
Bidentate Donor-Functionalized N-Heterocyclic Carbenes: Valuable Ligands for Ruthenium-Catalyzed Transfer Hydrogenation


Ecole Européenne de Chimie, Polymères et Matériaux, Université de Strasbourg, CNRS, LIMA, UMR 7042, 25 Rue Becquerel, 67087 Strasbourg, France
*
Author to whom correspondence should be addressed.
Molecules 2022, 27(15), 4703; https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27154703
Submission received: 8 July 2022
/
Revised: 20 July 2022
/
Accepted: 21 July 2022
/
Published: 23 July 2022
(This article belongs to the Special Issue Featured Reviews in Organometallic Chemistry)
Abstract
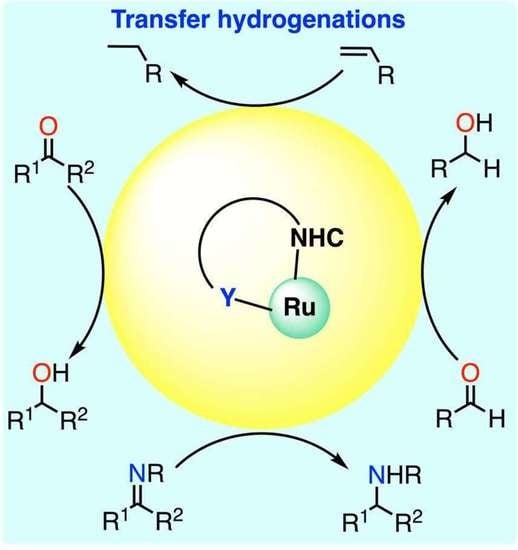
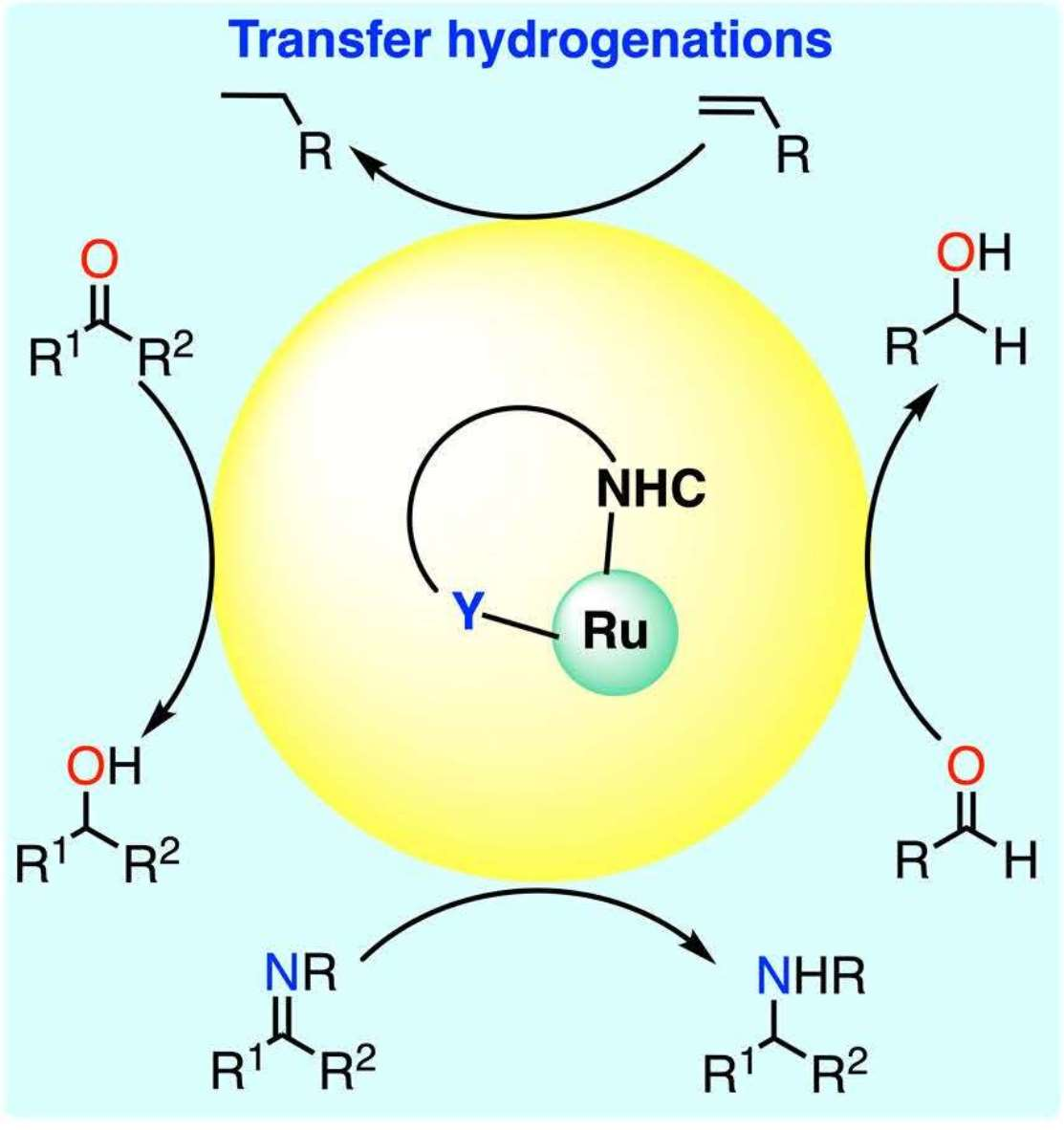
:Ruthenium complexes are by far the most studied compounds that catalyze hydrogen transfer reactions. In this review, we describe the use in this field of ruthenium complexes bearing bidentate donor-functionalized N-heterocyclic carbene ligands. The review specifically covers the application in transfer hydrogenations of (κ2-CNHC,Y)-ruthenacyclic compounds where the Y donor atom is a N, P, O, or S atom, and where the N-heterocyclic carbene ligand is a classical imidazol-2-ylidene, a benzimidazol-2-ylidene, a mesoionic 1,2,3-triazolylidene, or an imidazol-4-ylidene ligand. Tridentate donor-functionalized N-heterocyclic carbene complexes thus fall outside the scope of the review. Applications in (asymmetric) transfer hydrogenation of ketones, aldehydes, imines, alkenes, and nitrobenzene are discussed.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
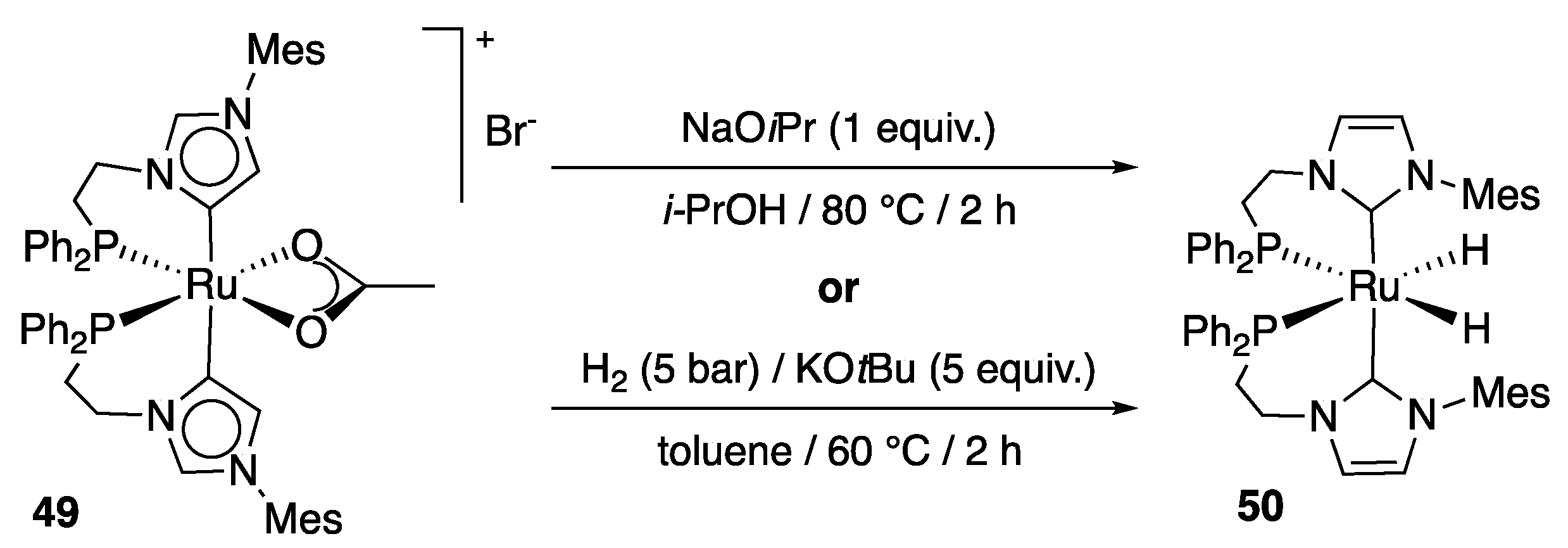{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
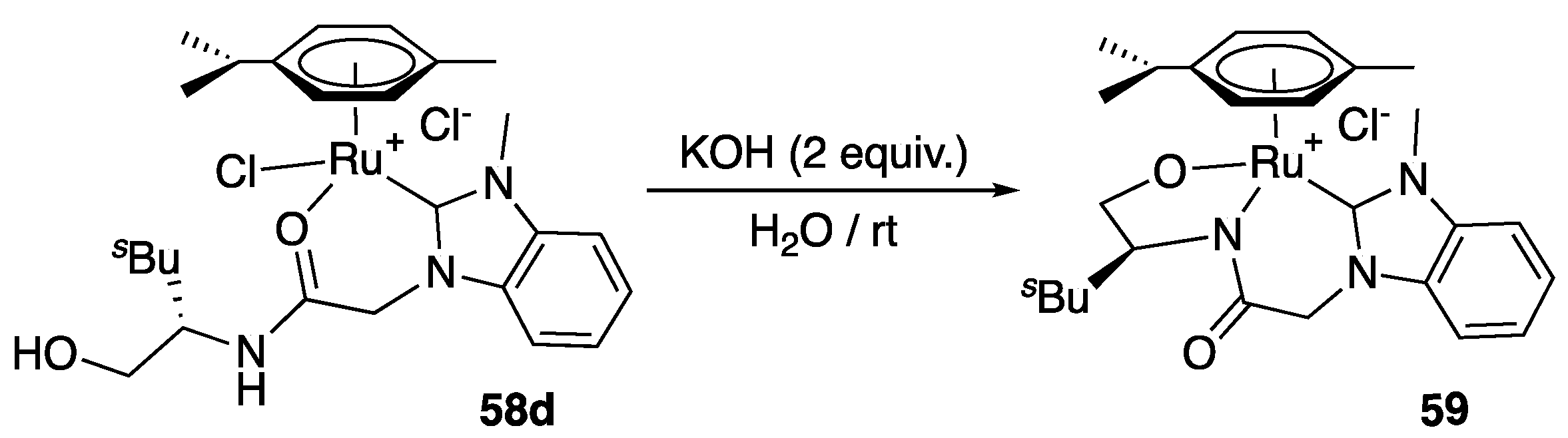{kind=link}
{kind=link}
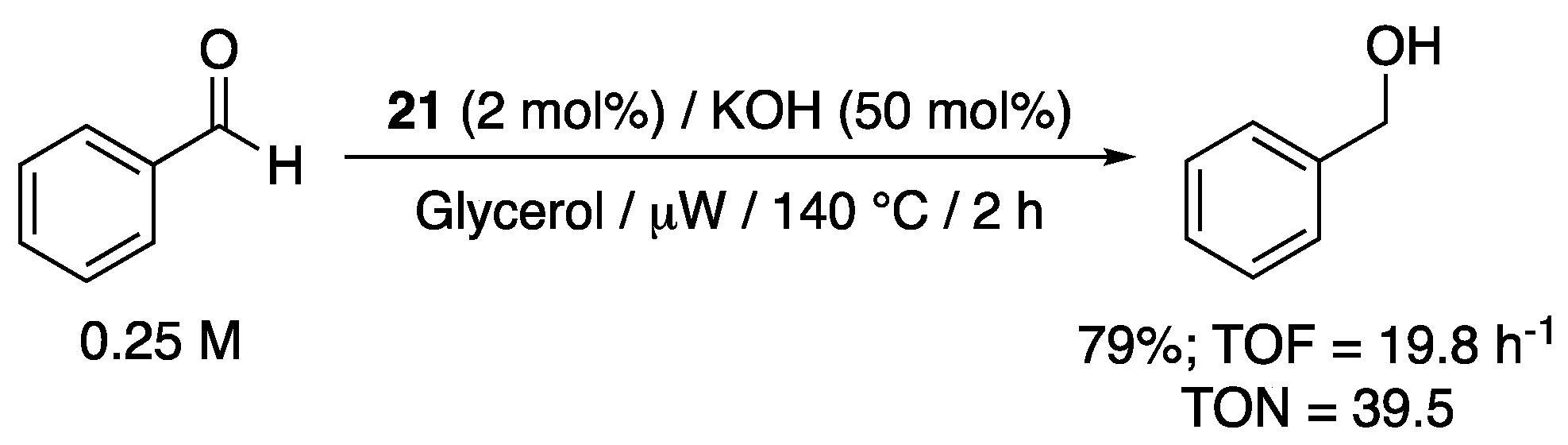{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Transition-metal catalyzed transfer hydrogenation (TH) is a key reaction for the fine chemical and pharmaceutical industries as a safer alternative to hydrogenation of multiple bonds with flammable hydrogen gas [1]. The TH of aldehydes, ketones, and imines in particular are industrially relevant. These reactions are usually carried out with isopropanol as both the solvent and the hydrogen source. Other common hydrogen donors include formic acid or, more recently, glycerol. Ruthenium is undoubtedly one of the most exploited transition metals, together with iridium and rhodium, for this transformation [2,3,4,5,6,7], and a wide variety of ligands have been used with it [3,5,6,7]. Among them, N-heterocyclic carbenes are interesting for affording high thermal stability and tunability of the steric and electronic properties of the metal [6]. Nevertheless, several common decomposition pathways exist for metal–NHC complexes [8], which limits the ability of monodentate NHC ligands to effectively stabilize the ruthenium centers at the high temperatures often employed in TH reactions, i.e., in refluxing 2-propanol or formic acid. In this context, the ability of bidentate donor-functionalized NHCs to stabilize metal centers by preventing decomposition and offering the possibility to act as a hemilabile ligand with reversible dissociation of the non-carbon atom is quite valuable [9,10] and has been largely exploited in ruthenium-catalyzed TH reactions. This review provides a comprehensive overview of the advances in this field with (κ2-CNHC,Y)-ruthenacyclic precatalysts where the Y donor atom is a N, P, O, or S atom, and where the N-heterocyclic carbene ligand comprises (benz)imidazol-2-ylidenes, mesoionic 1,2,3-triazolylidenes, and imidazol-4-ylidenes.
2. Transfer Hydrogenation of Ketones
As it will appear to the reader, the large majority of the literature for this reaction deals with cationic half-sandwich complexes that bear chelating N-functionalized imidazole-2-ylidene or mesoionic triazolylidene ligands.
2.1. Ketones’ Transfer Hydrogenations with (κ2-CNHC,N)-Complexes
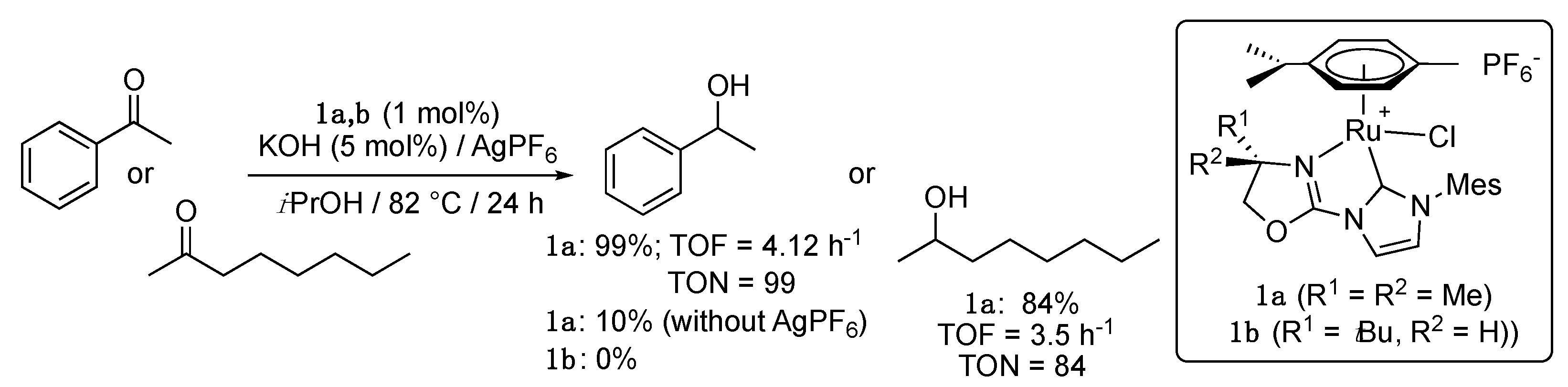
The first example of this long series was reported in 2006 by Gade et al. with the description of the synthesis—by transmetalation from the corresponding silver NHC complexes—and catalytic activity of the p-cymene-ruthenium complexes 1a,b bearing an oxazolinyl-functionalized imidazole-2-ylidene ligand [11]. Complex 1a showed moderate activity in the TH of acetophenone and 2-octanone from iPrOH/KOH at 82 °C in the presence of AgPF6, achieving turnover numbers (TON; expressed as the yield or conversion (%) after a given time/catalyst loading (mol%)) from 84 to 99, and turnover frequencies (TOF; expressed as the TON/reaction time (h)) from 3.5 to 4.12 h−1 (Scheme 1). However, the reaction failed with the more sterically demanding pinacolone. Interestingly, the formation of a dicationic species seemed to be crucial, since the use of 1a in the absence of AgPF6 led to very poor yields. On the other hand, the chiral complex 1b bearing a bulky t-butyl group proved completely unreactive under these conditions.
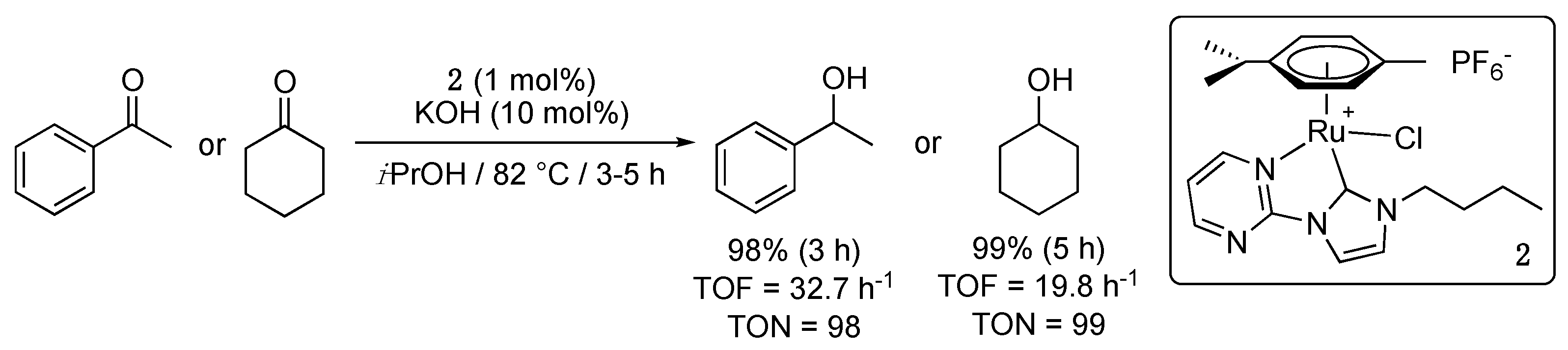
The second example only appeared three years later with a contribution of Crabtree et al. that disclosed the pyrimidine-functionalized imidazol-2-ylidene-Ru(II) complex 2 [12]. This air-stable complex, which was similarly prepared by transmetalation from the corresponding silver carbene complex, was found to be relatively more active than 1a achieving almost complete reduction in acetophenone and cyclohexanone in only 3 to 5 h in 2-propanol at 82 °C. However, it is noteworthy that 10 mol% of KOH as base were required (Scheme 2).
