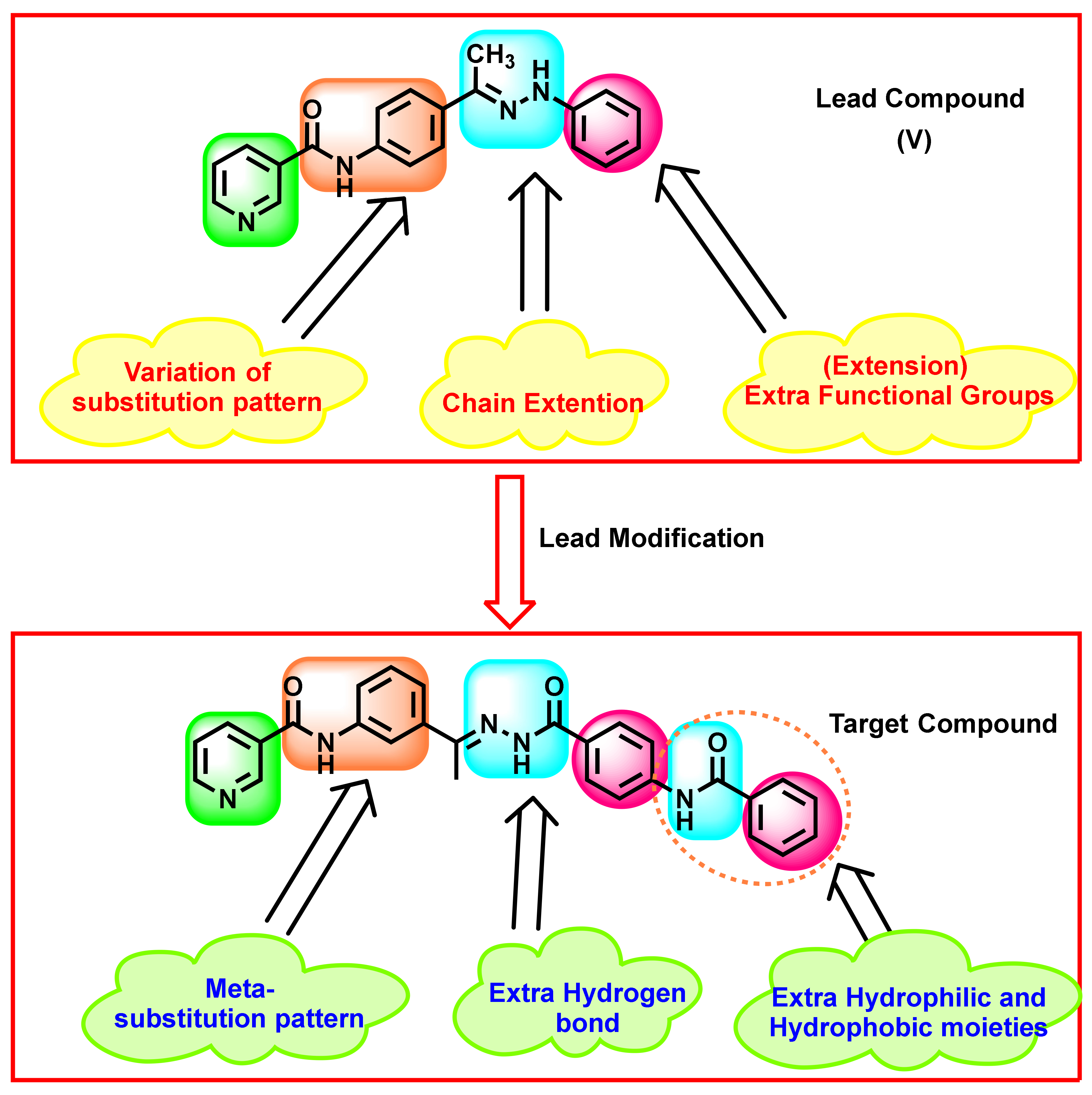
Rationale
2. Results
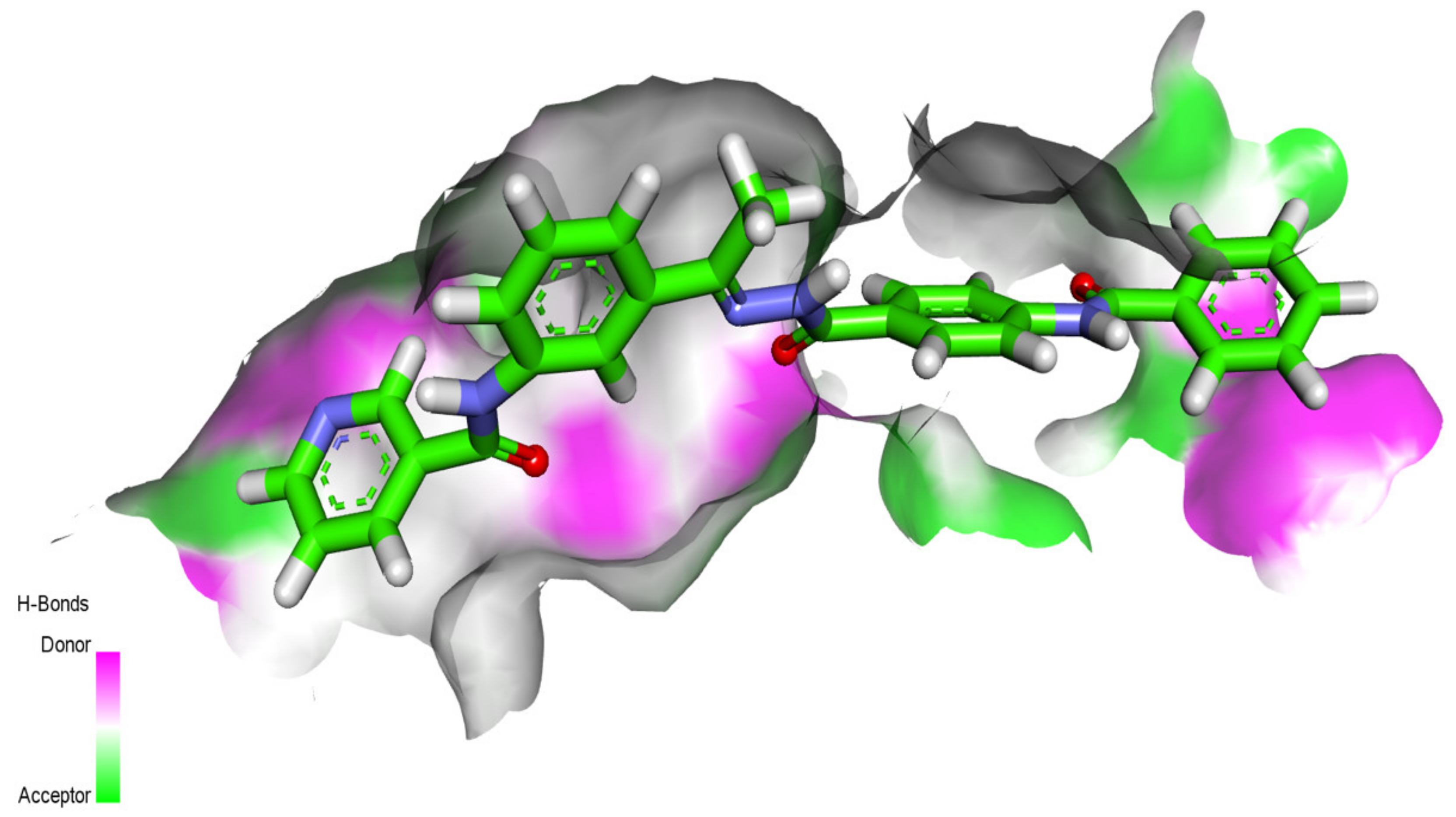
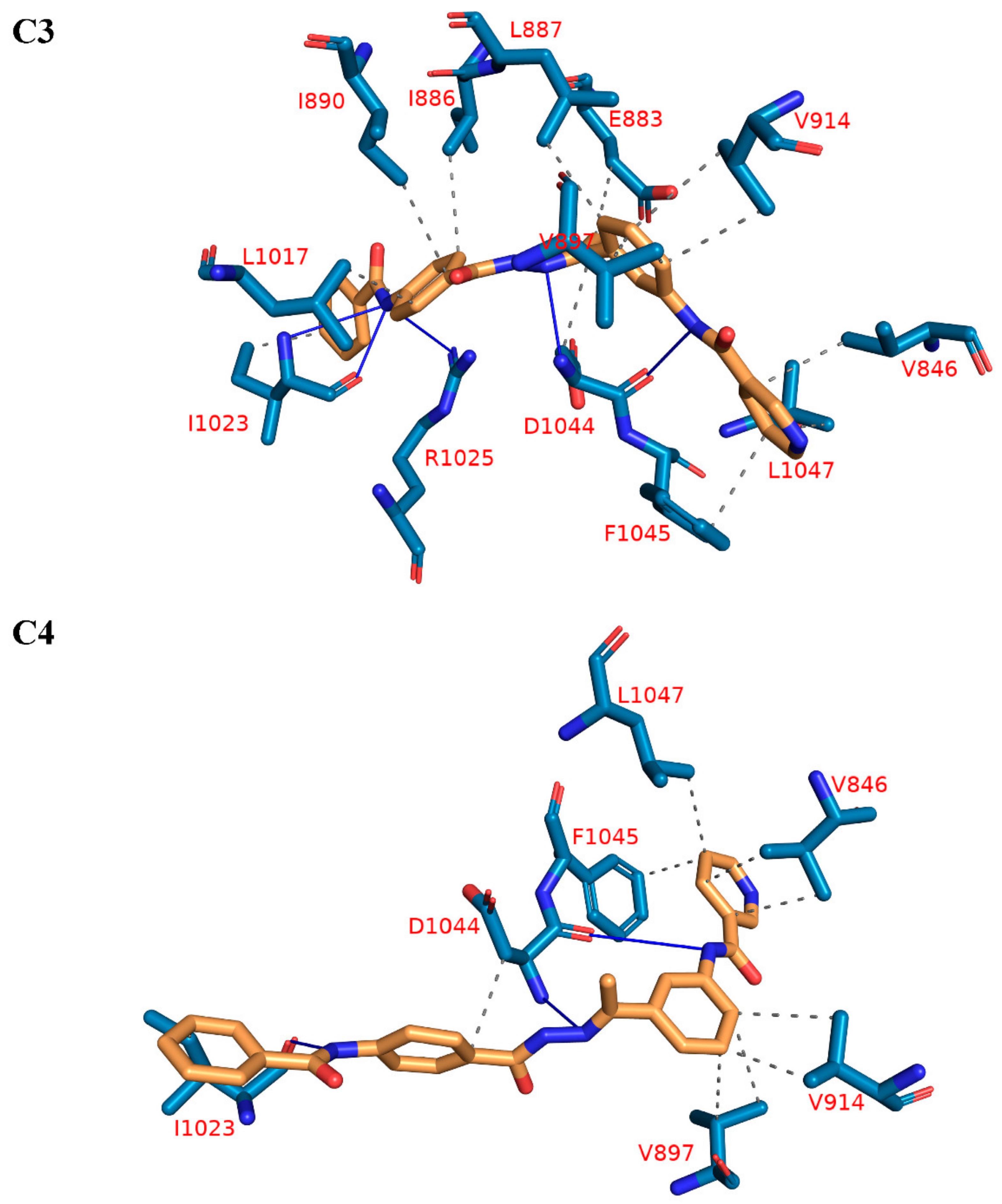
2.1. Molecular Docking Simulations against VEGFR-2
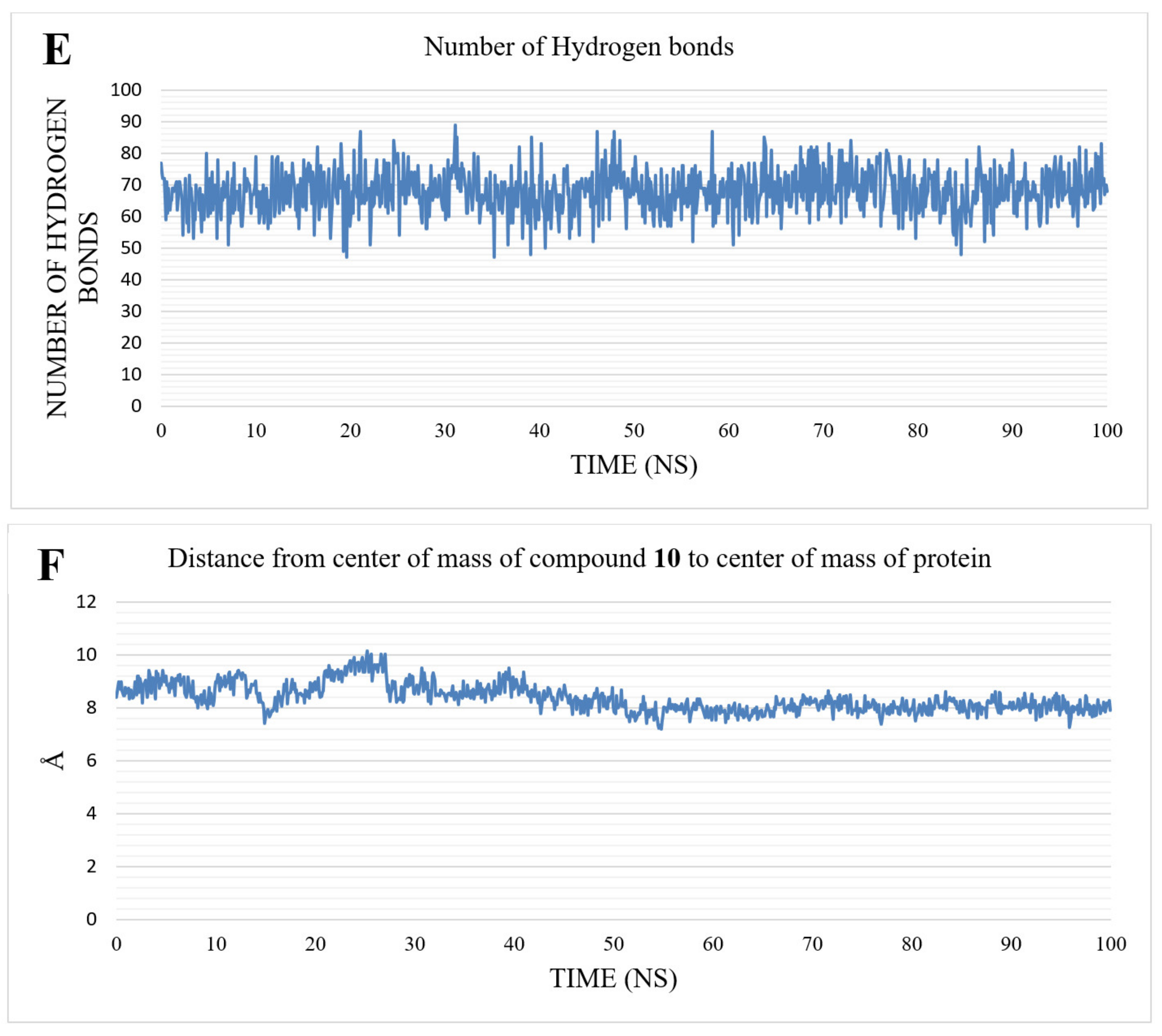