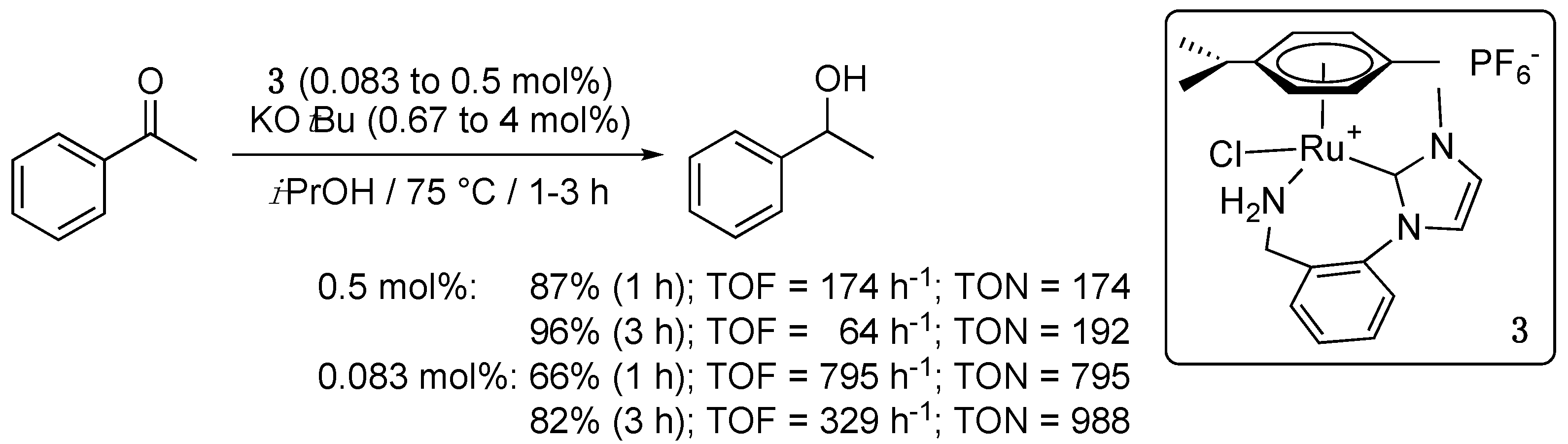
At about the same period, Morris et al. reported the first paper of a long series dealing with the use of half-sandwich Ru complexes bearing primary amino-functionalized NHC ligands in both TH and H2-hydrogenation catalysis [13]. By transmetalation of the amino-functionalized NHC ligand from the corresponding homoleptic bis-NHC-nickel(II) complex to [Ru(η6-p-cymene)Cl2]2, they prepared the cationic air-stable Ru(II) complex 3 in 60% yield. The latter proved inactive at room temperature, but efficiently catalyzed the TH of acetophenone at 75 °C in the presence KOtBu and 2-propanol, allowing 87% conversion in 1 h with a catalyst loading of 0.5 mol% (TOF = 174 h−1), and 66% in 1 h with a loading of only 0.083 mol% (TOF = 795 h−1) (Scheme 3). Notably, the catalytic activity of 3 was barely affected by the nature of the strong base used (KOtBu, NaOiPr, or KOH), or the presence of small amounts of water, which were generated when KOH was used as a base.
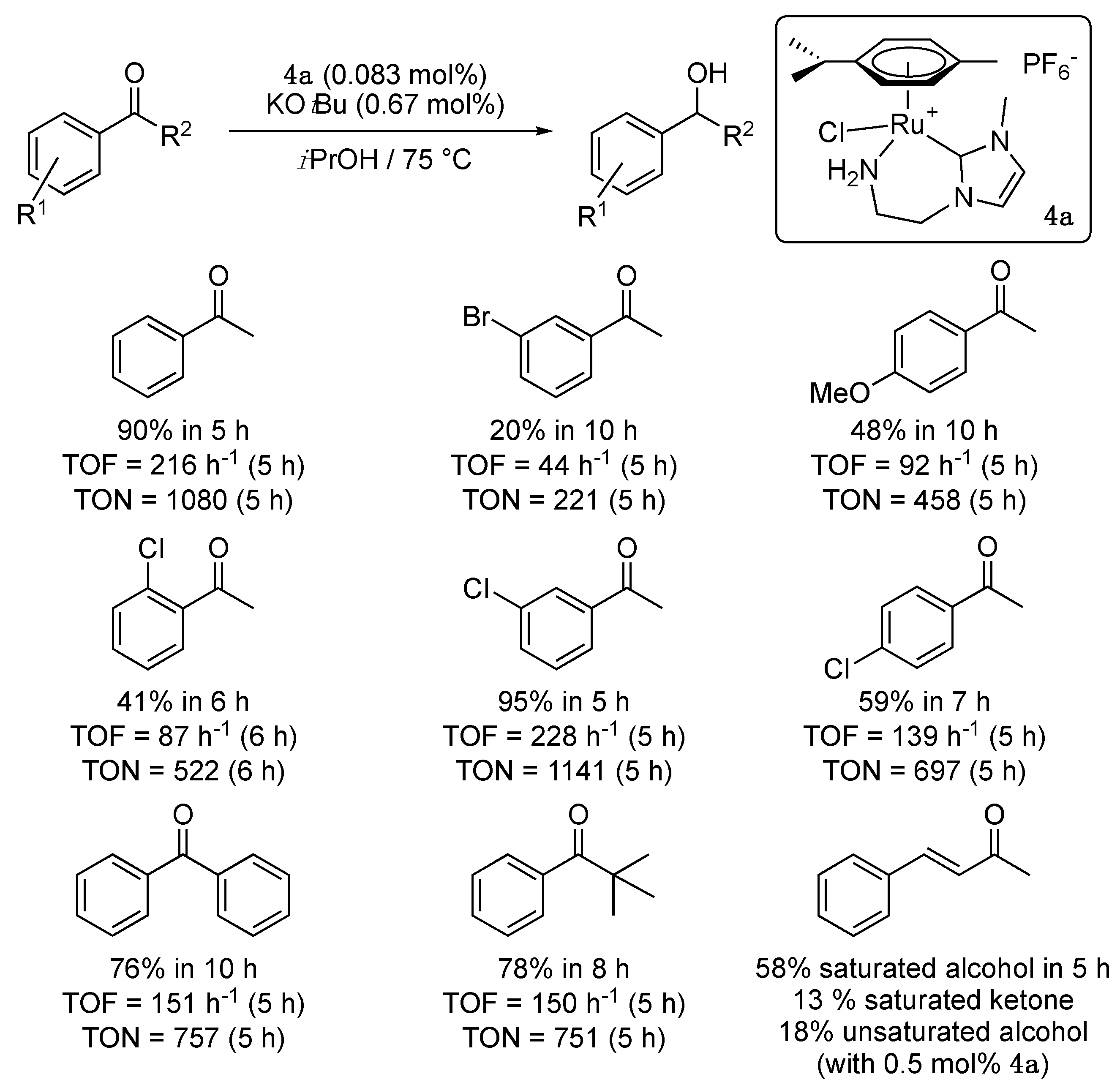
A couple of years later the same group disclosed another cationic amino-functionalized imidazol-2-ylidene-Ru(II) complex 4a, in which the chelating carbene ligand formed a six-membered ring instead of a seven-membered ring in complex 3 [14]. This complex, which was also synthesized by transmetalation from the adequate silver carbene species, showed a slightly lower activity than 3 under the same conditions (0.5 mol% 4a, 4 mol% KOtBu, 75 °C, in 2-propanol), with 49% conversion of acetophenone to 1-phenylethanol in 1 h (TOF = 98 h−1) and 92% conversion in 3 h (TOF = 61.3 h−1). A more comparable activity was observed with a catalyst loading of 0.083 mol%, with 53% conversion after 1 h (TOF = 636 h−1) and 90% after 5 h (TOF = 216 h−1).
A brief study of the reaction scope, carried out with a catalyst loading of 0.083 mol%, showed that TH was difficult for acetophenones that contained an electron-donating 4′-methoxy or a reactive bromide group on the phenyl group (Scheme 4). The 2′- and 4′-chloroacetophenones proved difficult to hydrogenate compared to 3′-chloroacetophenone. Good conversions could be achieved with benzophenone and 2,2-dimethylpropiophenone, yet after long reaction times. Finally, the reduction in trans-4-phenylbut-3-ene-2-one was unselective, giving predominantly the saturated alcohol, 4-phenylbutan-2-ol, which is characteristic of an inner-sphere mechanism involving the decoordination of the amine side arm.
The corresponding hydride complex 4b was prepared by reaction of 4a with 3 equiv. of NaOiPr in 2-propanol at 50 °C. Interestingly, it showed little reactivity in the absence of a base, giving only 22% conversion of acetophenone to 1-phenylethanol in 4 h in the presence of a 0.5 mol% catalyst loading, and about 10% conversion under stoichiometric conditions in THF-d8 at 50 °C. This low reactivity, together with the large free energy barrier (ΔG≠) that was computed by DFT for a H+/H− couple to transfer to acetophenone from a model amine-hydride complex militated against an outer-sphere bifunctional mechanism. In contrast, the favorable energy barriers computed for the two crucial steps of an inner-sphere mechanism involving the decoordination of the amine side arm, namely (i) the β-hydride elimination of a coordinated 2-propoxide ligand, and (ii) an attack of the resulting hydride on the coordinated acetophenone, strongly militated in favor of the latter [14].
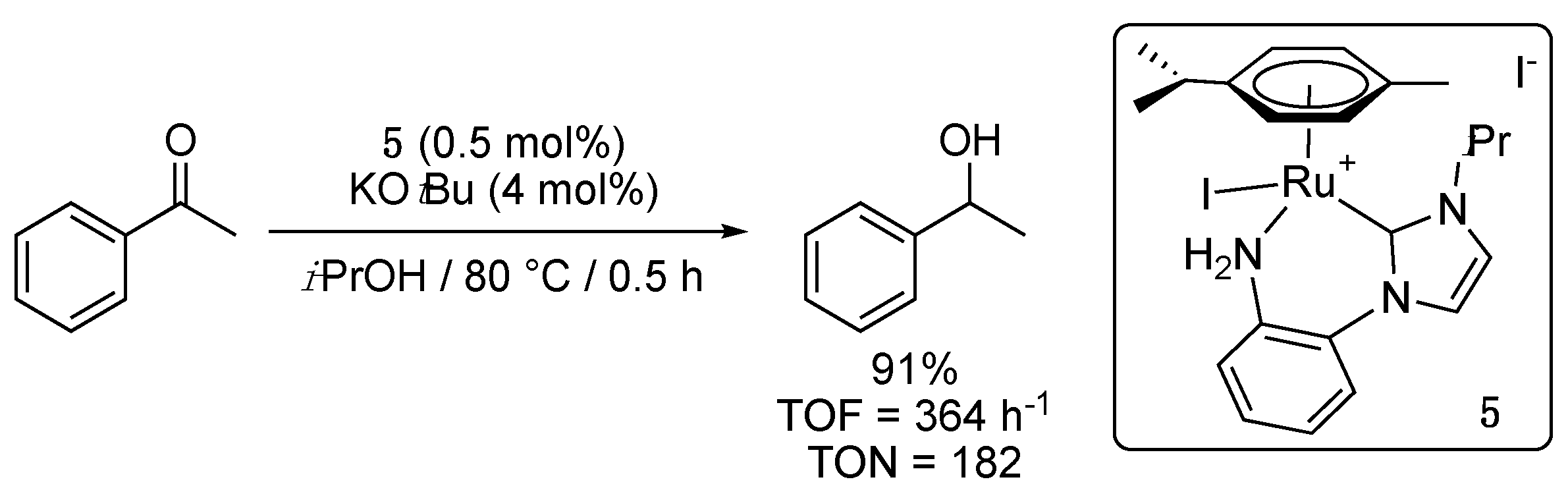
During the same period, Cross and his collaborators communicated about the related aniline-functionalized NHC-Ru(II) complex 5 [15], displaying also a six-membered metalacyclic ring. The latter demonstrated higher catalytic activity than 3 and 4a with 93% conversion of acetophenone to 1-phenylethanol in half an hour only, under similar conditions, i.e., 0.5 mol% 5, 4 mol% KOtBu, 80 °C, in 2-propanol (Scheme 5). Furthermore, 5 proved much more active than the corresponding Cp* (Cp* = η5-C5Me5) Rh complex, and the related Cp*Ir-amide complex. Similarly to 3 and 4a, 5 however showed no activity at room temperature.
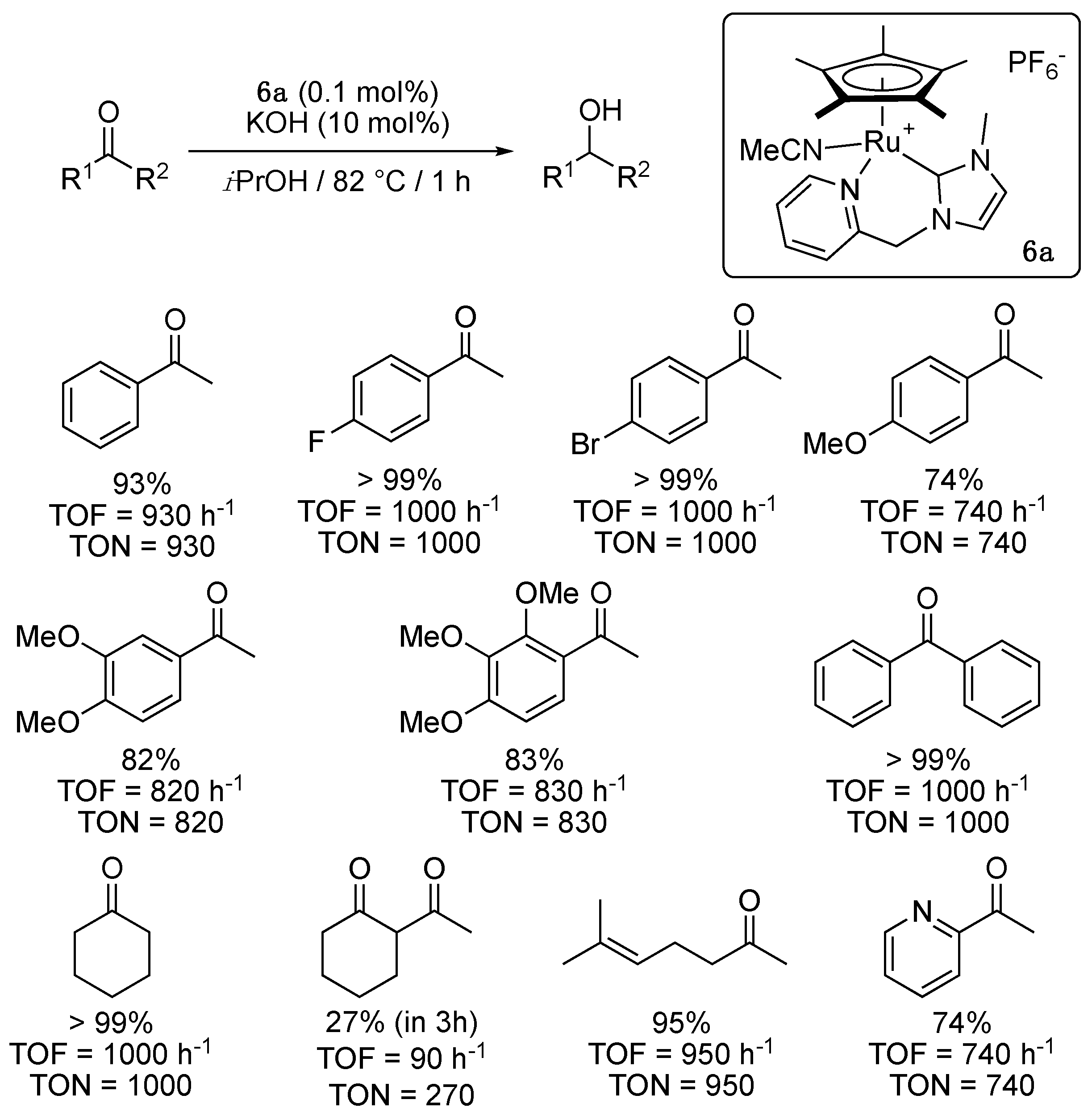
In 2011, Valerga and Puerta reported the synthesis and characterization of a series of picolyl-functionalized NHC-Ru(II) complexes 6 bearing a Cp* (η5-C5R5) ligand [16]. The cationic complexes were synthesized by treatment of [RuCp*(CH3CN)3][PF6] with a THF solution of the appropriate ligand, in situ generated by the reaction of the picolylimidazolium bromide and an excess of KOtBu. All tested picolylcarbene complexes were shown to catalyze the TH of acetophenone from isopropanol at 82 °C with KOH as base. Notably, the complex 6a containing a methyl group as a wingtip substituent and hydrogen atoms on the imidazole backbone was found to be more active than those containing an isopropyl (6b) or mesityl group (6c) as a wingtip substituent and those containing chloro substituents on the backbone and a methyl group as a wingtip substituent (6d), with a turnover frequency value at 50% conversion (TOF50) of 62 min−1 vs. TOF50 of 0.38 to 20 min−1 for 6b–d with a catalyst loading of 1 mol%. This showed that both steric and electronic have a non-negligible influence on the catalytic activity of these family of complexes. A maximum TOF50 of 100 min−1, i.e., 6000 h−1, could even be observed for the TH of acetophenone with a catalyst loading of 0.1 mol%, and 6a was able to reduce a variety of aromatic and aliphatic ketones with a remarkable efficacy compared to the previously described complexes 1–5 (Scheme 6).
Regarding the reaction mechanism, no experimental data was acquired [16], but some insight to the TH of acetophenone catalyzed by 6a was provided by a DFT study at the B3LYP level of theory conducted by Shang and Xu [17]. From the results of the calculations, it was concluded that a stepwise inner-sphere monohydride mechanism involving decoordination of the pycolyl arm as an entry point should be favored from an energetic point of view.