2.2. MD Simulations Results
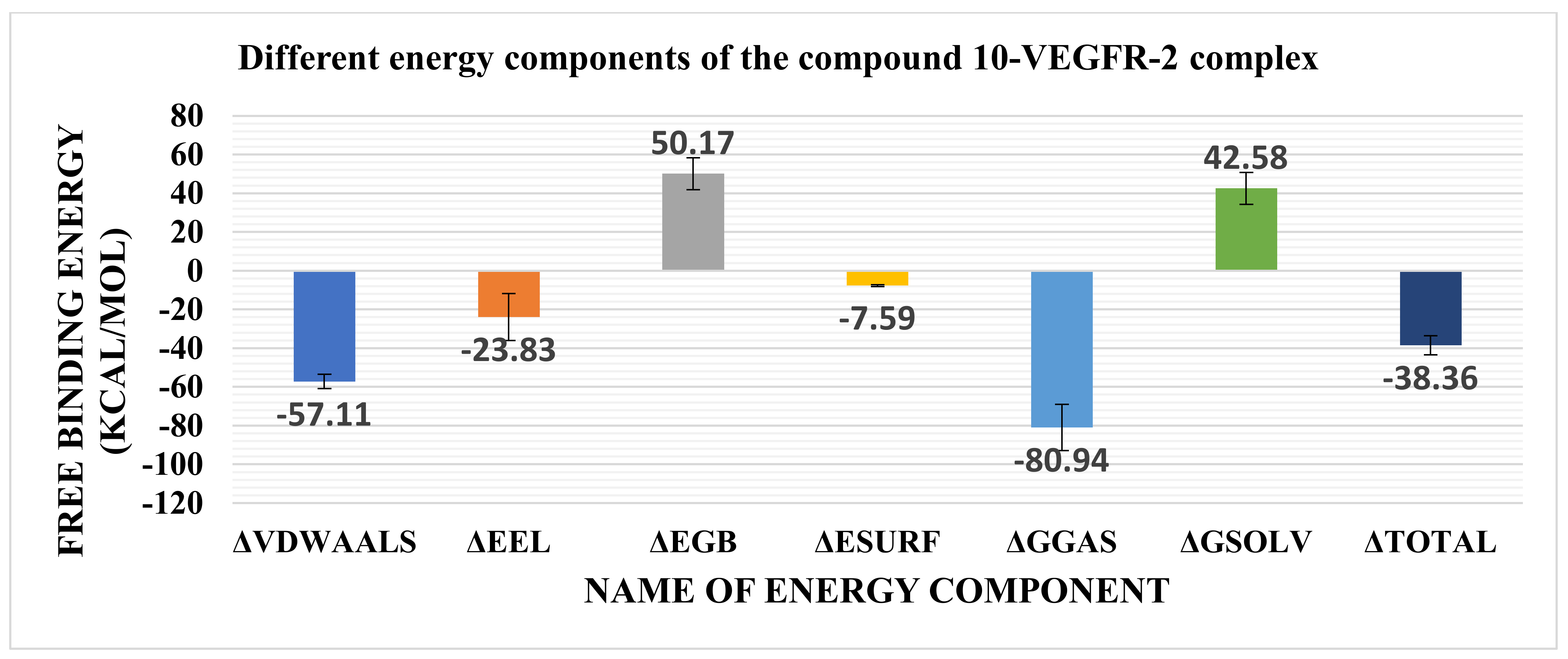
2.3. MMGBSA
2.4. Density Function Theory (DFT)
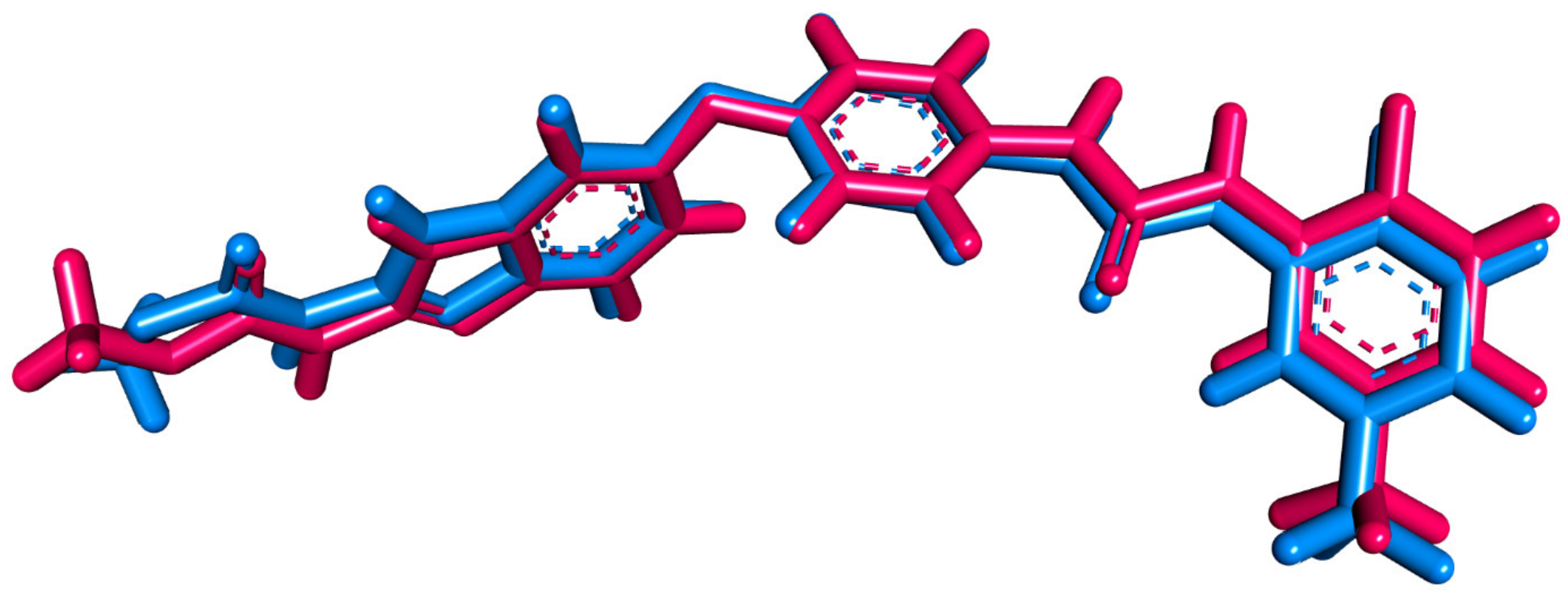
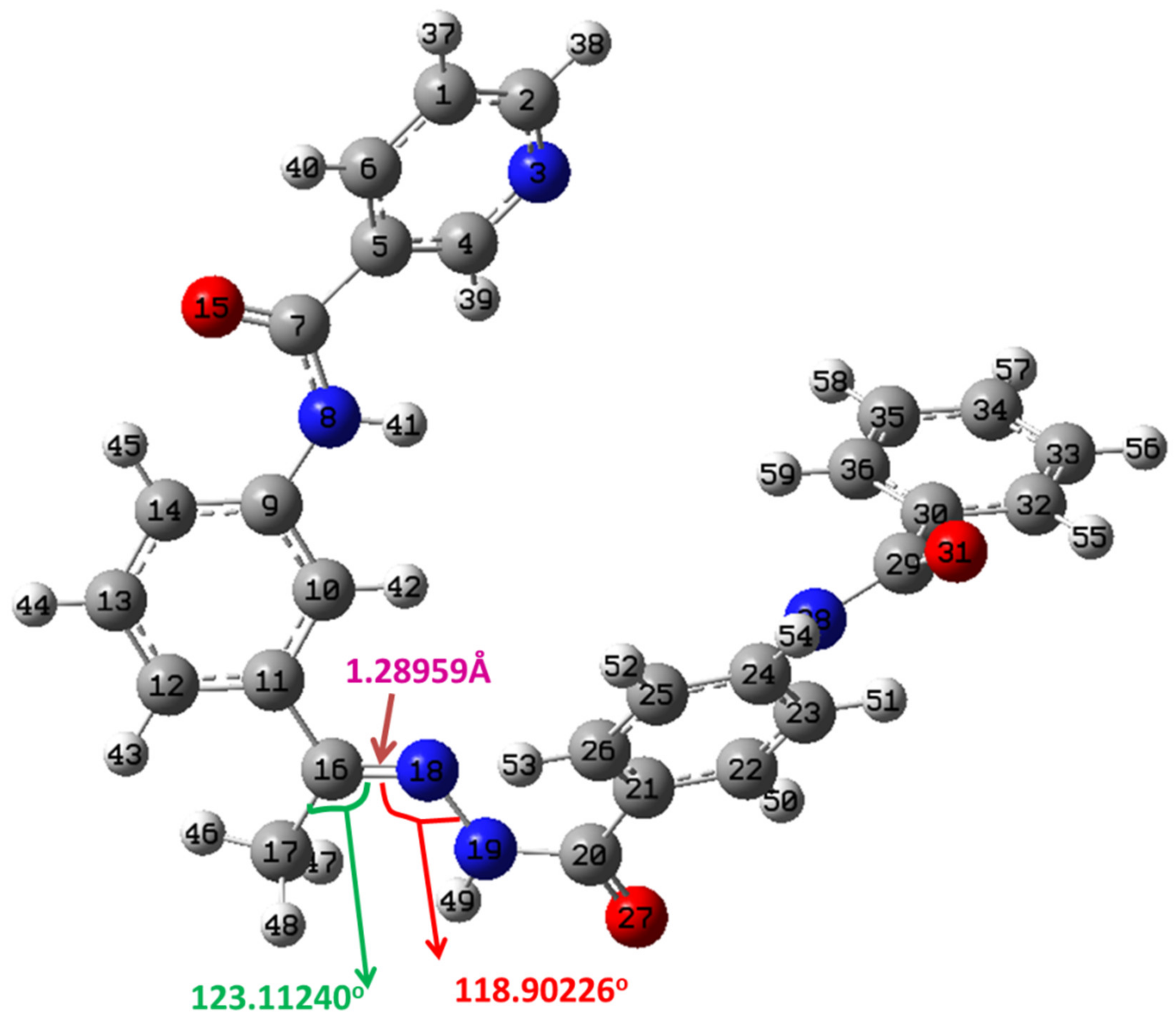
2.4.1. Structure Optimization
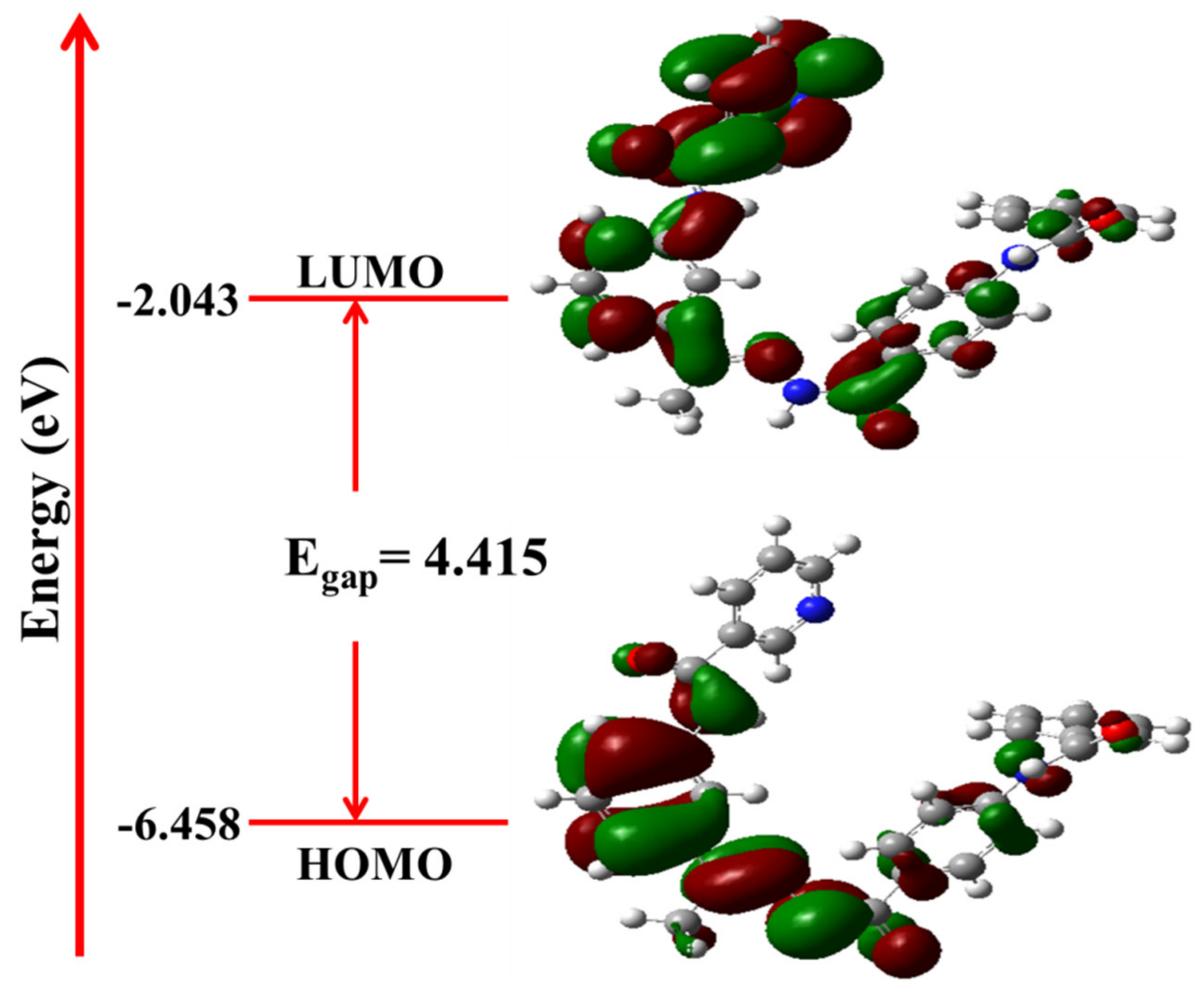
2.4.2. Frontier Molecular Orbital Analysis and Global Chemical Descriptors
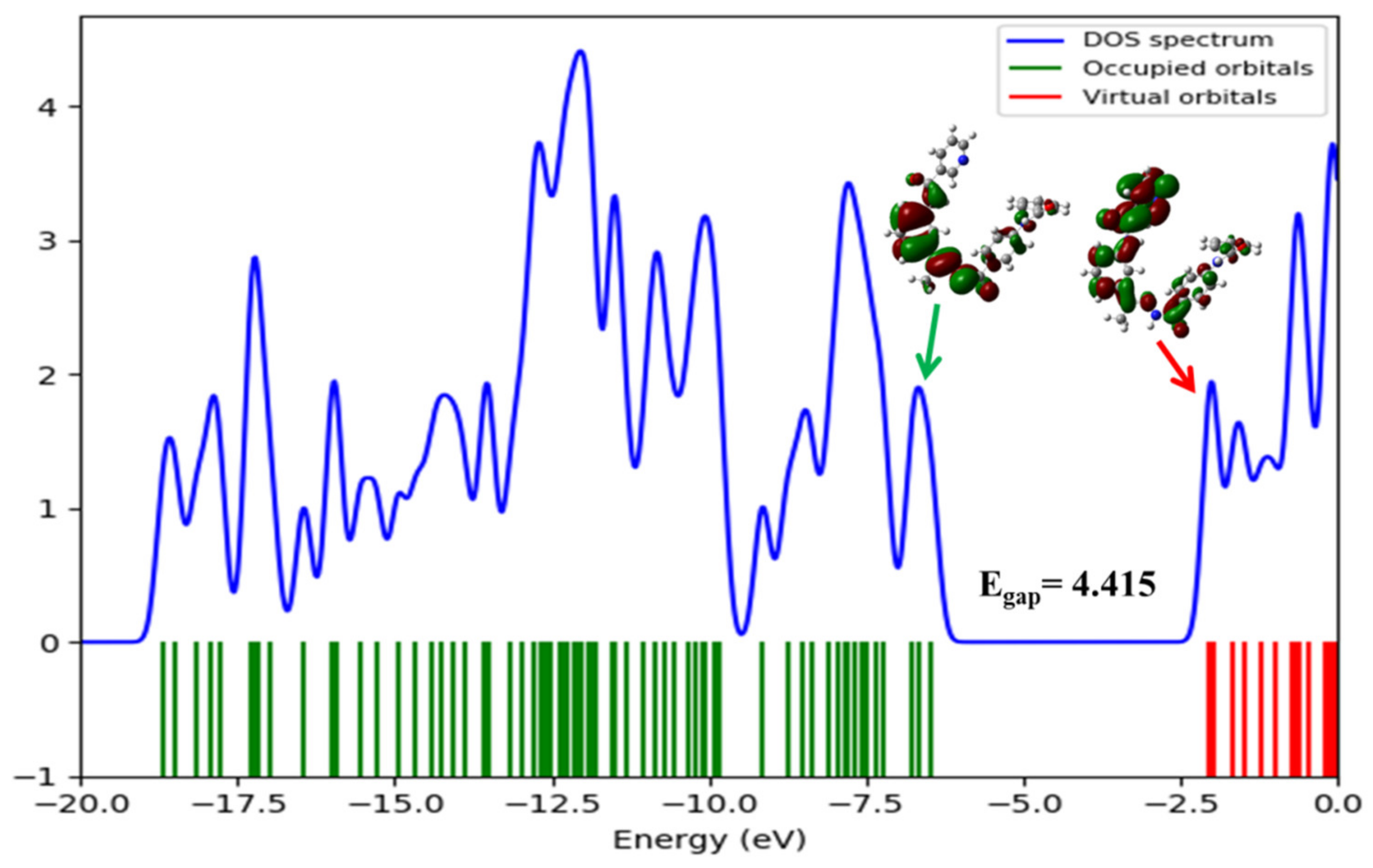
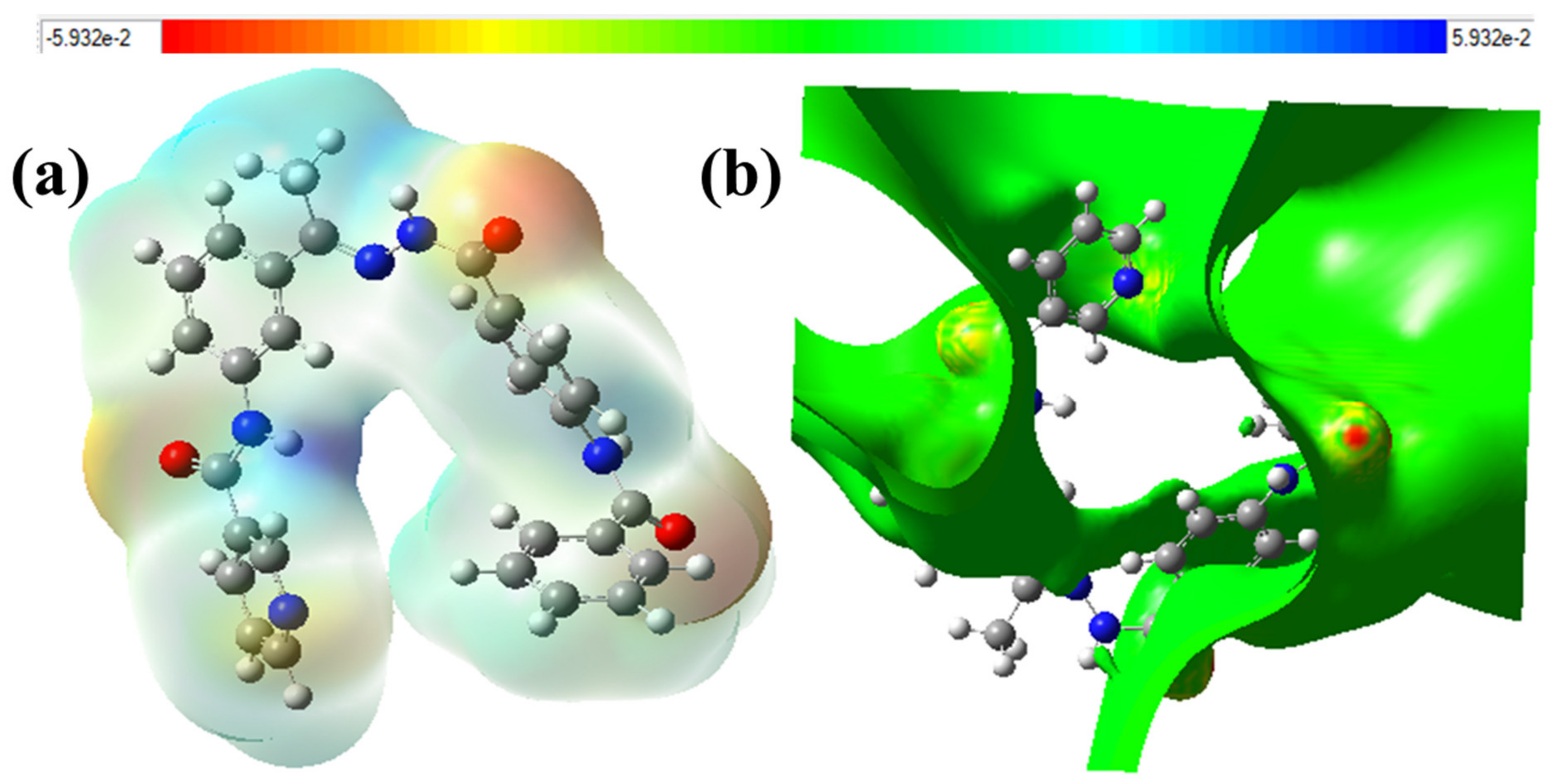
2.4.3. The Total Density of State (DOS) and Electron Density Maps