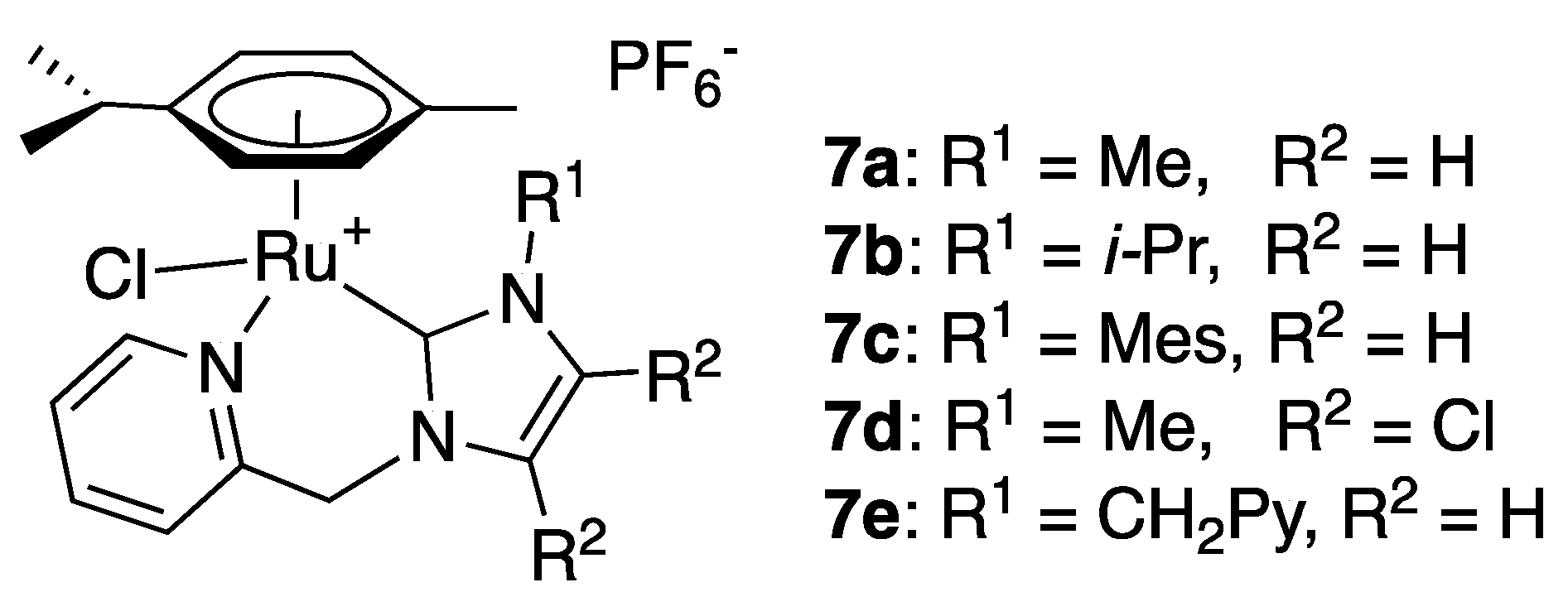
A year after their first study, Puerta and Valerga published a second article dealing with picolyl-imidazol-2-ylidene complexes, but this time with a η6-p-cymene instead of a Cp* as the capping ligand [18]. The results were in the same vein as those obtained with complexes 6a–d regarding the effect of the wingtip and imidazole backbone substituents on the comparative activity of complexes 7a–d (Figure 1) and the reaction scope. The activity was slightly lower though, as the most active complex 7a required a catalyst loading of 0.2 mol% and reaction times of 6–24 h to reach similar yields as those obtained with 5a in 1 h with a catalytic loading of only 0.1 mol% (Scheme 6). The more recently reported complex 7e bearing a second (uncoordinated) picolyl side arm as wingtip substituent proved, in contrast, to be almost unreactive, as only a 15% reduction in acetophenone was achieved after 16 h reaction with a catalyst loading of 1 mol% in the presence of KOtBu (5 mol%) in isopropanol at 80 °C [19].
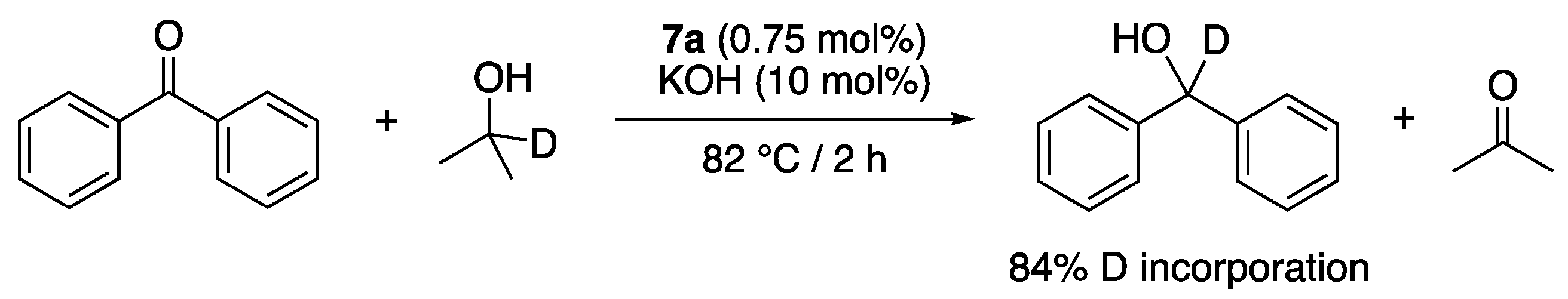
The reasons for the lower activity of complexes 7a–d compared to their Cp* analogues 6a–d were not discussed. However, the application of the deuterium labeling protocol—first described by Bäckvall [20] and Crabtree [21]—to distinguish between possible monohydride and dihydride reaction mechanisms, allowed the authors to establish a monohydride mechanism with 7a [18], in line with the results of the DFT calculations conducted by Shang and Xu with 6a [17]. Indeed, when a monohydride mechanism is operating, the C–H bond of the hydrogen donor should end up exclusively as the C–H bond on the carbinol carbon of the product, whereas when a dihydride mechanism is operating, the donor C–H bond should be scrambled between the carbinol C–H and the O–H bonds, and the TH of benzophenone from 2-propanol-2-d in the presence of 7a resulted in the formation of mostly monodeuterated 1,1-diphenylmethanol-1-d with 84% deuterium incorporation at the carbinol position (Scheme 7) [18].
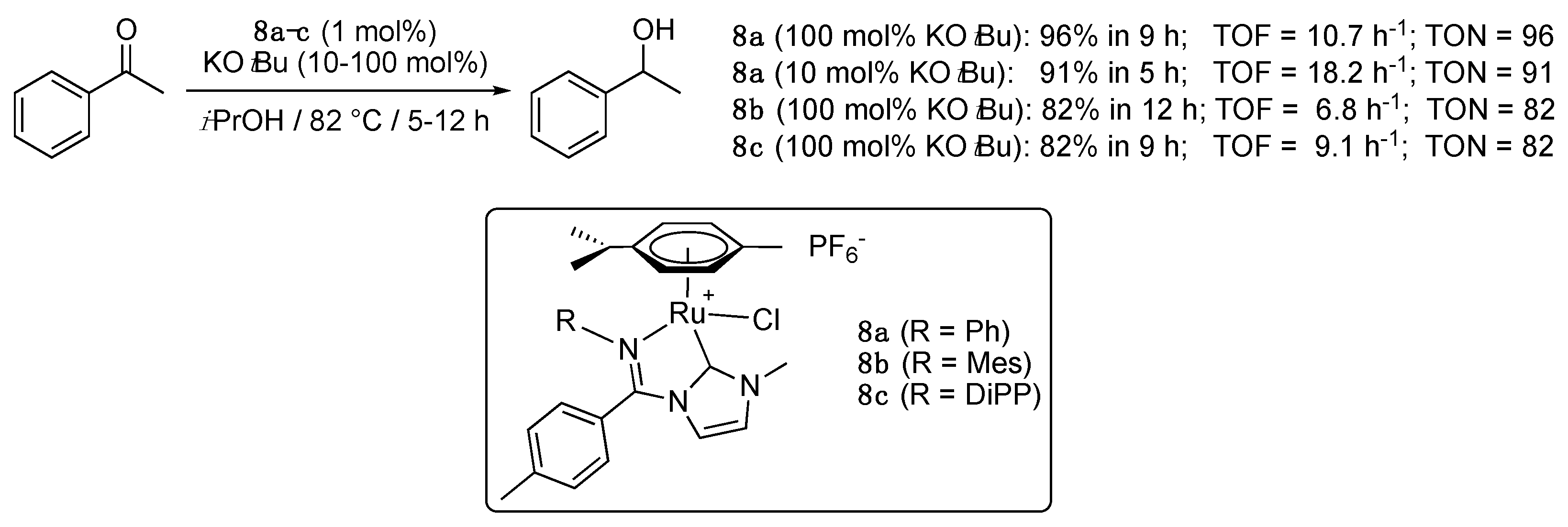
The synthesis, via transmetalation from the corresponding silver complexes, and characterization of a series of three five-membered imino-functionalized imidazol-2-ylidene-Ru(II) complexes 8a–c bearing a η6-p-cymene ligand, and of the corresponding Cp* iridacycles was reported by Hou et al. in 2012 [22]. All complexes showed relatively modest activity (>80% in 9–12 h) for the TH of acetophenone from isopropanol at reflux with a catalyst loading of 1 mol% in the presence of 1 equiv. of KOtBu as base. As observed with complexes 6a–d and 7a–d, the complex 8a with the smallest steric footprint proved to be the most effective with a yield of up to 96% in 9 h under these conditions, and of up to 91% in 5 h in the presence of 10 mol% KOtBu (Scheme 8). Under these optimized conditions (10 mol% KOtBu), 8a reduced a small range of seven aromatic ketones, as well as the aliphatic ketone, 2-decanone, with good to excellent yields. No mechanistic details were given.
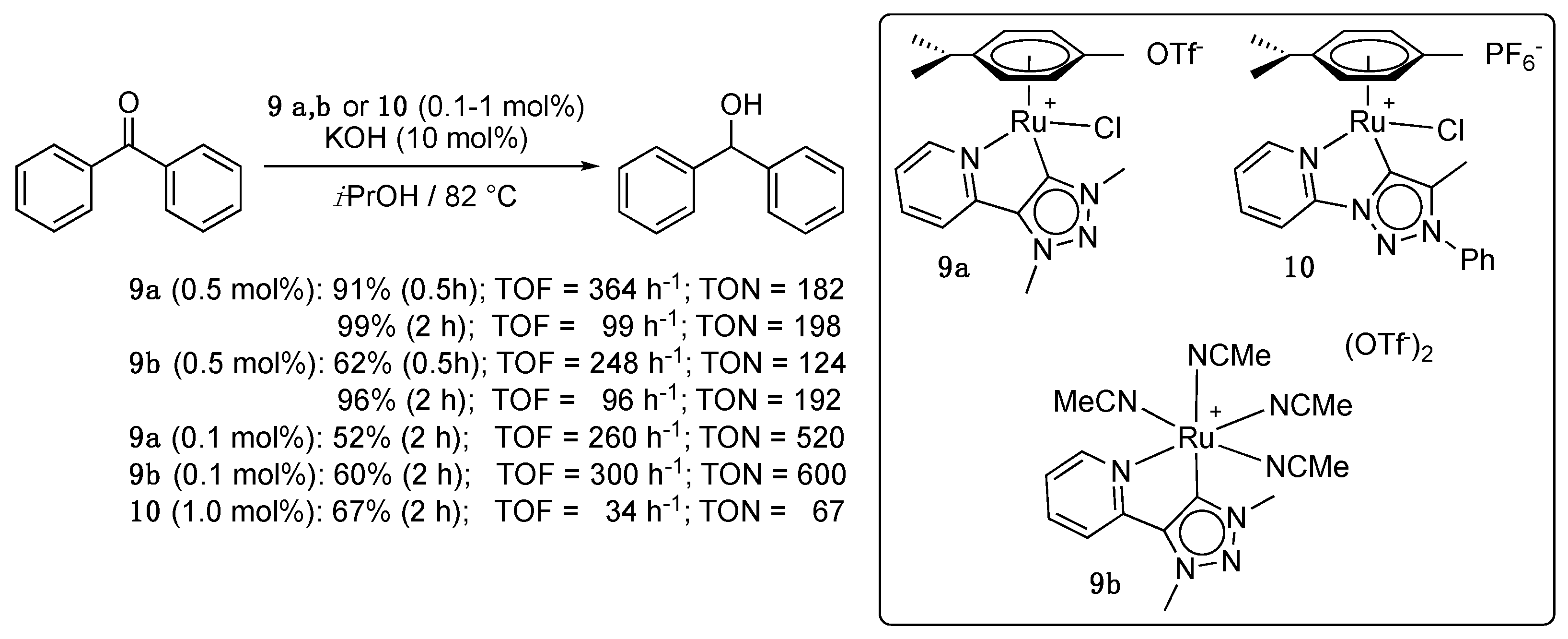
The first examples of donor-functionalized mesoionic triazolylidene ruthenium precatalysts for ketone TH appeared in 2014 [23]. As a subclass of NHCs, mesoionic 1,2,3-triazolylidene ligands generally present slightly stronger σ-donor properties compared to classical imidazolium-derived systems and are easily accessed by click chemistry. In this early report, two pyridyl-functionalized triazolylidene (η6-p-cymene)Ru complexes 9a and 10, that differ by the bonding of pyridyl unit to either the triazole nitrogen or carbon, were synthesized by transmetalation from the corresponding silver carbenes and shown by Albrecht et al. to catalyze the TH of benzophenone with low to good efficiency depending on their structure (Scheme 9). Hence, the C-bonded pyridyl complex 9a achieved a TON as high as 520 in 2 h in refluxing isopropanol whereas the N-bonded derivative 10 gave at most a TON of 67 in the same amount of time. The octahedral derivative of 9a, the p-cymene-free complex 9b bearing four acetonitrile ligands, showed a comparable activity to this latter. The relatively high activity of 9a/b was attributed to the relatively low electron density of the ruthenium center, which would facilitate the isopropoxide anion coordination. Time-dependent monitoring of the reaction conducted with a 9a loading of 0.5 mol% revealed a TOF50 of 680 h−1, a value that is lower by a 10-fold order to that observed with the picolyl-NHC complex 6a [16], and also lower to the values reported for the amino-NHC complexes 3 [13] and 4a [14]. No induction period was revealed by these kinetic measurements, and pseudo-first order kinetics was observed for both substrate consumption and product formation. A short reaction scope study demonstrated that diaryl, aryl-alkyl, and dialkyl ketones (7 examples) are readily hydrogenated under these conditions [23].
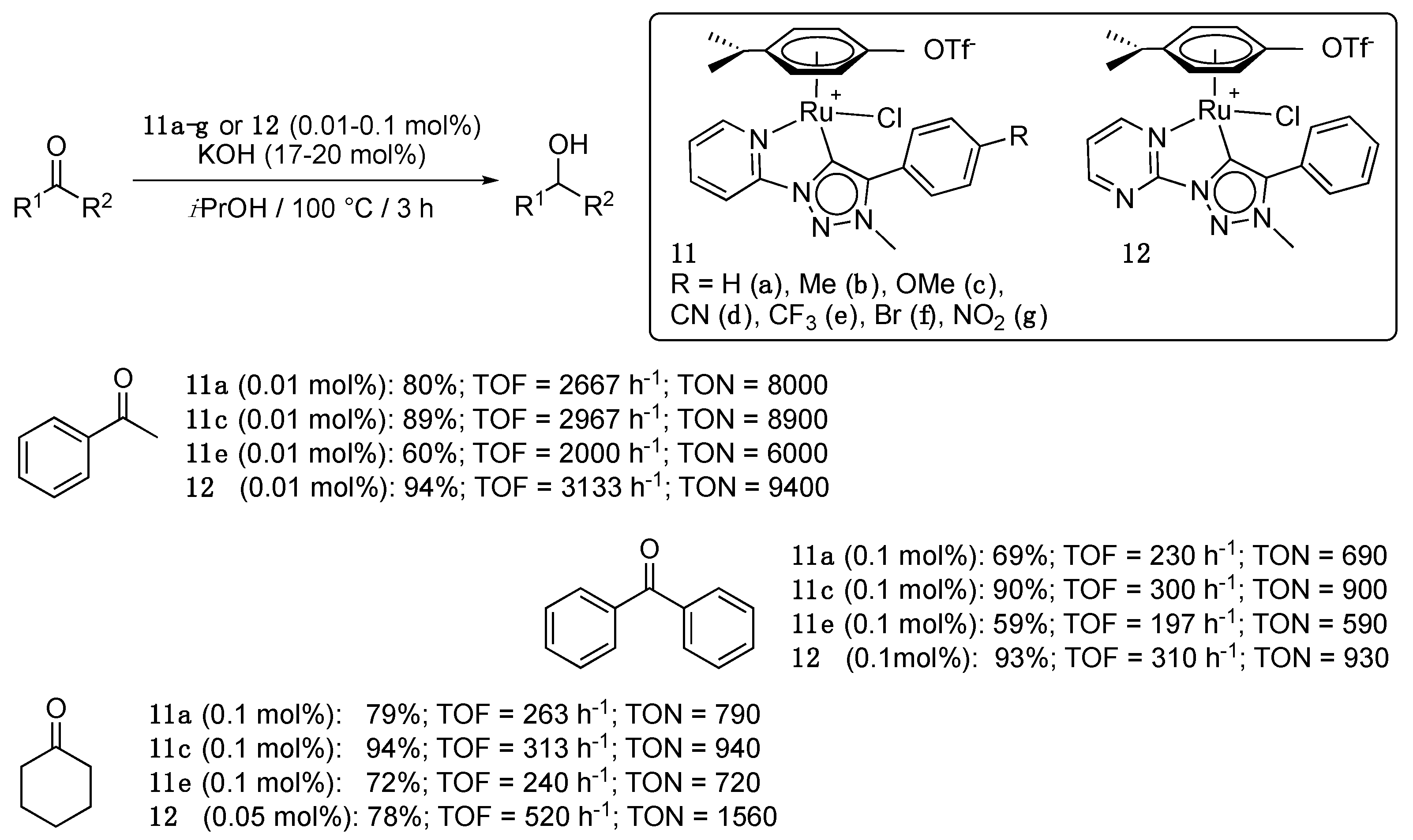
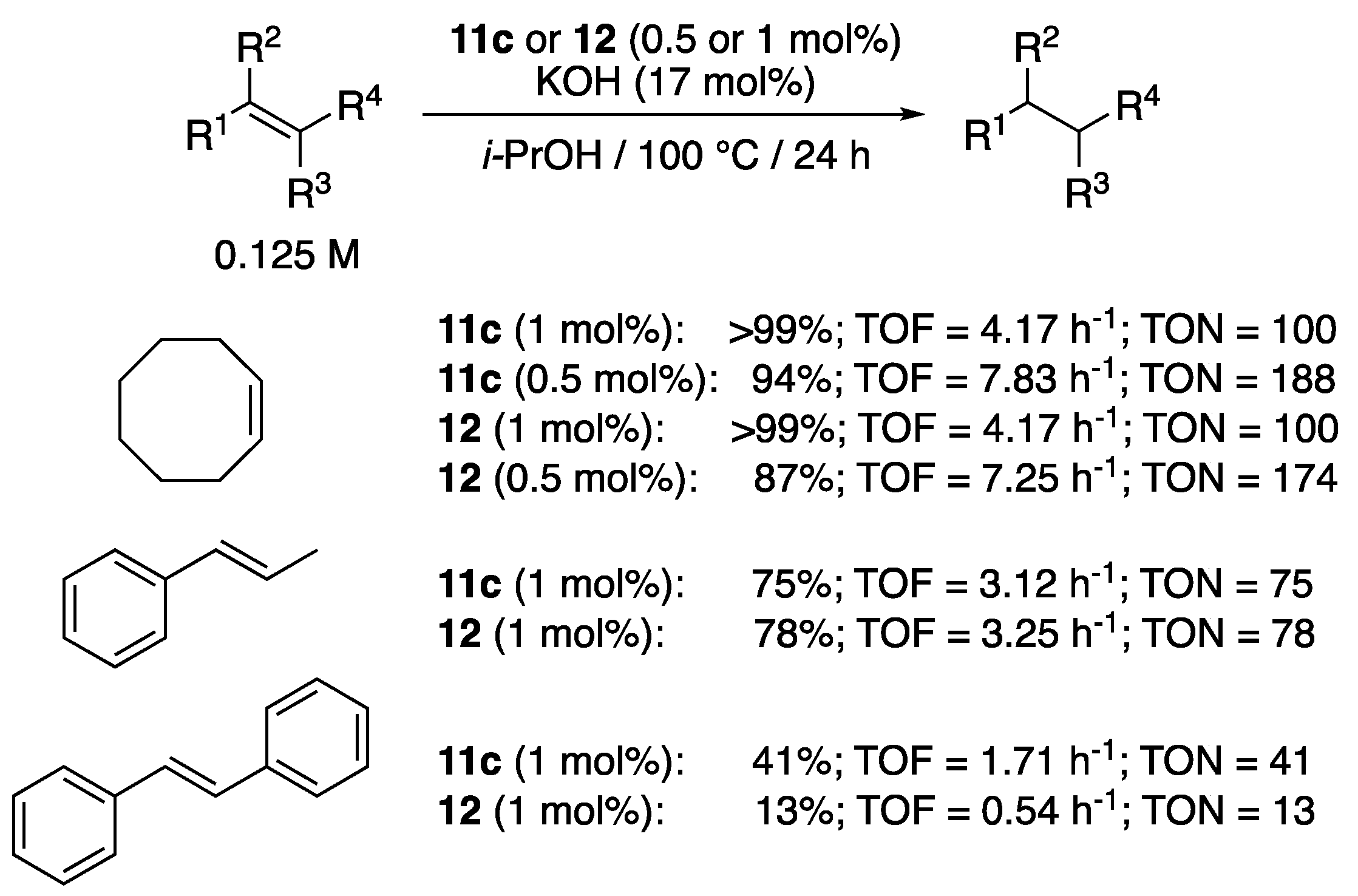
Shortly after Albrecht’s initial report, Sarkar et al. described a series of eight analogues of 10 with varying electronic properties, that showed greatly improved catalytic properties compared to 9a,b and 10, though under harsher conditions (100 °C, KOH (17–20 mol%)) [24,25].The complex 11c (0.01 mol%) bearing an electron-donating methoxy substituent [24] and the pyrimidine derivative 12 [25], in particular, gave outstanding results with TON as high as 8900 and 9400, respectively, for the TH of acetophenone, while complexes 11a and 11e displayed slightly lower activities (Scheme 10), and complexes 11b,d,f,g required a loading of 0.5 mol% to achieve high yields. The same trend was observed with the more sterically hindered benzophenone, with TON of 900 and 930, and with the aliphatic cyclohexanone, with TON of 940 and 1560, respectively, (Scheme 10). Notably, the catalytic activity of Cp*-iridium and osmium analogues of 11a–g and 12 was also studied, and ruthenium worked better in all cases. Similarly to what was observed with the ruthenium complexes 11a–g, osmium complexes bearing electron-donating substituents were found to be more active than those bearing electron-withdrawing substituents. For the iridium complexes, the electronic effects were reversed. The authors suggested that this trend could be related to the difference between the p-cymene and Cp* ligands in both systems. Finally, attempts to detect a Ru-hydride species by 1H NMR spectroscopy after the reaction of 11a in the absence of substrate, but under otherwise similar conditions, failed. In contrast, the same procedure with the Os and Cp*-Ir species allowed detecting two hydride species in each case, therefore suggesting the intermediacy of such species in the reaction mechanism [24].
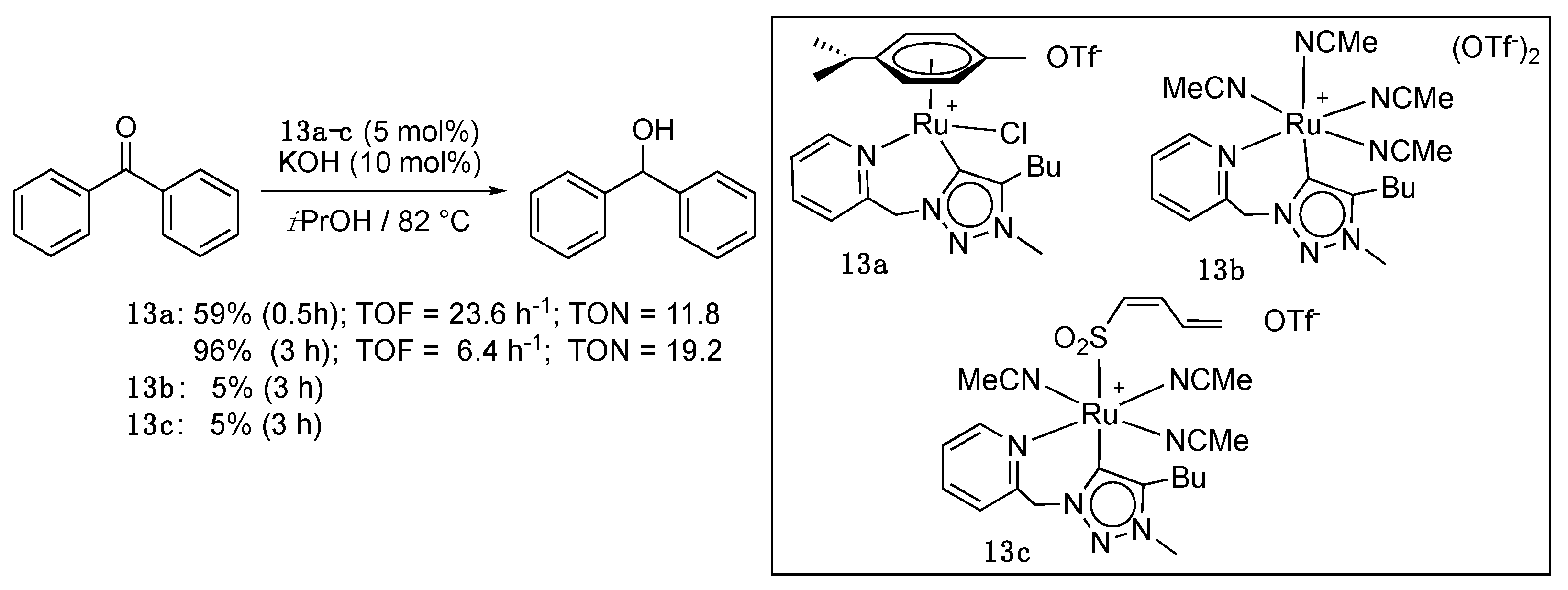
The closely related picolyl-functionalized triazolylidene (η6-p-cymene)Ru complex 13a (5 mol%), which was prepared via the usual transmetalation procedure from the adequate silver carbene, showed a much lower activity in comparison to complexes 9–12 with a maximum TON of 19.2 after 3 h reaction for the TH of benzophenone in the presence of 10 mol% KOH in refluxing isopropanol (Scheme 11) [26]. In addition, in contrast to 9b vs. 9a, the octahedral derivatives 13b and 13c bearing, respectively, four acetonitrile ligands and three acetonitrile and one butadienesulfonyl ligand showed almost no activity.
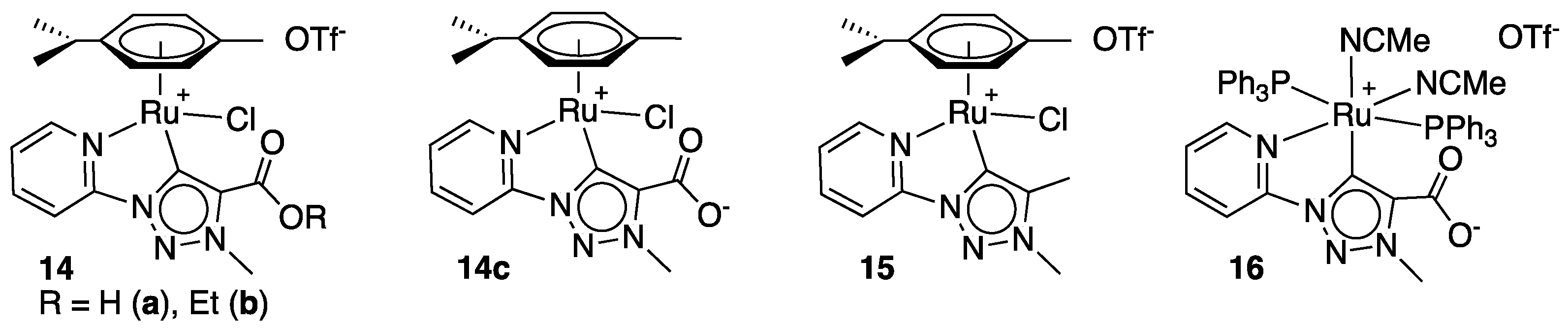
Having in mind the idea of bringing a non-coordinating Lewis base in close proximity to a Lewis acidic metal center in order to possibly access metal–ligand synergies, in particular for reversible substrate binding and proton release, Albrecht et al. introduced carboxylate groups to a pyridyl-triazolylidene ligand, closely resembling those found in complexes 10–12 [27]. The resulting complexes 14a–c bearing an ester, carboxylic acid, or carboxylate functionality (Figure 2), were fully characterized and tested for the TH of acetophenone from isopropanol at reflux in the presence of KOH (10 mol%). The observed activity, although modest (95 to 97% conversion in 1 h with a catalytic loading of 1 mol%), suggested that the presence of a pendant carboxylic acid (14a) or ester group (14b) is beneficial for enhancing the catalyst activity when compared to that of the complex 15 (1 mol%) that does not bear a coordinating functionality (29% conversion in 1 h). The carboxylate derivative 14c proved slightly less effective (75% conversion in 1 h), probably because this complex undergoes faster decarboxylation under basic conditions in comparison to 14a and 14b. Notably, the TH of acetophenone under base-free conditions with 14a or 14c led only to 0 and 9% conversion after 1 h, respectively, indicating that the pendant carboxylic group is not sufficiently basic for the formation of the reactive isopropoxide. The octahedral derivative 16 of the carboxylate complex 14c, containing two labile acetonitrile and two triphenylphosphine ligands, displayed very low activity compared to the latter with only 16% conversion after 1 h. Furthermore, a TH experiment run with benzophenone in heptadeuterated isopropanol (CD3)2CDOH resulted in the exclusive and almost complete deuteration of the reaction product at the carbinol carbon, thus suggesting a monohydride mechanism [20]. Attempts to detect a ruthenium hydride species by reaction of 14b in isopropanol in the presence of KOtBu were however unsuccessful.
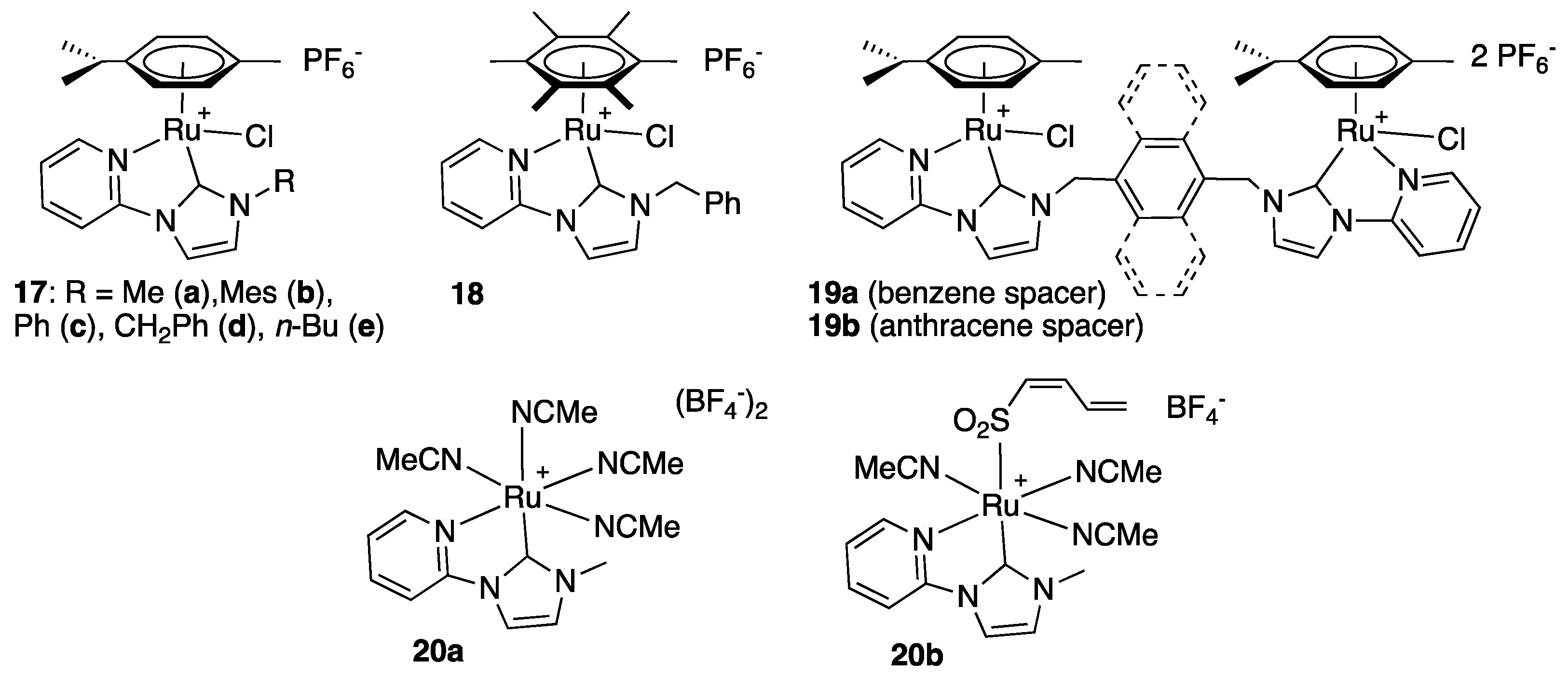
A number of η6-arene and octahedral ruthenium complexes bearing pyridyl-functionalized imidazole-2-ylidenes (Figure 3) was reported by several groups to catalyze the TH of acetophenone from isopropanol at reflux [28,29,30,31]. Relatively modest activities were observed in all cases as maximum TON ranged from 10 to 19 in the cases of 17a and 20a,b [28] and from 198 to 200 in the cases of 17d, 18, and 19a,b [30], and as rather hard conditions were necessary to reach these values, i.e., iPrOH at 82 °C for 2 h with catalyst loadings as high as 5 mol% in the former case [28], and base (KOH or NaOH) loadings as high 1 equivalent in iPrOH at 80 °C for 2 h in the latter case [30]. Notably, complexes 17d, 18, and 19a,b also reduced a couple of electron-deficient aromatic ketones with comparable efficiency. Electron-rich aromatic ketones required slightly longer reaction times (4–5 h instead of 1–2 h). The authors of this study proposed a classical monohydride mechanism that was checked by DFT calculation at the B3LYP level. Interestingly, all postulated intermediates could be optimized and were found to be energetically favorable [30]. Finally, it is worth mentioning that the activity of 17e (1 mol%) in the presence of KOH (10 mol%) was compared to that of its Os, Cp*Rh, and Cp*Ir counterparts, and that it was found slightly more active than them [31].
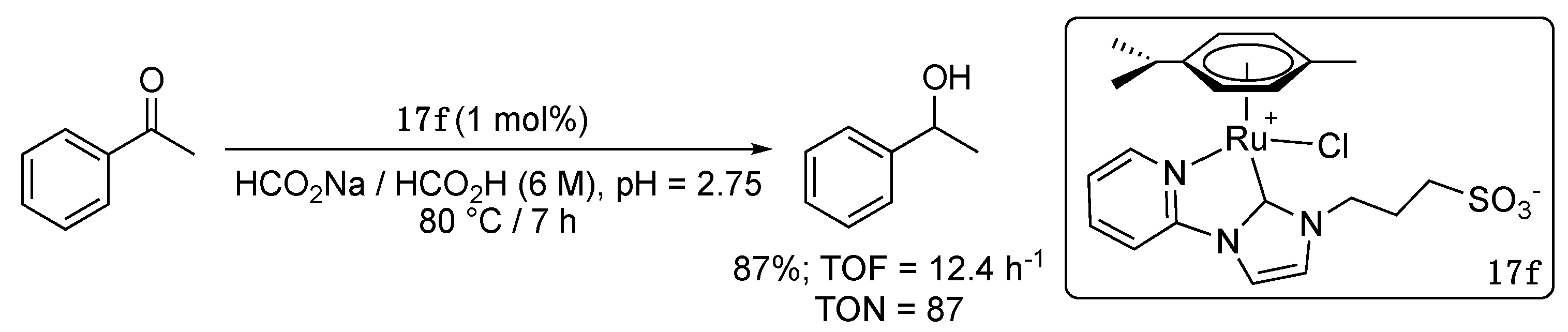
The water-soluble derivative 17f of complex 17e, bearing a propylsulfonate substituent instead of a n-butyl group, was prepared by transmetalation from the corresponding silver carbene complex and applied as catalyst for the TH of acetophenone in aqueous medium using HCO2Na/HCO2H as buffer (6 M, pH = 2.75) and hydrogen source [31]. A moderate yield of 87% was obtained after 7 h reaction (TOF = 12.4 h−1) at 80 °C with a catalytic charge of 1 mol% (Scheme 12). Notably, the Cp*Rh analogue proved much more active in this case, affording a 96% yield in only 10 min (TOF = 576 h−1) under these conditions.
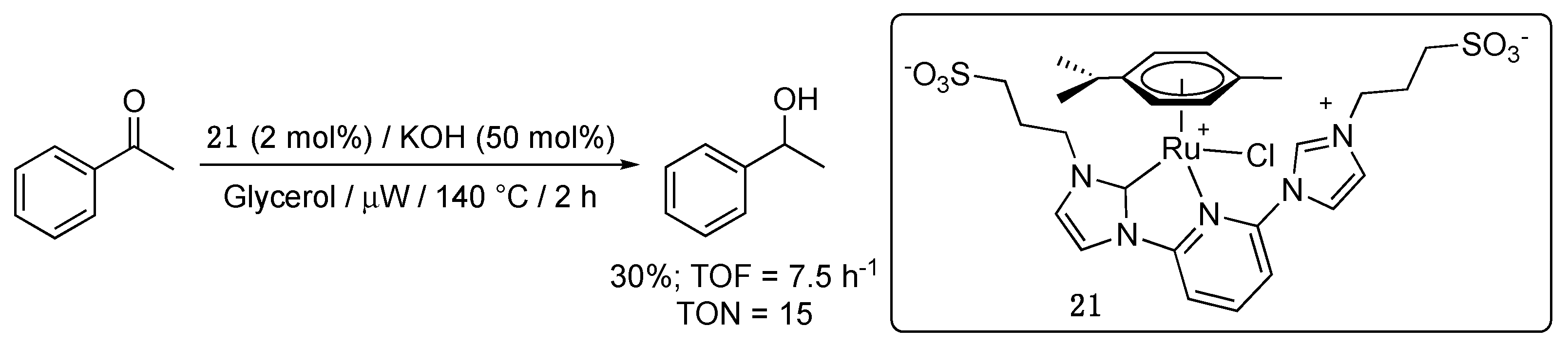
In the same vein, the related pyridyl-functionalized imidazole-2-ylidene complex 21, that bears two propylsulfonate groups, including one that is bound to a pendant imidazolium, was shown by Voutchkova-Kostal to be moderately active for the TH of acetophenone from glycerol, an inexpensive, renewable, non-toxic, and fully biodegradable solvent and hydrogen donor [32]. Thus, in glycerol at 140 °C under microwave heating, 30% yield to 1-phenylethanol was achieved after 2 h reaction with 2 mol% of 21 and 50 mol% KOH (Scheme 13).