2.5. ADMET Profiling Study
2.6. In Silico Toxicity Studies
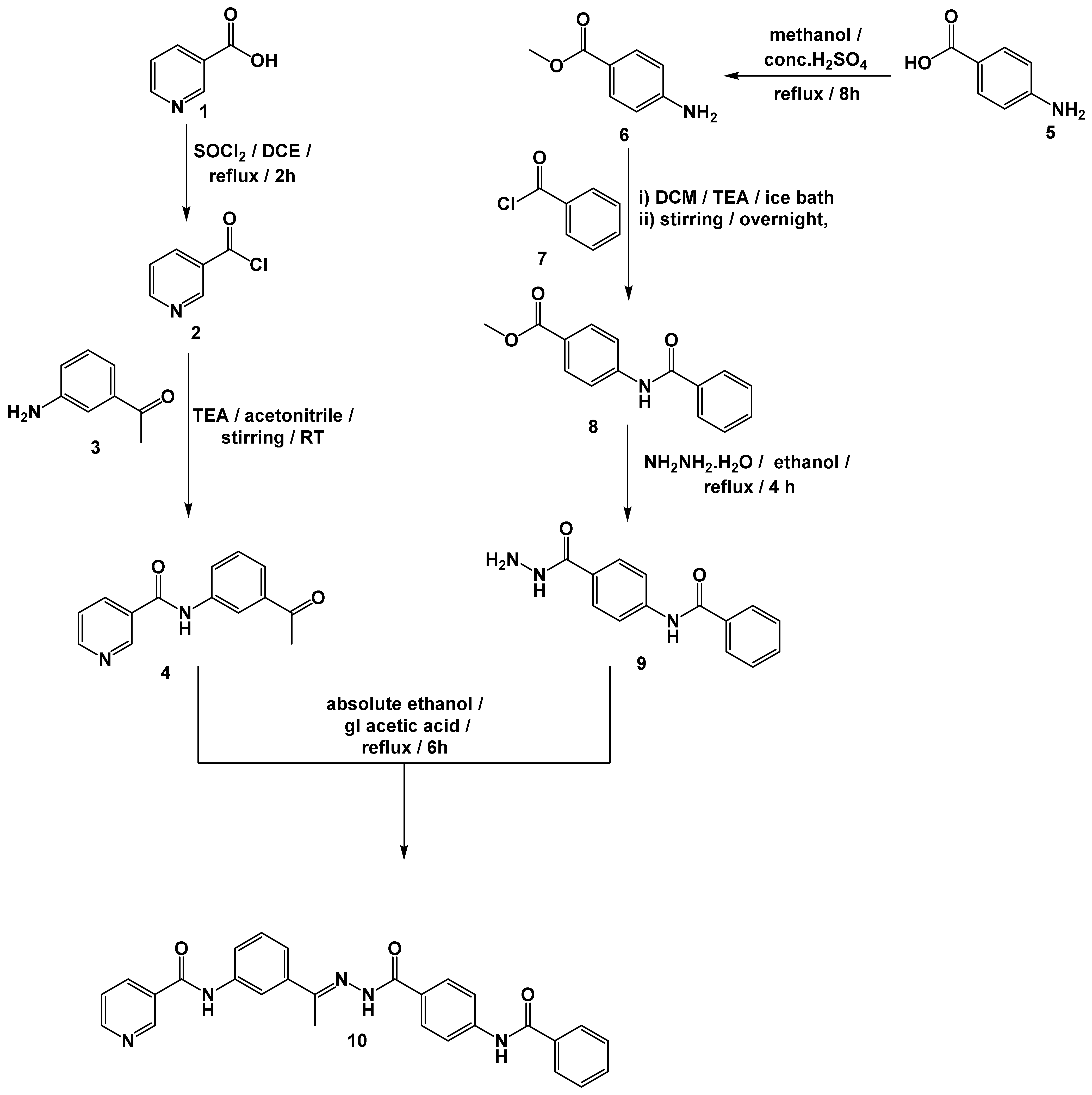
2.7. Chemistry
2.8. Biological Results
2.8.1. VEGFR-2 Prohibition
2.8.2. Cytotoxicity
2.8.3. Safety Assessment
3. Experimental
3.1. Docking Studies
3.2. MD Simulations
3.3. MM-GBSA
3.4. DFT
3.5. ADMET Studies
3.6. Toxicity Studies
3.7. General Procedure for the Synthesis of Compound 10

3.8. Biological Studies
3.8.1. In Vitro VEGFR-2 Inhibition
3.8.2. In Vitro Antiproliferative Activity
3.8.3. Safety Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- WHO. Cancer, Fact Sheet. Available online: https://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 13 November 2021).