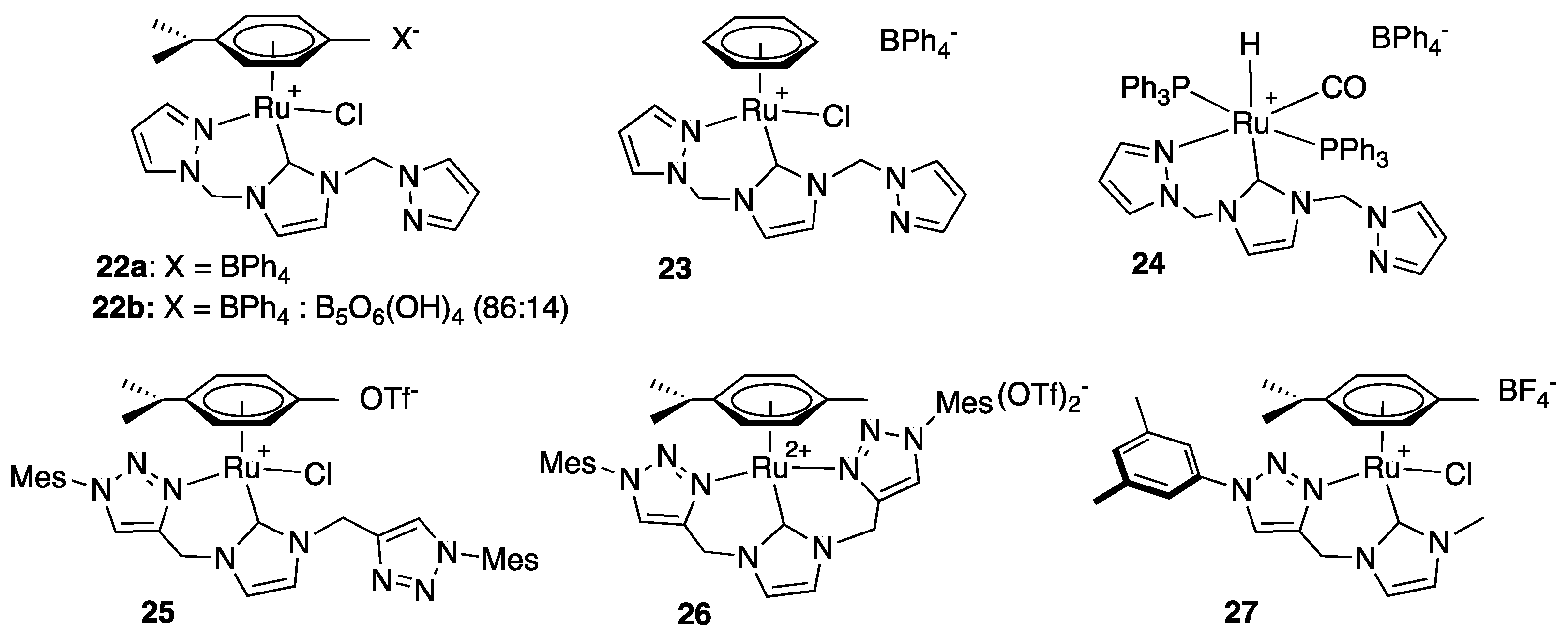
Rare examples of pyrazole- and triazole-functionalized imidazole-2-ylidene-Ru(II) complexes were reported by the groups of Messerle [33], Kühn [34], and Rit [35]. Similarly to the pyridine-functionalized NHC complexes 17–20, relatively modest activities were observed for the TH of acetophenone with the pyrazole derivatives 22a, 23, and 24 that bear both a coordinated and an uncoordinated pyrazole moiety (Figure 4). They indeed required 4 h reaction at reflux with catalyst loadings of 1.5 mol% and base loadings of 18 mol% (KOH) to achieve conversions of 80% (22a: TOF = 13.3 h−1; TON = 53), >99% (23: TOF = 16.6 h−1; TON = 66.7) and 24% (24: TOF = 4 h−1; TON = 16) [33]. The very modest activity observed with the hydride complex 24, in particular, was surprising, as the pre-existing hydride ligand was expected to enable a more efficient TH reaction. The triazole derivatives 25, 26, and 27 proved slightly more active with maximum TON of 170, 168, and 500, respectively, under milder conditions, i.e., 0.1 (27) to 0.5 (25, 26) mol% catalyst and only 5 (25, 26) to 10 (27) mol% NaOiPr or KOtBu as base in isopropanol at reflux for 2–3 h [34,35]. It is noteworthy, however, that full conversion could not be attained in the cases of 25 and 26 under these conditions. Thus, although the tridentate complex 26, with both triazole moieties coordinated to the ruthenium center, exhibits the highest TOF of these two triazole derivatives, with a TOF as high as 1100 h−1 at t = 2 min (19% conversion), it slows down significantly after 60% conversion to reach a maximum conversion of 84% after 2 h reaction (TOF = 84 h−1), suggesting an instability of the active species. Unfortunately, no mechanism insight was provided.
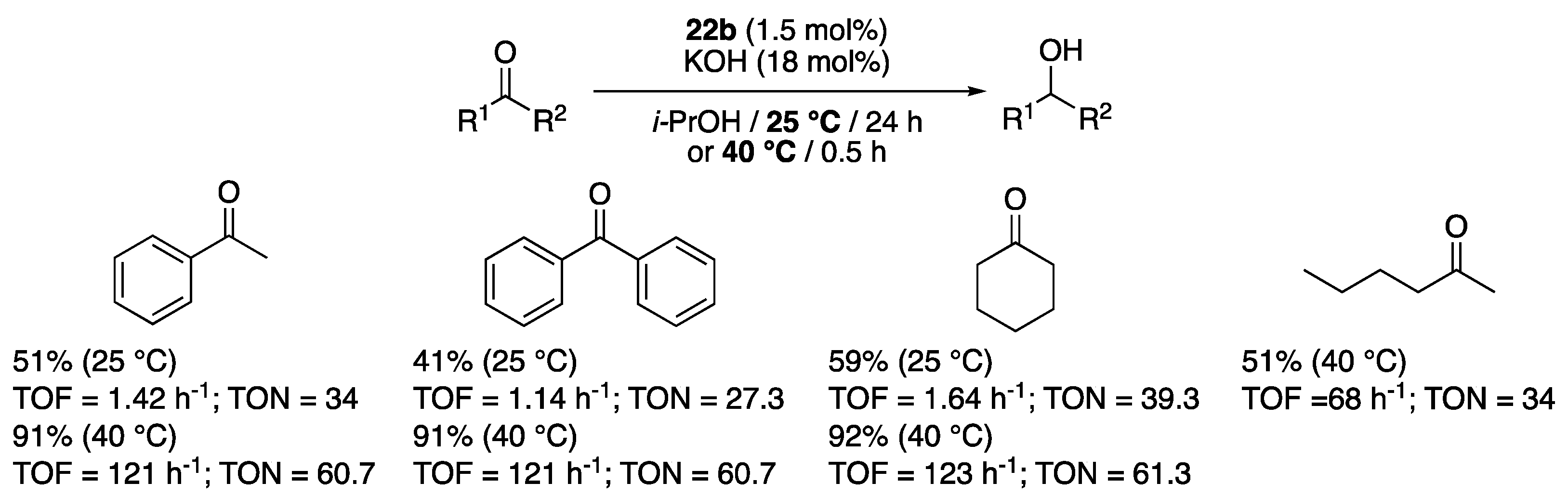
The mixed anion complex 22b, that contains an unusual 86:14 mixture of BPh4− and [B5O6(OH)4]− as a counter-anion deserves a particular attention. It indeed proved much more active than 22a containing pure BPh4−, and was demonstrated to be a very rare example of Ru(II) complex able to catalyze the TH of various ketones at low temperatures (25 to 40 °C, Scheme 14) [33] (see the works of Bera [36], Sakaguchi [37], and Pfeffer and de Vries [38,39] for other rare known examples of low-temperature efficient Ru(II)-based TH catalysts). The difference of activity between 22a and 22b, suggests a non-innocent role of the pentaborate anion, which most certainly acts as a catalyst in its own right, as has been shown in the literature for various polyborate anions [40,41].
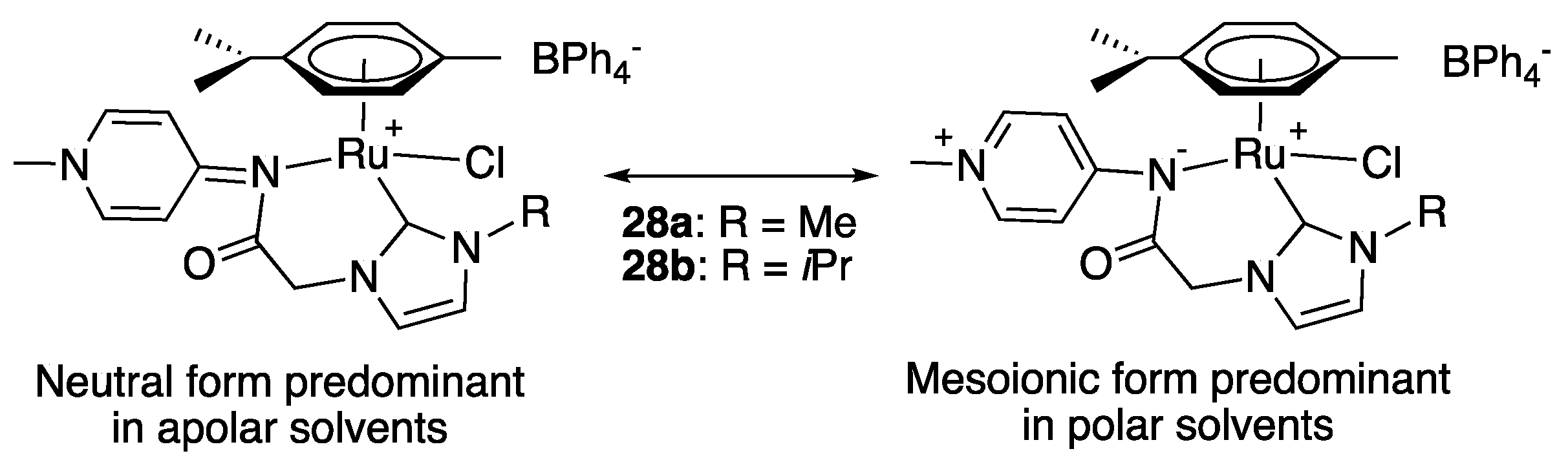
In 2014, Albrecht and Wright introduced an interesting class of hybrid pyridylideneamide-imidazol-2-ylidene ligands that has two dissimilar strong σ donors, comprised of a relatively soft C donor with significant covalent bonding preference and of a harder N donor that favors ionic interactions [42]. Interestingly, the resulting half-sandwich ruthenium (II) complexes 28a,b, which were prepared by transmetalation of the adequate hybrid ligand from silver to [Ru(η6-p-cymene)Cl2]2, displayed two solvent-dependent limiting resonance structures. While the neutral pyridylidene imine form was shown by cyclic voltammetry, NMR and UV-visible spectroscopy to be predominant in apolar solvents such as dichloromethane, the mesoionic pyridinium amidate form was found predominant in polar solvents such as methanol and DMSO (Scheme 15). This property was demonstrated to have a direct influence on the catalytic activity of 28a for the dehydrogenation of benzyl alcohol to benzaldehyde in various solvents, the best results being observed in the very polar DMSO. Nevertheless, both 28a and 28b (5 mol%, 10 mol% KOH) behaved as very poor catalysts for the TH of benzophenone to diphenylethanol in isopropanol at reflux, with maximum conversions of 20% (TON = 4) after 6 h (TOF = 0.67 h−1), presumably because of a massive decomposition of the complexes under these conditions, as shown by the presence of black particles in the medium at the end of the reactions [42].
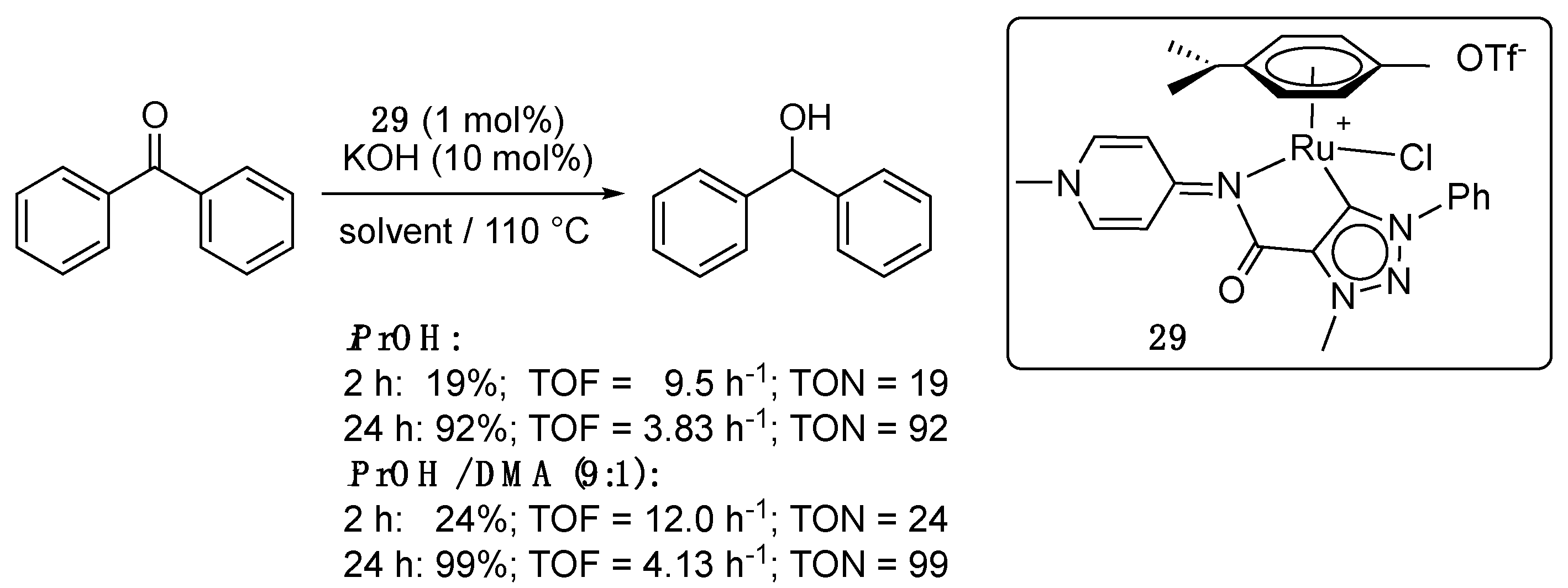
More recently, Albrecht completed his study with the synthesis, via a similar transmetalation route, and catalytic investigation in TH of the complex 29 bearing a hybrid pyridylideneamide-triazolylidene ligand [43,44]. The latter achieved 92% reduction in benzophenone in 24 h with 1 mol% loading (TOF = 3.83 h−1) in the presence of 10 mol% KOH in refluxing isopropanol (Scheme 16). Interestingly, increasing the polarity of the reaction medium by the addition of 10% dimethylacetamide (DMA) as a co-solvent to isopropanol had a notable impact on the catalytic activity of 29 [43]. Under these conditions, it achieved essentially full conversion after 24 h, and 24% conversion after 2 h (vs. 19% in pure isopropanol, Scheme 16). This increased activity in a more polar medium was attributed to an increased relevance of the mesoinonic resonance form. Furthermore, no decomposition was observed with 29 under the basic conditions of the TH reaction. This enhanced stability of the triazolylidene derivative 29 under basic conditions when compared to the imidazol-2-ylidene derivatives 28a,b was attributed to the absence of an acidic CH2 group between the pyridylideneamide and the triazolylidene [43].
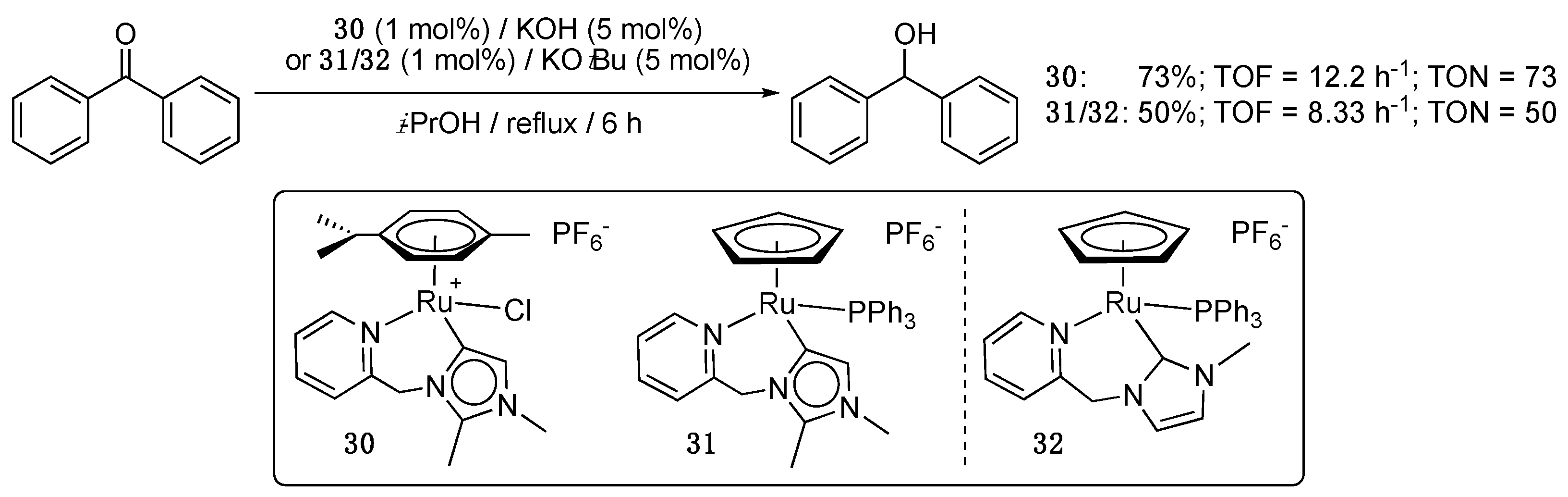
Albrecht and Landman recently reported the synthesis and catalytic activity in ketone TH of rare examples of picolyl-functionalized imidazol-4-ylidene ruthenium(II) complexes [45]. C(4)/C(5)-bound imidazolylidenes are known to be significantly stronger donors compared to their C(2)-bound counterparts [46]. Their synthetic accessibility is however limited by the low acidity of the imidazolium C(4)/C(5) protons, which requires specific protocols to achieve metalation at this position, such as the protection of the C(2)-position with alkyl or aryl groups [47] or the chelation assistance strategy [48]. The authors used a combination of these two strategies to prepare η6-p-cymene and η5-cyclopentadienyl (Cp) derivatives bearing a 1,2-dimethyl-3-pycolyl-imidazol-4-ylidene ligand. While the p-cymene complex 30 was isolated in a pure form, the Cp derivative 31 was isolated as a 1:1 mixture with the 1-methyl-3-pycolyl-imidazol-2-ylidene complex 32 resulting from C(2)–CH3 bond cleavage (Scheme 17). Both complexes (1 mol%) showed a relatively moderate activity with initial TOF values measured after 15 min of reaction of 34 h−1 (30) and 22 h−1 (31/32), and TON of 73 (30) and 50 (31/32) after 6 h reaction in isopropanol at reflux with a base loading of 5 mol%. Interestingly, closely related derivatives of 30 and 31 bearing a N-2-methylprop-2-ene tether instead of a N-picolyl group displayed slightly higher activity, notably in the case of the p-cymene derivative, which showed an initial TOF of 60 h−1 and a TON of 93 after 6 h. The higher activity observed with the p-cymene complexes was attributed to an easier substitution by the base of the chloride ligand in the p-cymene complexes than of any of the ligands in the Cp complexes.
Interestingly, complexes 30 and 31/32 proved to be also moderately active in the anaerobic oxidation of 1-phenylethanol to acetophenone in the presence of KOtBu (5 mol%) in o-dichlorobenzene at 150 °C, with conversions of 73 and 56% after 24 h reaction, respectively [45]. As no hydride signals of either the p-cymene or Cp catalysts could be observed by 1H NMR spectroscopy in both the TH and alcohol oxidation reactions, the authors proposed a monohydride mechanism where the monohydride intermediate catalytic species would not be the resting-state intermediate species.
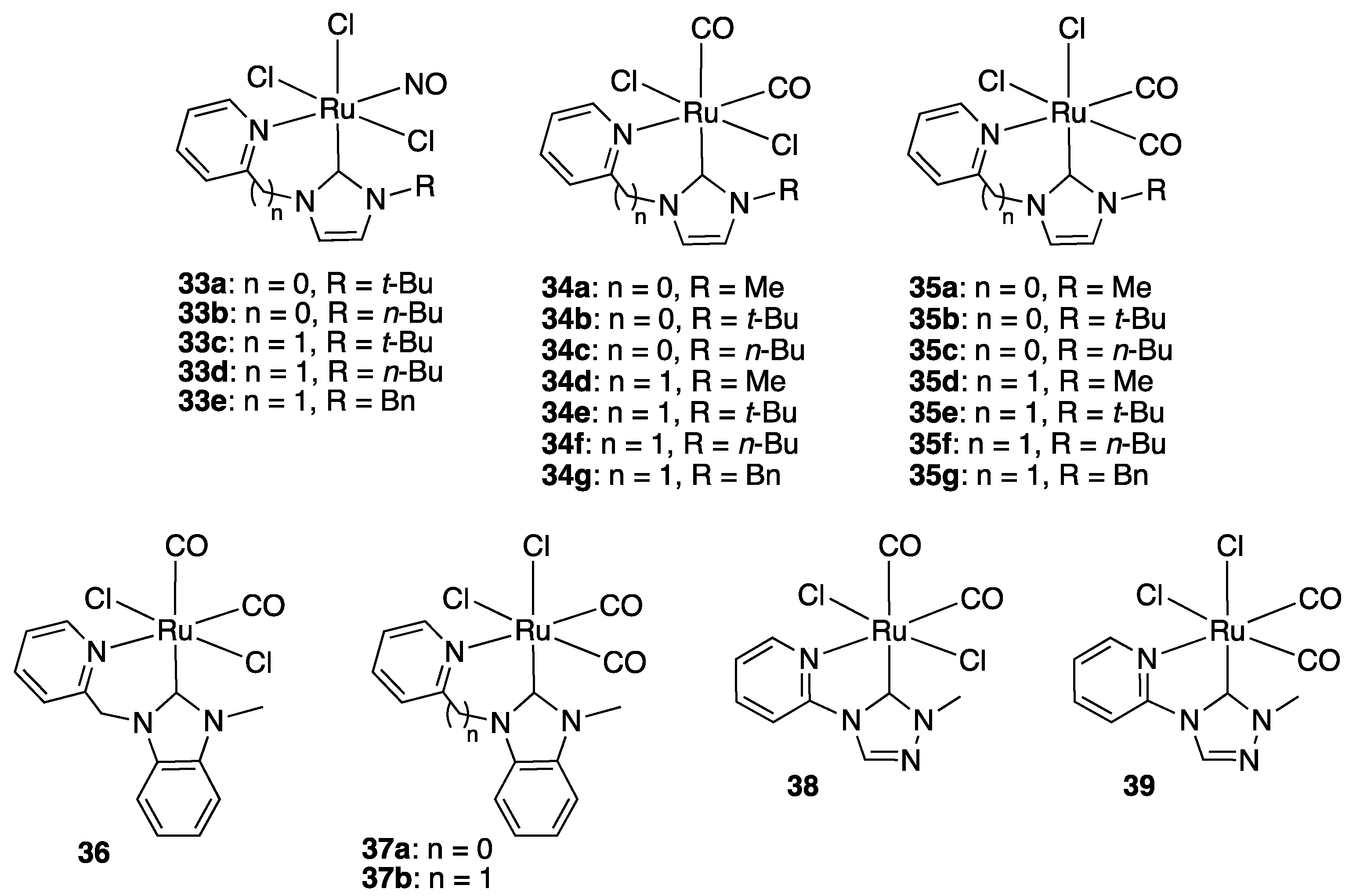
Aside from all these half-sandwich complexes, a number of octahedral complexes were also reported as ketone TH catalysts, notably a series of ruthenium(II) nitrosyl trichloride (33a–e) and dicarbonyl dichloride (34–39) complexes bearing pyridyl- or picolyl-functionalized imidazol-, benzimidazol-, or triazol-2-ylidenes (Figure 5) [49,50,51]. With the exception of the picolyl-benzimidazol-2-ylidene complex 37a, which was synthesized by direct reaction of the corresponding benzimidazolium hexafluorophosphate with [Ru(CO)2Cl2] in the presence of NEt3 [51], they were all prepared by transmetalation from the adequate in situ prepared silver carbene complex with either [Ru(NO)Cl2] or [Ru(CO)2Cl]n. The observed activity in TH decreased in the order of the σ-donor ability of the NHC ligand: imidazolylidene (33a–35g) > benzimidazolylidene (36–37b) > triazolylidene (38, 39). Among the imidazol-2-ylidene derivatives, the methyl- and t-butyl-substituted complexes 33a, 33c, 34a, 35a, and 35b proved to be the most active. With the exception of the nitrosyl species 33c, all these were comprised of a five-membered ring. In particular, the 1-methyl-3-pyridyl-imidazol-2-ylidene complex 34a showed the highest TOFs (from 87.5 to 248 h−1 with a small range of 6 aromatic and aliphatic ketones), and the 1-t-butyl-3-pyridyl-imidazol-2-ylidene derivative 35b, which could work with a loading as low as 0.2 mol% in the presence of 5 mol% KOH in isopropanol at 82 °C, the highest TONs (from 50 to 455).
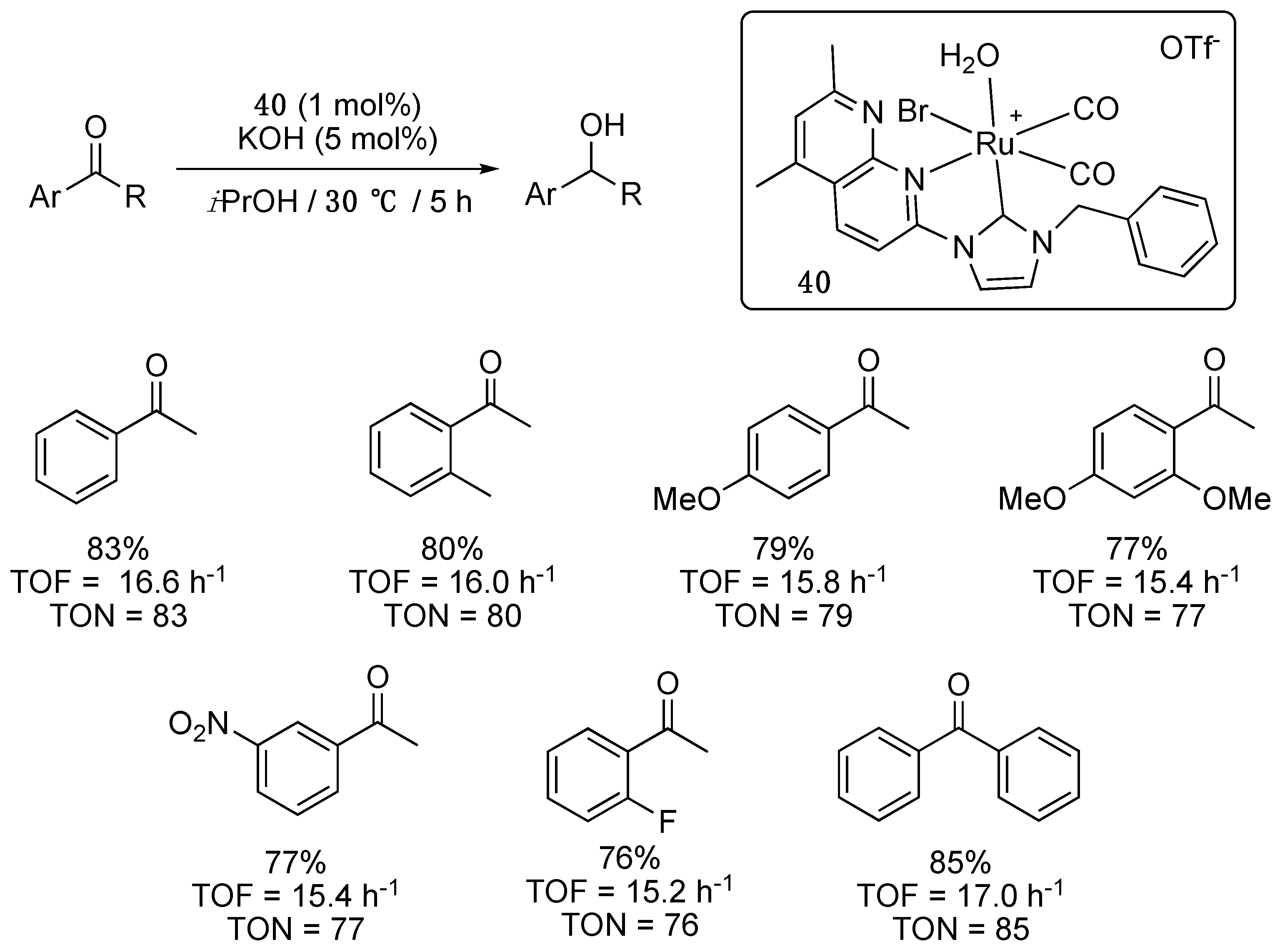
Room temperature treatment of [Ru2(CO)4(CH3CN)6](OTf)2 with two equivalents of 1-benzyl-3-(5,7-dimethyl-1,8-naphtyrid-2-yl)imidazolium bromide provided the related octahedral aqua-bromo-dicarbonyl-ruthenium(II) complex 40 which was reported by Bera et al. to act as a rare example of low-temperature-efficient catalyst for the TH of a short series of aromatic ketones in the presence of 5 mol% KOH [36]. With a catalyst loading of 1 mol%, TON ranging from 76 to 85 were achieved in 5 h (TOF = 15.2–17 h−1) at 30 °C with electron-neutral, as well as electron-poor and electron-rich aromatic ketones (Scheme 18). Heteroaromatic ketones such as acetylthiazole and acetylpyridine, however, required 12 h at 80 °C to reach moderate yields: 65% (TOF = 5.41 h−1) and 59% (4.92 h−1), respectively. No conversion was observed for acetylpyrrole even after prolonged heating.
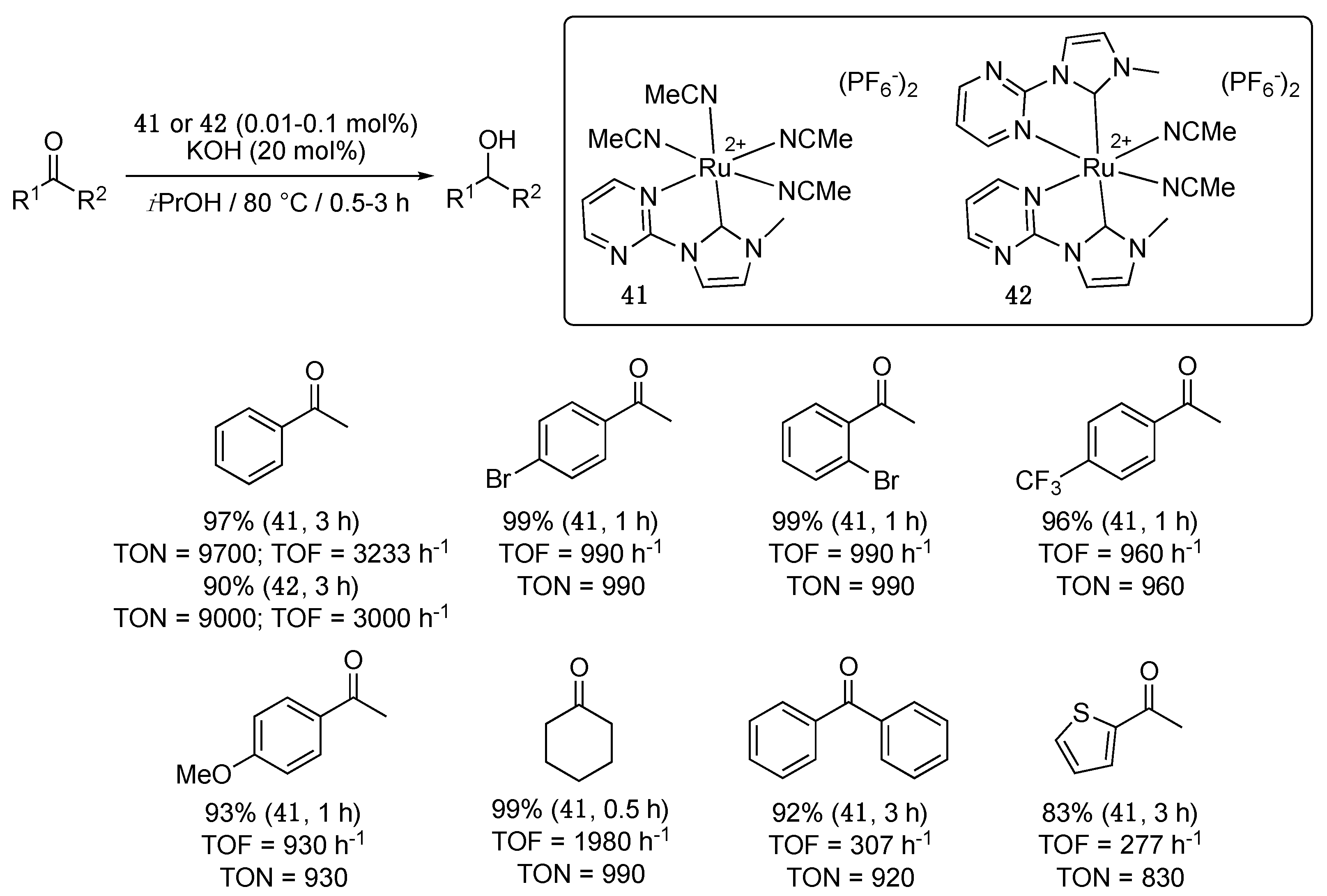
Finally, a pyrimidine-functionalized imidazol-2-ylidene-Ru(II) motif similar to that found in the p-cymene complex 2 was used in the octahedral dicationic complexes 41 and 42 reported by W. Chen et al. in 2015 [52]. The complexes bearing either one or two chelating units and, respectively, four or two acetonitrile ligands were obtained by transmetalation from the corresponding nickel-NHC complexes with 0.5 or 0.25 equiv. of [Ru(η6-p-cymene)Cl2]2 in acetonitrile at reflux. Both complexes showed greatly improved catalytic performances compared to the half-sandwich complex 2, and to the related octahedral complexes 9b and 13b, bearing a pyridyl- or pycolyl-functionalized triazolylidene and four acetonitrile ligands, with TON and TOF values for the TH of acetophenone as high as 9700 and 3233 h−1 (41) and 9000 and 3000 h−1 (42) (Scheme 19). Much more basic conditions than with 2, 9b, and 13b were however required; 20 mol% KOH vs. 10 mol% with 2, 9b, and 13b.
A brief study of the reaction scope, carried out with 0.1 mol% 41, showed that it was very active in the TH of cyclohexanone and aromatic ketones bearing either electron-withdrawing or electron-donating substituents. Longer reaction times were however required with benzophenone and 2-acetylthiophene to observe high reduction yields (Scheme 19).
2.2. Ketones’ Transfer Hydrogenations with (κ2-CNHC,P)-Complexes
Examples of (κ2-CNHC,P)-complexes as ketone TH catalysts are surprisingly rare and involve mainly abnormal NHC-phosphine complexes, described by the group of Baratta. The first examples, however, arose from the groups of Grotjahn and Domsky in 2011 and 2013, and involved protic and classic NHCs, respectively.
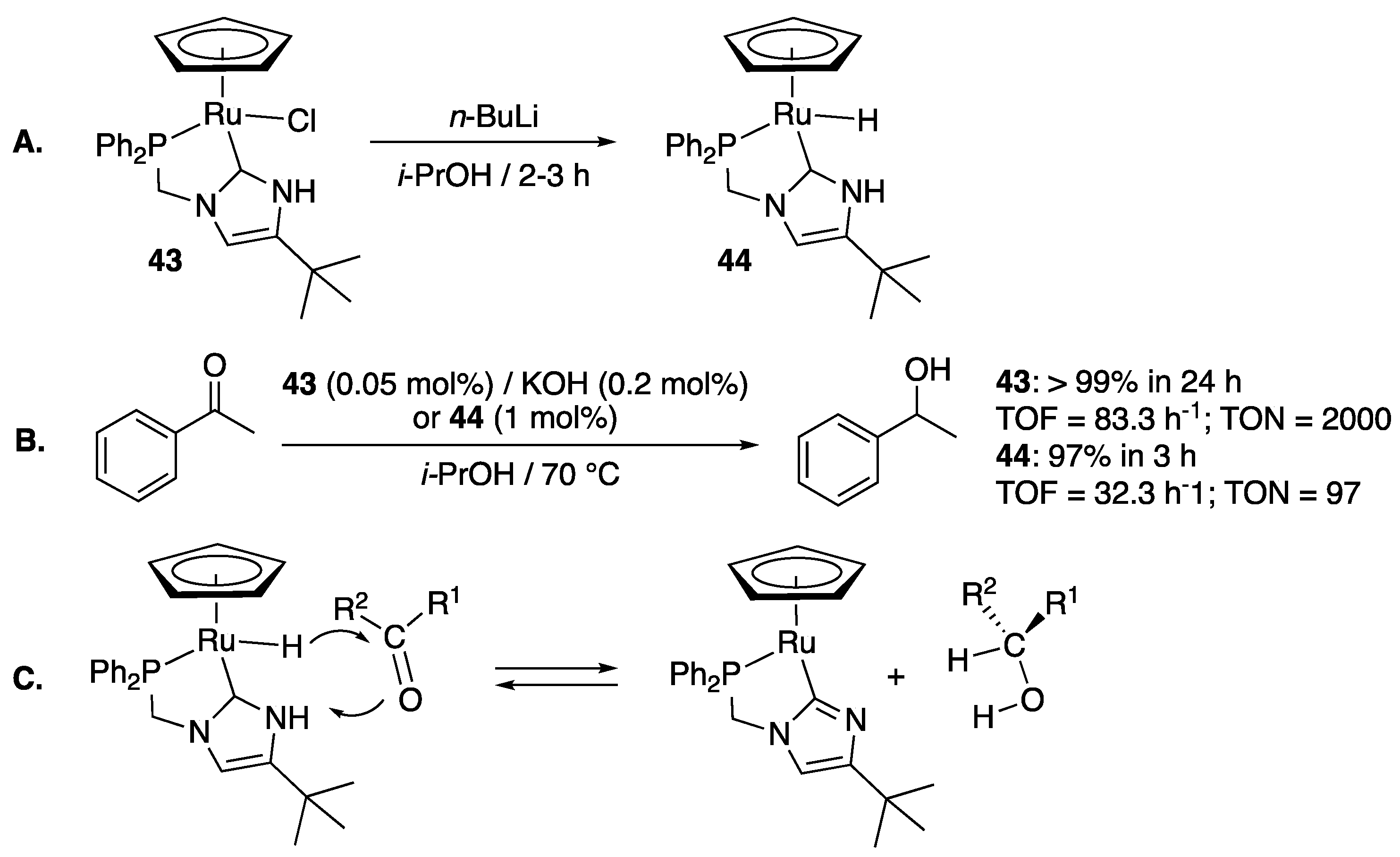
In the first example, Grotjahn et al. thus described the synthesis—via direct reaction of the corresponding imidazole with [RuCpCl(cod)] (cod = 1,5-cyclooctadiene) in THF at 100 °C—and reactivity of the CpRu complex 43 bearing a diphenylphosphino-functionalized protic imidazol-2-ylidene ligand [53]. Its reaction with n-BuLi in isopropanol provided the corresponding hydride complex 44 (Scheme 20A). Both complexes proved active for the TH of acetophenone (2 M) to 1-phenylethanol in isopropanol at 70 °C. Significantly, 44 (1 mol%) was efficient in the absence of a base and achieved 97% yield in 3 h, whereas 43 (0.050 mol%) required the presence of KOH (0.2 mol%) to produce 44 in situ and achieve full reduction in 24 h (Scheme 20B). The catalytic process is believed to involve the N-H function of the protic NHC as shown in Scheme 20C, and thus constitute a rare example of a cooperative hydrogenation–dehydrogenation process involving both a metal center and one ring nitrogen atom of a NHC ligand [54].