- El-Dash, Y.; Elzayat, E.; Abdou, A.M.; Hassan, R.A. Novel thienopyrimidine-aminothiazole hybrids: Design, synthesis, antimicrobial screening, anticancer activity, effects on cell cycle profile, caspase-3 mediated apoptosis and VEGFR-2 inhibition. Bioorg. Chem. 2021, 114, 105137. [Google Scholar] [CrossRef] [PubMed]
- Nishida, N.; Yano, H.; Nishida, T.; Kamura, T.; Kojiro, M. Angiogenesis in cancer. Vasc. Health Risk Manag. 2006, 2, 213. [Google Scholar] [CrossRef] [PubMed]
- Quesada, A.R.; Muñoz-Chápuli, R.; Medina, M.A. Anti-angiogenic drugs: From bench to clinical trials. Med. Res. Rev. 2006, 26, 483–530. [Google Scholar] [CrossRef] [PubMed]
- Olsson, A.-K.; Dimberg, A.; Kreuger, J.; Claesson-Welsh, L. VEGF receptor signalling? In control of vascular function. Nat. Rev. Mol. Cell Biol. 2006, 7, 359–371. [Google Scholar] [CrossRef]
- Carmeliet, P.J.O. VEGF as a key mediator of angiogenesis in cancer. Oncology 2005, 69, 4–10. [Google Scholar] [CrossRef]
- Alsaif, N.A.; Taghour, M.S.; Alanazi, M.M.; Obaidullah, A.J.; Alanazi, W.A.; Alasmari, A.; Albassam, H.; Dahab, M.A.; Mahdy, H.A. Identification of new [1, 2, 4] triazolo [4,3-a] quinoxalines as potent VEGFR-2 tyrosine kinase inhibitors: Design, synthesis, anticancer evaluation, and in silico studies. Bioorg. Med. Chem. 2021, 46, 116384. [Google Scholar] [CrossRef]
- Shah, A.A.; Kamal, M.A.; Akhtar, S.J.C.D.M. Tumor angiogenesis and VEGFR-2: Mechanism, pathways and current biological therapeutic interventions. Curr. Drug Metab. 2021, 22, 50–59. [Google Scholar] [CrossRef]
- Elrazaz, E.Z.; Serya, R.A.; Ismail, N.S.; Albohy, A.; Abou El Ella, D.A.; Abouzid, K.A. Discovery of Potent Thieno [2, 3-d] pyrimidine VEGFR-2 Inhibitors: Design, Synthesis and Enzyme Inhibitory Evaluation Supported by Molecular Dynamics Simulations. Bioorg. Chem. 2021, 113, 105019. [Google Scholar] [CrossRef]
- Sana, S.; Reddy, V.G.; Bhandari, S.; Reddy, T.S.; Tokala, R.; Sakla, A.P.; Bhargava, S.K.; Shankaraiah, N. Exploration of carbamide derived pyrimidine—Thioindole conjugates as potential VEGFR-2 inhibitors with anti-angiogenesis effect. Eur. J. Med. Chem. 2020, 200, 112457. [Google Scholar] [CrossRef]
- Peng, F.-W.; Liu, D.-K.; Zhang, Q.-W.; Xu, Y.-G.; Shi, L. VEGFR-2 inhibitors and the therapeutic applications thereof: A patent review (2012–2016). Expert Opin. Ther. Patents 2017, 27, 987–1004. [Google Scholar] [CrossRef]
- Sun, S.; Zhang, J.; Wang, N.; Kong, X.; Fu, F.; Wang, H.; Yao, J. Design and discovery of quinazoline-and thiourea-containing sorafenib analogs as EGFR and VEGFR-2 dual TK inhibitors. Molecules 2017, 23, 24. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Mohsen, H.T.; Abdullaziz, M.A.; El Kerdawy, A.M.; Ragab, F.A.; Flanagan, K.J.; Mahmoud, A.E.; Ali, M.M.; El Diwani, H.I.; Senge, M.O. Targeting receptor tyrosine kinase VEGFR-2 in hepatocellular cancer: Rational design, synthesis and biological evaluation of 1, 2-disubstituted benzimidazoles. Molecules 2020, 25, 770. [Google Scholar] [CrossRef] [Green Version]
- Cavasotto, C.N.; Aucar, M.G.; Adler, N.S. Computational chemistry in drug lead discovery and design. Int. J. Quantum Chem. 2019, 119, e25678. [Google Scholar] [CrossRef] [Green Version]
- Filipe, H.A.; Loura, L.M. Molecular Dynamics Simulations: Advances and Applications. Molecules 2022, 27, 2105. [Google Scholar] [CrossRef]
- Ojo, O.A.; Ojo, A.B.; Okolie, C.; Nwakama, M.-A.C.; Iyobhebhe, M.; Evbuomwan, I.O.; Nwonuma, C.O.; Maimako, R.F.; Adegboyega, A.E.; Taiwo, O.A. Deciphering the interactions of bioactive compounds in selected traditional medicinal plants against Alzheimer’s diseases via pharmacophore modeling, auto-QSAR, and molecular docking approaches. Molecules 2021, 26, 1996. [Google Scholar] [CrossRef]
- Elton, D.C.; Boukouvalas, Z.; Fuge, M.D.; Chung, P.W. Deep learning for molecular design—A review of the state of the art. Mol. Syst. Des. Eng. 2019, 4, 828–849. [Google Scholar] [CrossRef] [Green Version]
- Keith, J.A.; Vassilev-Galindo, V.; Cheng, B.; Chmiela, S.; Gastegger, M.; Muüller, K.-R.; Tkatchenko, A. Combining machine learning and computational chemistry for predictive insights into chemical systems. Chem. Rev. 2021, 121, 9816–9872. [Google Scholar] [CrossRef]
- Cáceres, E.L.; Tudor, M.; Cheng, A.C. Deep learning approaches in predicting ADMET properties. Futur. Med. Chem. 2020, 12, 1995–1999. [Google Scholar] [CrossRef]
- Ciallella, H.L.; Zhu, H. Advancing computational toxicology in the big data era by artificial intelligence: Data-Driven and mechanism-driven modeling for chemical toxicity. Chem. Res. Toxicol. 2019, 32, 536–547. [Google Scholar] [CrossRef]
- Tielens, F.; Gierada, M.; Handzlik, J.; Calatayud, M. Characterization of amorphous silica based catalysts using DFT computational methods. Catal. Today 2020, 354, 3–18. [Google Scholar] [CrossRef]
- Pracht, P.; Bohle, F.; Grimme, S. Automated exploration of the low-energy chemical space with fast quantum chemical methods. Phys. Chem. Chem. Phys. 2020, 22, 7169–7192. [Google Scholar] [CrossRef]
- Seidel, T.; Schuetz, D.A.; Garon, A.; Langer, T. The pharmacophore concept and its applications in computer-aided drug design. Prog. Chem. Org. Nat. Prod. 2019, 110, 99–141. [Google Scholar]
- Adnane, L.; Trail, P.A.; Taylor, I.; Wilhelm, S.M. Sorafenib (BAY 43-9006, Nexavar®), a dual-action inhibitor that targets RAF/MEK/ERK pathway in tumor cells and tyrosine kinases VEGFR/PDGFR in tumor vasculature. Methods Enzymol. 2006, 407, 597–612. [Google Scholar]
- Yousef, R.G.; Sakr, H.M.; Eissa, I.H.; Mehany, A.B.; Metwaly, A.M.; Elhendawy, M.A.; Radwan, M.M.; ElSohly, M.A.; Abulkhair, H.S.; El-Adl, K. New quinoxaline-2 (1 H)-ones as potential VEGFR-2 inhibitors: Design, synthesis, molecular docking, ADMET profile and anti-proliferative evaluations. New J. Chem. 2021, 45, 16949–16964. [Google Scholar] [CrossRef]
- Ran, F.; Li, W.; Qin, Y.; Yu, T.; Liu, Z.; Zhou, M.; Liu, C.; Qiao, T.; Li, X.; Yousef, R.G. Inhibition of vascular smooth muscle and cancer cell proliferation by new VEGFR inhibitors and their immunomodulator effect: Design, synthesis, and biological evaluation. Oxidative Med. Cell. Longev. 2021, 2021, 1–21. [Google Scholar] [CrossRef]