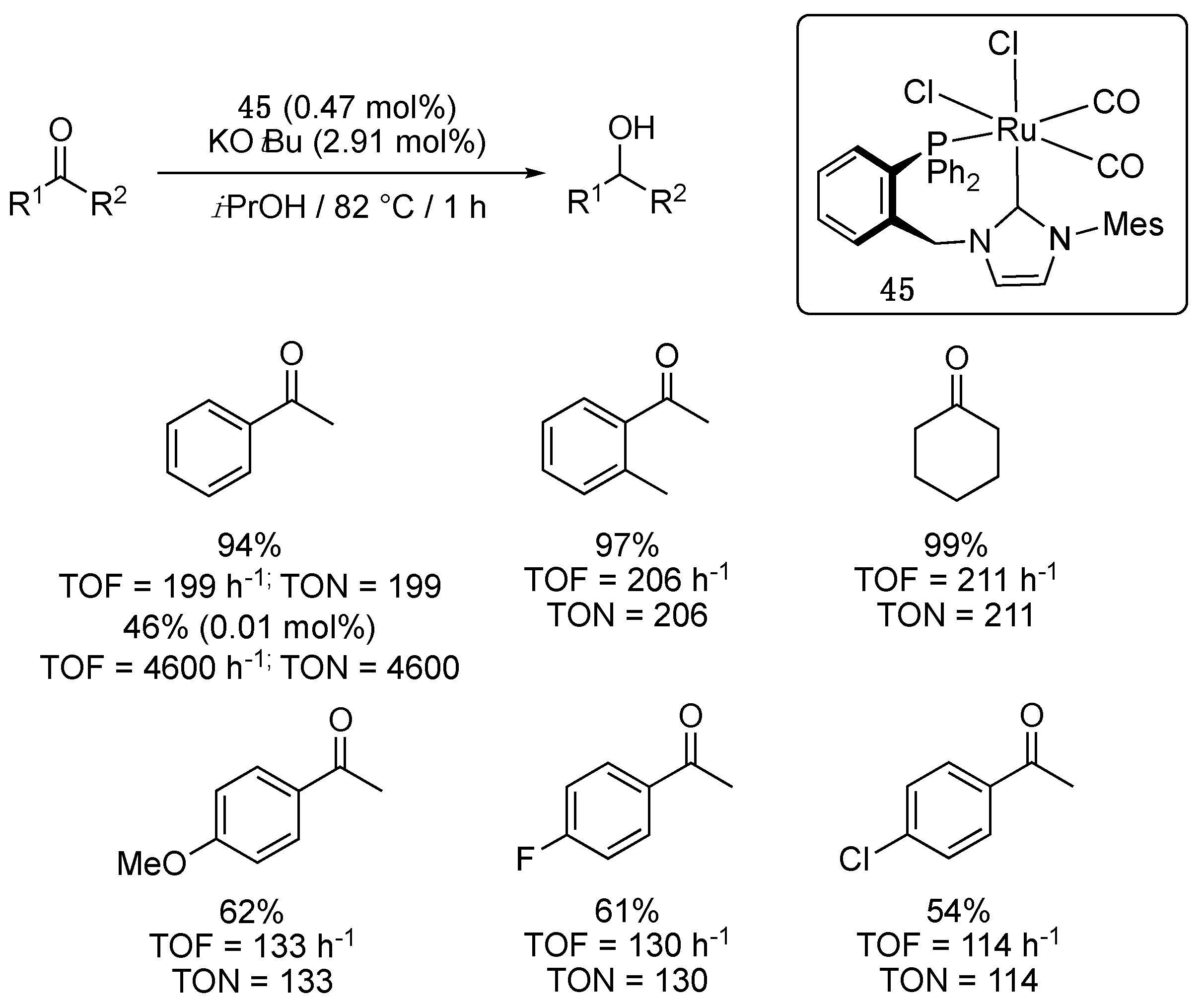
Two years later, Domski et al. reported the synthesis via a classical transmetalation procedure from silver of an octahedral ruthenium dicarbonyl dichloride complex 45, bearing an ortho-(diphenylphosphino)benzyl-functionalized imidazol-2-ylidene ligand, that proved catalytically effective for the TH of a small number of ketones in refluxing isopropanol [55]. With a loading of 0.47 mol% in the presence of KOtBu (2.91 mol%) as base, acetophenone, 2-methylacetophenone, and cyclohexanone were all almost fully reduced in 1 h, giving TOF ranging from 199 to 211 h−1 (Scheme 21). A TOF of 4600 h−1 could even be reached for acetophenone when a catalytic loading of 0.01 mol% was used under otherwise similar conditions. Substitution at the para-position, however, led to lower yields with both electron-donating and electron-withdrawing substituents.
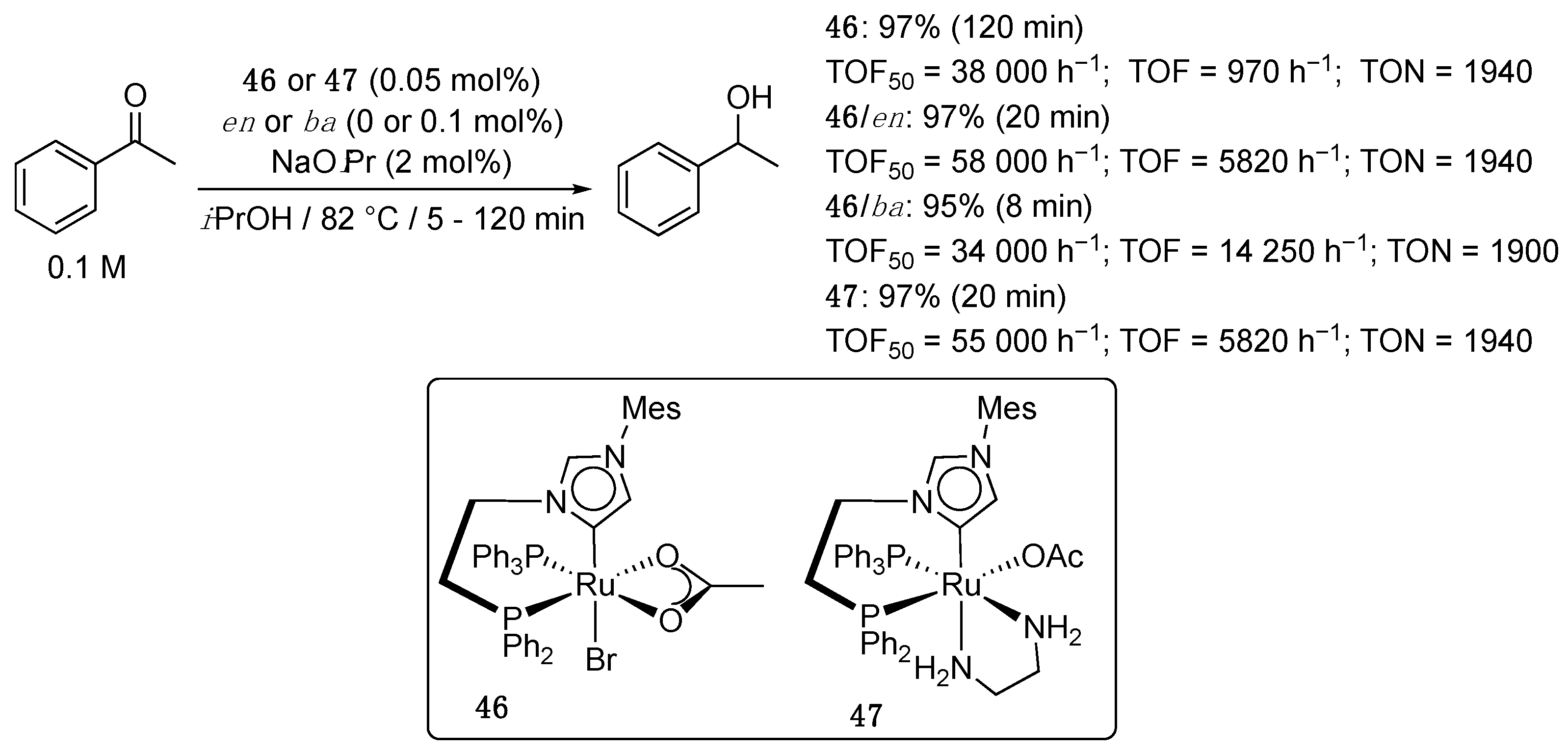
At about the same period, Baratta et al. reported the first of a series of four articles on ruthenium(II) complexes bearing a bifunctional phosphine-imidazol-4-ylidene ligand [56]. In this initial account, the [RuBr(κ2-OAc)(PPh3)(Ph2P-(CH2)2-aNHC)] complex 46 was prepared by treatment of [Ru(OAc)2(PPh3)2] with 1.2 equivalents of the corresponding phosphine-imidazolium bromide in the presence of sodium acetate in THF under reflux. Complex 46 (0.05 mol%) showed high initial activity for the TH of acetophenone (0.1 M) to 1-phenylethanol in refluxing 2-propanol with a TOF50 as high as 38,000 h−1, but deactivated quickly to reach full conversion after 2 h and a final TOF of only 970 (Scheme 22), which was still about five times higher than the TOF observed with complex 45. The addition of ethylenediamine (en) or benzylamine (ba) (0.1 mol%) as nitrogen ligands containing a N–H function [57] gave more active and productive catalysts. Thus, in the presence of these ligands, acetophenone was reduced quantitatively in 20 or 8 min, respectively, leading to TOF of 5820 and 14 250 h−1. Interestingly, the isolated complex, [Ru(κ1-OAc)(PPh3){Ph2P-(CH2)2-aNHC}{H2N-(CH2)2-NH2}] (47), showed the same activity as the in situ generated system, 46/en, and underwent fast proton exchange of the four NH and the carbene NCHN protons in basic D2O, unveiling a possible active role in bifunctional catalysis [57]. The study was completed by a brief overview of the reaction scope that revealed that hex-5-en-2-one, 2-methylcyclohexanone and benzophenone could all be fully and selectively reduced to the corresponding alcohols in 2 to 60 min in the presence of 46/en under similar reaction conditions.
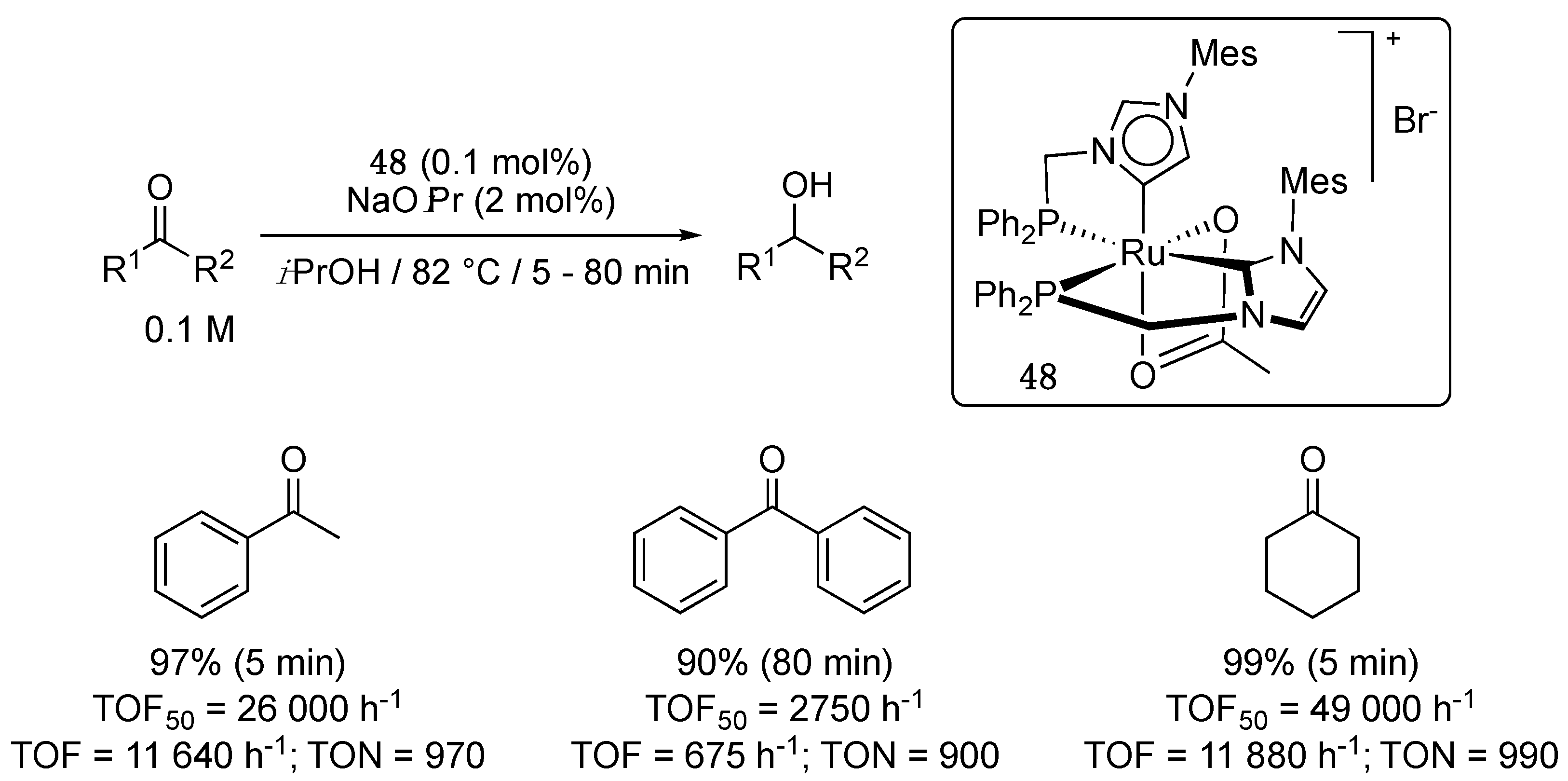
In 2015, the same group described a rare example of ruthenium bis-carbene complex 48 bearing both a (diphenylphosphino)methylene-functionalized imidazol-2-ylidene and imidazol-4-ylidene ligand, that may form on account of steric factors involving the bulky mesityl substituents carried by the N-heterocyclic carbenes [58]. It could be prepared either in one or two steps by the reaction of [Ru(OAc)2(PPh3)2] with 1 + 1 or directly 2 equivalents of the corresponding phosphine–imidazolium bromide in the presence of sodium acetate in THF at reflux. Catalytic studies revealed high activities for the TH of a couple of ketones with TOF50 and final TOF values of up to 49,000 h−1 and 11 880 h−1, respectively (Scheme 23) [58,59]. These values make of 48 a TH catalyst of comparable activity to 46/en [56], although it does not comprise an N–H function, which often entails high activity through bifunctional catalysis [57].
Derivatization of complexes 46 and 48 by coordination of Ag or of an IrCl(η4-cyclooctadiene) fragment to the abnormal C2 carbon generated bimetallic TH catalysts that displayed much lower activity than 46 and 48 [59], indirectly suggesting that the NCHN proton of the imidazol-4-ylidene ligand could play an active role in the TH catalysis, in agreement with the fast proton exchange observed with 47 in basic D2O (vide supra) [56].