- Legler, C.; Brown, N.; Dunbar, R.; Harness, M.; Nguyen, K.; Oyewole, O.; Collier, W.; Spectroscopy, B. Scaled quantum mechanical scale factors for vibrational calculations using alternate polarized and augmented basis sets with the B3LYP density functional calculation model. Acta Part A Mol. Biomol. Spectrosc. 2015, 145, 15–24. [Google Scholar] [CrossRef]
- Husein, D.Z.; Hassanien, R.; Khamis, M. Cadmium oxide nanoparticles/graphene composite: Synthesis, theoretical insights into reactivity and adsorption study. RSC Adv. 2021, 11, 27027–27041. [Google Scholar] [CrossRef]
- Elkaeed, E.B.; Yousef, R.G.; Elkady, H.; Gobaara, I.M.M.; Alsfouk, A.A.; Husein, D.Z.; Ibrahim, I.M.; Metwaly, A.M.; Eissa, I.H. The Assessment of Anticancer and VEGFR-2 Inhibitory Activities of a New 1H-Indole Derivative: In Silico and In Vitro Approaches. Processes 2022, 10, 1391. [Google Scholar] [CrossRef]
- Wang, T.; Husein, D.Z. Novel synthesis of multicomponent porous nano-hybrid composite, theoretical investigation using DFT and dye adsorption applications: Disposing of waste with waste. Environ. Sci. Pollut. Res. 2022, 23, 1–28. [Google Scholar] [CrossRef]
- El-Zahabi, M.A.; Sakr, H.; El-Adl, K.; Zayed, M.; Abdelraheem, A.S.; Eissa, S.I.; Elkady, H.; Eissa, I.H. Design, synthesis, and biological evaluation of new challenging thalidomide analogs as potential anticancer immunomodulatory agents. Bioorg. Chem. 2020, 104, 104218. [Google Scholar] [CrossRef]
- Li, X.; Jiang, Y.; Peterson, Y.K.; Xu, T.; Himes, R.A.; Luo, X.; Yin, G.; Inks, E.S.; Dolloff, N.; Halene, S. Design of hydrazide-bearing HDACIs based on panobinostat and their p53 and FLT3-ITD dependency in antileukemia activity. J. Med. Chem. 2020, 63, 5501–5525. [Google Scholar] [CrossRef] [PubMed]
- El-Adl, K.; El-Helby, A.-G.A.; Sakr, H.; Elwan, A. [1, 2, 4] Triazolo [4, 3-a] quinoxaline and [1, 2, 4] triazolo [4, 3-a] quinoxaline-1-thiol-derived DNA intercalators: Design, synthesis, molecular docking, in silico ADMET profiles and anti-proliferative evaluations. New J. Chem. 2021, 45, 881–897. [Google Scholar] [CrossRef]
- Parmar, D.R.; Soni, J.Y.; Guduru, R.; Rayani, R.H.; Kusurkar, R.V.; Vala, A.G.; Talukdar, S.N.; Eissa, I.H.; Metwaly, A.M.; Khalil, A.; et al. Discovery of new anticancer thiourea-azetidine hybrids: Design, synthesis, in vitro antiproliferative, SAR, in silico molecular docking against VEGFR-2, ADMET, toxicity, and DFT studies. Bioorg. Chem. 2021, 115, 105206. [Google Scholar] [CrossRef] [PubMed]
- Elkady, H.; Elwan, A.; El-Mahdy, H.A.; Doghish, A.S.; Ismail, A.; Taghour, M.S.; Elkaeed, E.B.; Eissa, I.H.; Dahab, M.A.; Mahdy, H.A. New benzoxazole derivatives as potential VEGFR-2 inhibitors and apoptosis inducers: Design, synthesis, anti-proliferative evaluation, flowcytometric analysis, and in silico studies. J. Enzym. Inhib. Med. Chem. 2022, 37, 397–410. [Google Scholar] [CrossRef]
- Elkaeed, E.B.; Youssef, F.S.; Eissa, I.H.; Elkady, H.; Alsfouk, A.A.; Ashour, M.L.; El Hassab, M.A.; Abou-Seri, S.M.; Metwaly, A.M. Multi-Step In Silico Discovery of Natural Drugs against COVID-19 Targeting Main Protease. Int. J. Mol. Sci. 2022, 23, 6912. [Google Scholar] [CrossRef]
- El-Adl, K.; Sakr, H.M.; Yousef, R.G.; Mehany, A.B.; Metwaly, A.M.; Elhendawy, M.A.; Radwan, M.M.; ElSohly, M.A.; Abulkhair, H.S.; Eissa, I.H. Discovery of new quinoxaline-2 (1H)-one-based anticancer agents targeting VEGFR-2 as inhibitors: Design, synthesis, and anti-proliferative evaluation. Bioorg. Chem. 2021, 114, 105105. [Google Scholar] [CrossRef]





















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cluster | No. of H Interactions | Amino Acids in the VEGFR-2 | No. of H Bonds | Amino Acids in the VEGFR-2 |
|---|---|---|---|---|
| C1 | 8 | H814-V846-K866-I886-V914-D1044-F1045 (2) | 2 | E883-D1044 |
| C2 | 7 | I886-I890-V897 (2)-V914-1023-F1045 | 3 | I1023 (2)-D1044 |
| C3 | 14 | V846-E883-I886-L887-I890-V897 (2)-V914 (2)-L1017-I1023-D1044-F1045-L1047 | 5 | I1023 (2)-R1025-D1044 (2) |
| C4 | 10 | V846 (2)-V897 (2)-V914 (2)-I1023-D1044-F1045-L1047 | 3 | I1023-D1044 (2) |
| IP | EA | μ (eV) | χ (eV) | η (eV) | σ (eV) | ω (eV) | Dm (Debye) | TE (eV) | ∆Nmax | ∆E (eV) |
|---|---|---|---|---|---|---|---|---|---|---|
| −6.458 | −2.043 | −4.250 | 4.250 | 2.208 | 0.453 | 19.939 | 3.538 | −43013.1 | 1.925 | −19.939 |
| Comp. | TD50-R, mg/kg/day | Am-M, g/kg | DTP, g/kg | MFTD-R, g/kg | R-O-LD50, g/kg | LOAEL-R, g/kg | Skin Irritancy | Ocular Irritancy |
|---|---|---|---|---|---|---|---|---|
| 10 | 45.374 | Non-Mutagen | Non-Toxic | 0.117 | 2.637 | 0.117 | Non-Irritant | Mild |
| Sorafenib | 14.244 | Non-Mutagen | Toxic | 0.089 | 0.823 | 0.089 | Non-Irritant | Mild |
| MCF7 IC50 (μM) | HCT 116 IC50 (μM) | VEGFR IC50 (nM) | |
|---|---|---|---|
| Compound 10 | 8.25 | 6.48 | 51 |
| Sorafenib | 4.32 | 7.28 | 35 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elkaeed, E.B.; Yousef, R.G.; Elkady, H.; Gobaara, I.M.M.; Alsfouk, B.A.; Husein, D.Z.; Ibrahim, I.M.; Metwaly, A.M.; Eissa, I.H. Design, Synthesis, Docking, DFT, MD Simulation Studies of a New Nicotinamide-Based Derivative: In Vitro Anticancer and VEGFR-2 Inhibitory Effects. Molecules 2022, 27, 4606. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27144606
Elkaeed EB, Yousef RG, Elkady H, Gobaara IMM, Alsfouk BA, Husein DZ, Ibrahim IM, Metwaly AM, Eissa IH. Design, Synthesis, Docking, DFT, MD Simulation Studies of a New Nicotinamide-Based Derivative: In Vitro Anticancer and VEGFR-2 Inhibitory Effects. Molecules. 2022; 27(14):4606. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27144606
Chicago/Turabian StyleElkaeed, Eslam B., Reda G. Yousef, Hazem Elkady, Ibraheem M. M. Gobaara, Bshra A. Alsfouk, Dalal Z. Husein, Ibrahim M. Ibrahim, Ahmed M. Metwaly, and Ibrahim H. Eissa. 2022. "Design, Synthesis, Docking, DFT, MD Simulation Studies of a New Nicotinamide-Based Derivative: In Vitro Anticancer and VEGFR-2 Inhibitory Effects" Molecules 27, no. 14: 4606. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27144606