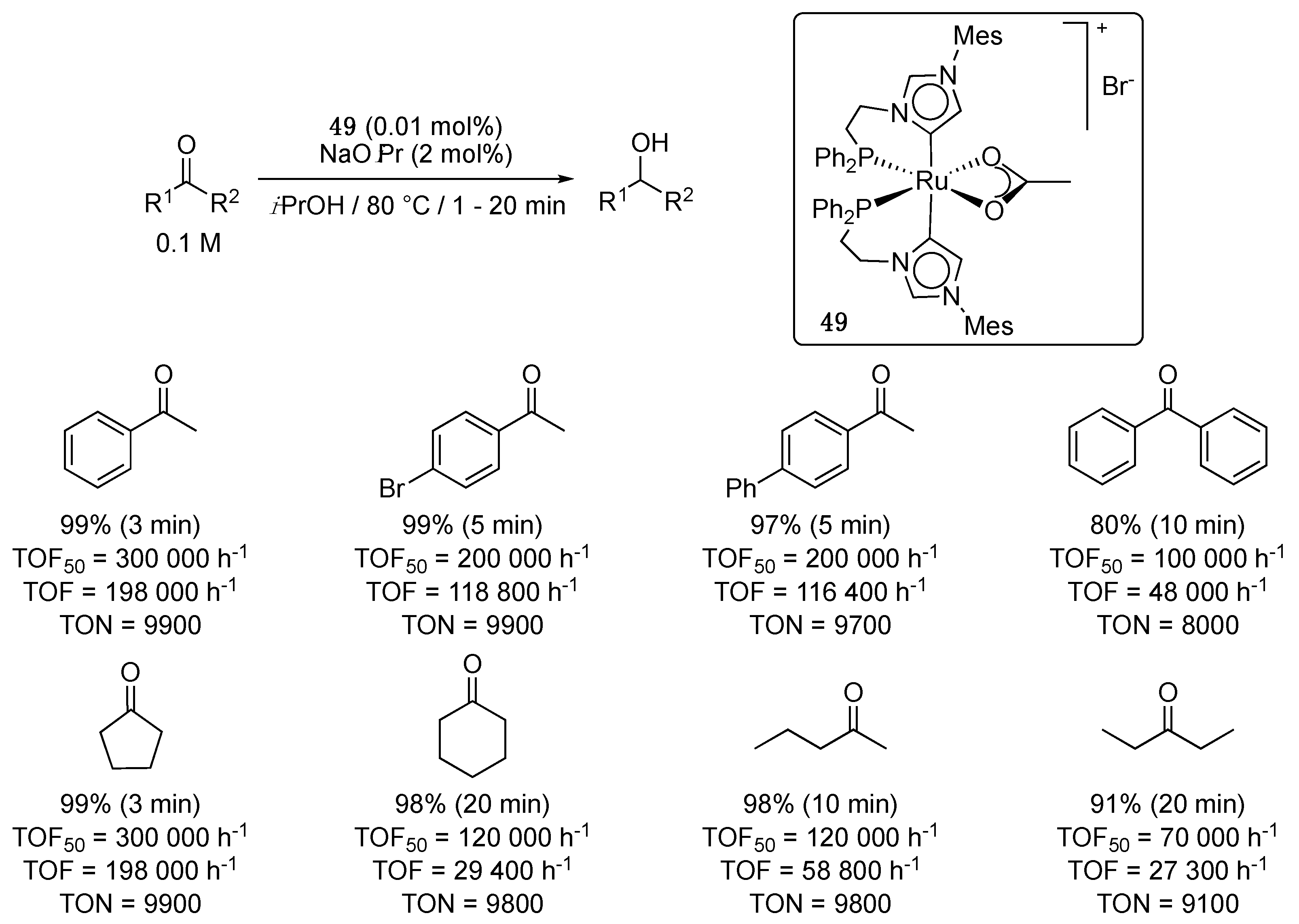
More recently, Baratta et al. disclosed the synthesis and characterization of the first ruthenium complex bearing two abnormal NHC ligands, 49 [60]. This (diphenylphosphino)ethylene-functionalized bis-imidazol-4-ylidene species, which was synthesized in 85% yield by reaction of complex 46 with two equivalents of 1-(diphenylphosphino)ethylene-3-mesitylene-imidazol-2-ylidene in the presence of sodium acetate as base in THF at 60 °C, displayed outstanding catalytic activity in both ketone TH and the reverse Oppenauer-type alcohol oxidation. At 80 °C in isopropanol, in the presence of NaOiPr (2 mol%), acetophenone was quantitatively reduced to 1-phenylethanol within 3 min using a catalytic loading of 0.01 mol%, giving TOF50 and TOF at full conversion of 300,000 and 198,000 h−1, respectively (Scheme 24). Even more impressively, at reflux, the reduction occurred in only 1 min, affording a TOF50 of 600,000 h−1, which is among the highest TOFs reported in TH [61], and makes of 49 the most active Ru-carbene TH catalyst to date [56,59,62,63,64]. 4-Bromo-acetophenone, 4-acetylbiphenyl, cyclopentanone, cyclohexanone and 2-pentanone were all fully reduced within 3 to 20 min with the same catalytic loading, giving TOF at full conversion ranging from 29 400 h−1 and 198,000 h−1 (Scheme 24). Benzophenone was converted to benzhydrol in 80% in 10 min (TOF = 48,000 h−1, TON = 8000) and 3-pentanone to 3-pentanol in 91% in 20 min (TOF = 27 300 h−1, TON = 9100).
Interestingly, VT NMR experiments revealed that a rapid H/D exchange occurred at the NCHN proton positions of the two imidazole-4-ylidene ligands when NaOiPr (1 equiv.) was added to 49 in 2-propanol-d8 at RT, similarly to what had been observed with complex 46 in basic D2O (vide supra) [56]. In addition, the reaction of 49 with NaOiPr (10 equiv.) in 2-propanol at 80 °C for 2 h led to the formation of the dihydride complex 50 bearing two (diphenylphosphino)methylene-functionalized imidazol-2-ylidenes (Scheme 25). The latter can also be obtained by reaction of 49 with H2 (5 bar) in the presence of KOtBu in toluene at 60 °C. Complex 50 proved, however, to be inactive in catalytic TH. The abnormal carbene coordination found in 49 is therefore considered by the authors to be crucial to observe high catalytic performances, on account of the smaller steric hindrance of the mesityl groups in this species, and possibly via deprotonation at the C2 carbene carbons [60]. Accordingly, the formation of 50 is regarded as a catalyst deactivation pathway.
2.3. Ketones’ Transfer Hydrogenations with (κ2-CNHC,O)-Complexes
Although the first report appeared a decade ago [65], examples of (κ2-CNHC,O)-complexes as ketone TH catalysts remain very rare and, with one exception, always comprise (benz)imidazol-2-ylidene or 1,2,3-triazolylidene ligands functionalized with a carboxylate side-arm.
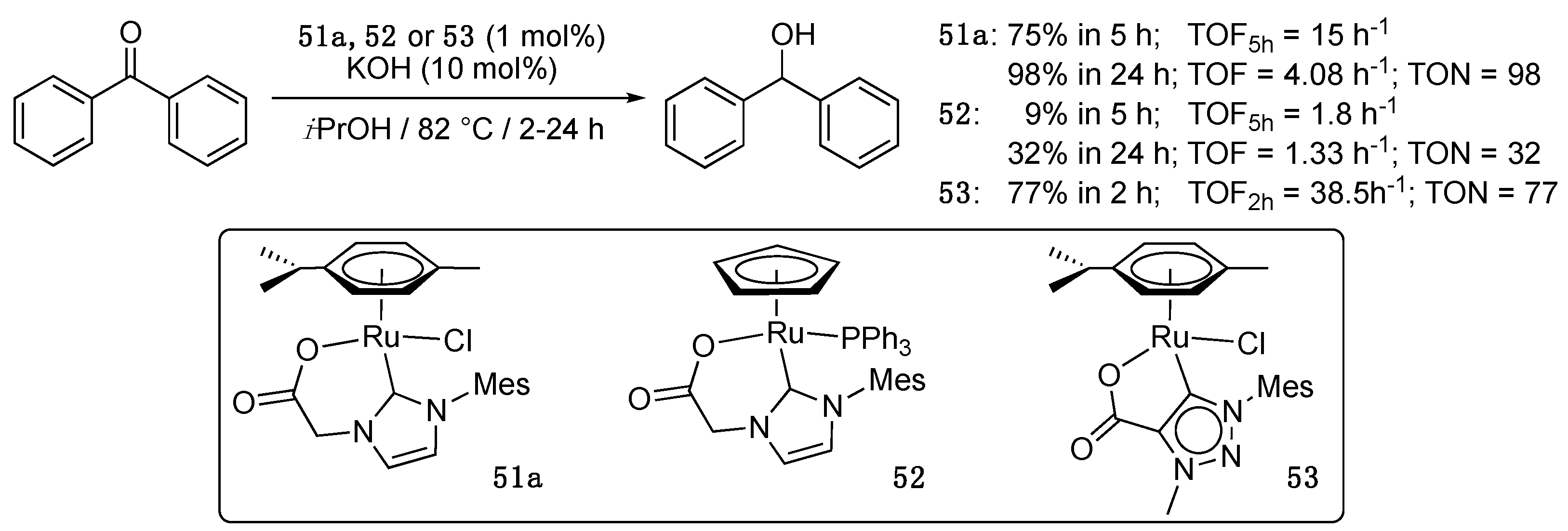
Thus, the first examples of this short series were reported in 2011 by Albrecht et al. with the description of the catalytic activity in the TH of benzophenone of two carboxylate-functionalized imidazole-2-ylidene complexes, 51a and 52, that bore the same functionalized carbene but differed by the nature of the ruthenium scaffold, “(η6-p-cymene)RuCl” vs. “CpRu(PPh3)”, and were prepared in a one-pot procedure via transmetalation from the corresponding silver carbene complex to [RuCl2(η6-p-cymene)]2 or [RuCpCl(PPh3)2], respectively [65,66]. Whereas the (η6-p-cymene)RuCl derivative 51a achieved full conversion in 24 h in refluxing 2-propanol with a loading of 1 mol% and 10 mol% of KOH as activator, the CpRu(PPh3) derivative 52 displayed very poor activity, reaching only 32% conversion after 24 h under the same conditions (Scheme 26). These two initial half-sandwich C,O-catalysts were joined a couple of years later by a novel (η6-p-cymene)RuCl derivative 53 bearing a mesoionic carbene C-functionalized with a carboxylate group [23]. The latter proved to be more than twice as reactive as 51a, achieving 77% conversion in 2 h under the same conditions, while 51a necessitated 5 h to reach a similar conversion (Scheme 26).
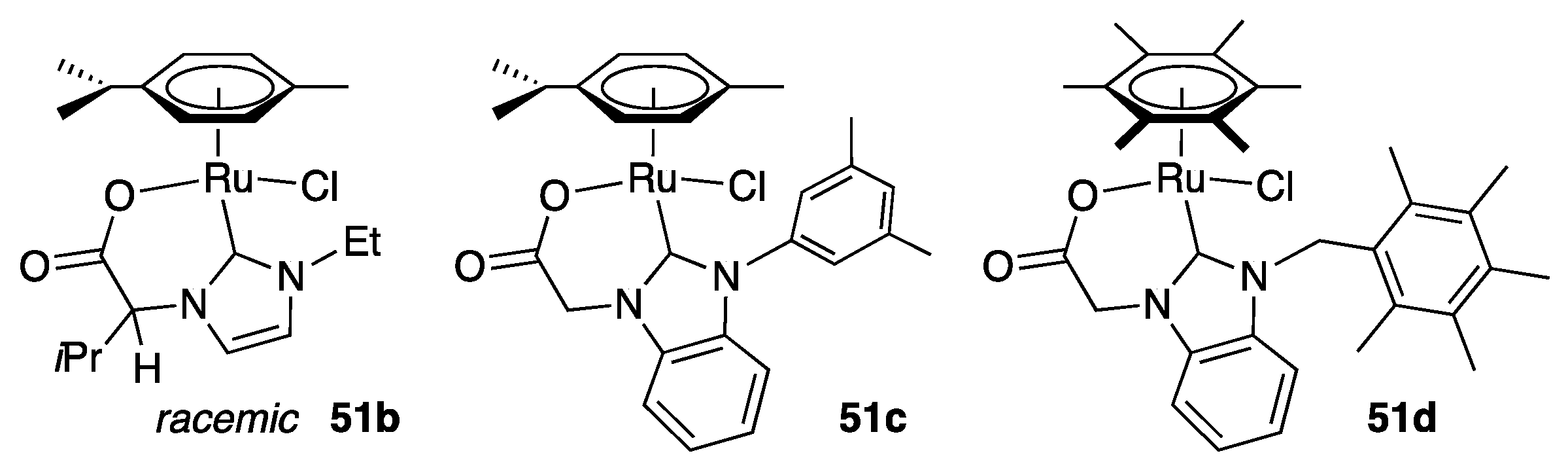
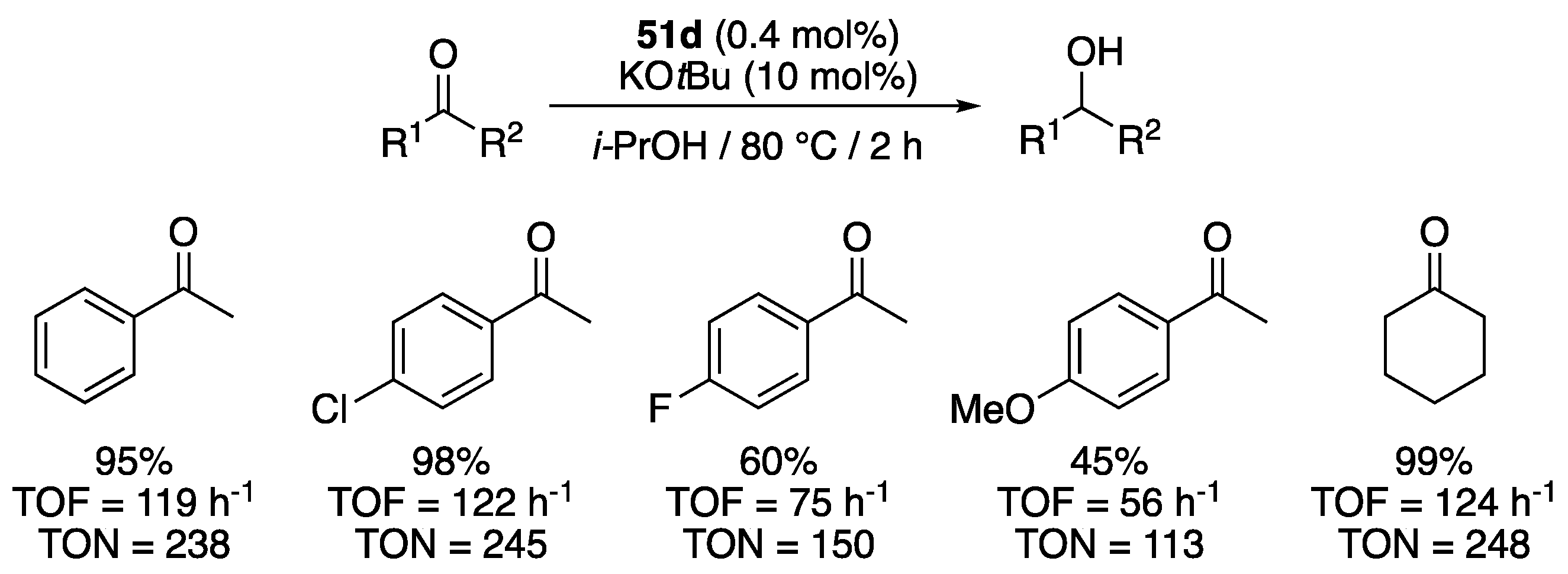
At the same period, Papish [67], Tennyson [68], and Altin and coworkers [69] described the syntheses, via similar means, and catalytic activities of complexes 51b, 51c, and 51d, respectively (Figure 6), which closely resemble 51a. Noteworthy, although 51b was prepared from the corresponding enantiopure proligand (in the form an ester), it was obtained as a racemic mixture. The activity of all three complexes were evaluated for the TH of acetophenone from isopropanol at reflux. Complex 51d proved to be most active of the three, as it achieved 95% conversion in 2 h with a catalyst loading of 0.4 mol% (and 10 mol% KOtBu), giving a TON of 238 and a TOF of 119 h−1 [69]. In contrast, complexes 51b (0.5 mol% with 12.5 mol% KOH) and 51c (1 mol% with 4.95 mol% KOtBu) only achieved initial TOF1h of 44 and 31 h−1, and TON of 190 and 96 after 6 and 24 h reaction, respectively [67,68]. However, 51b showed some inconsistency with anomalous conversions as high as 88% after 1 h, which would correspond to an initial TOF1h of 176 h−1 [67]. Papish et al. suggested that this could be due to decomposition to a highly active heterogeneous catalyst or to a reaction pathway that would be highly sensitive to trace contaminants, such as a radical mechanism, but no mechanistic investigations were carried out to sustain these assumptions.
Altin et al. briefly examined the scope of substrates that could undergo TH by the carboxylate-functionalized benzimidazolylidene complex 51d under the established conditions (0.4 mol% 51d, 10 mol% KOtBu, iPrOH at 80 °C) [69]. Similarly to acetophenone, p-chloroacetophenone and cyclohexanone, were converted to the corresponding alcohols in nearly quantitative yield in 2 h (Scheme 27). p-Fluoroacetophenone and p-methoxyacetophenone gave lower TOF, but still achieved TON of over 110 in 2 h.
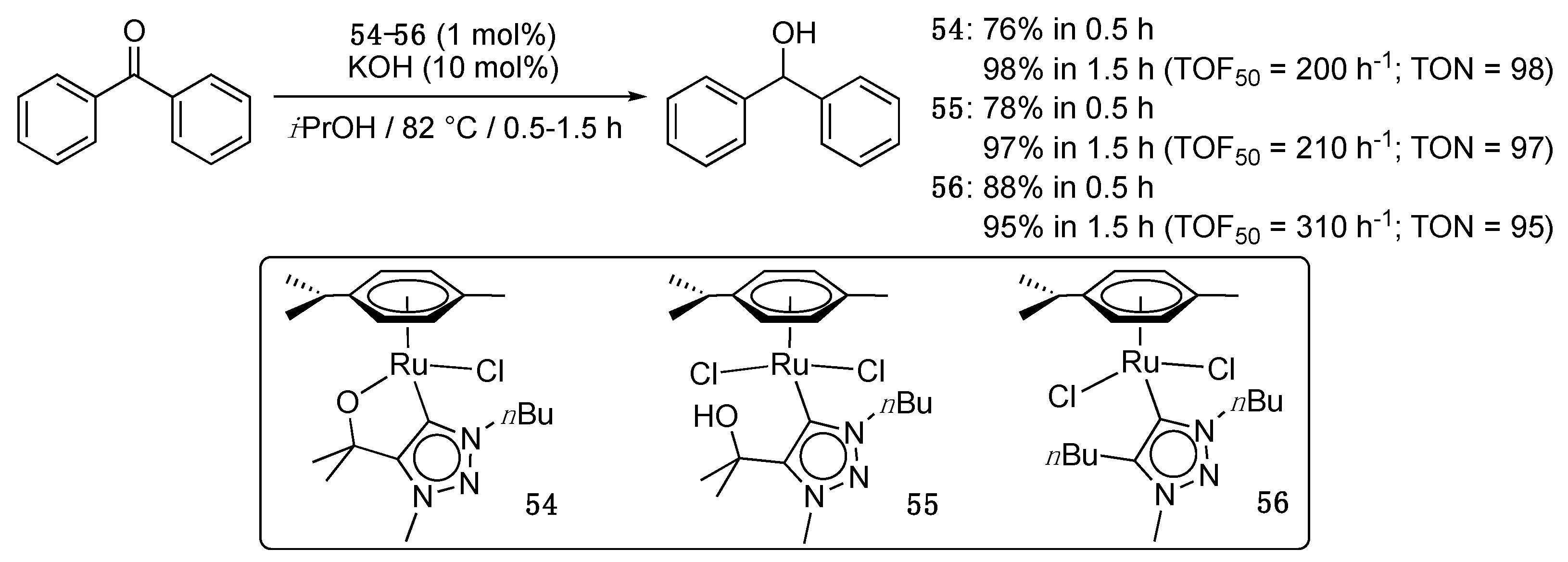
A last example deals with hydroxy-functionalized 1,2,3-triazolyildene complexes, in which the pendant hydroxyl group can be either chelating (54) or not (55) [70]. The monodentate and bidentate bonding modes are interconvertible by the addition of a chloride scavenger such as AgBF4 or NH4BF4, or conversely brine, which demonstrates the hemilabile character of the hydroxyl group. Both complexes (1 mol%) reached 76–78% conversion in 0.5 h and full conversion in 1.5 h for the TH of benzophenone in the presence of KOH (10 mol%) in isopropanol at reflux (Scheme 28). The activity of the analogous complex 56 bearing a 1,4-dibutyl-1,2,3-triazolylidene was also evaluated for comparison. It reached the 78% conversion in 15 min and 88% conversion in 0.5 h. The higher activity of the latter (TOF50 = 310 h−1 vs. 200 and 210 h−1 for 54 and 55, respectively) clearly shows the absence of beneficial role of the hydroxy group, whether it is chelating or not. As stated by the authors, these data militate in favor of an inner sphere monohydride mechanism [4], in which the intermediate ruthenium-hydride species is formed via β-H-elimination from the previous ruthenium-isopropoxide species without any help of the hydroxy-functionality, in contrast to what was initially hoped.
3. Asymmetric Transfer Hydrogenation (ATH) of Ketones
To the best of our knowledge, donor-functionalized NHC-ruthenium complexes have, surprisingly, almost never been used in ATH of ketones. We are indeed aware of only two reports of chiral catalysts, including one that gave almost no enantiomeric excess (ee).
3.1. Ketones’ Asymmetric Transfer Hydrogenation with (κ2-CNHC,N)-Complexes
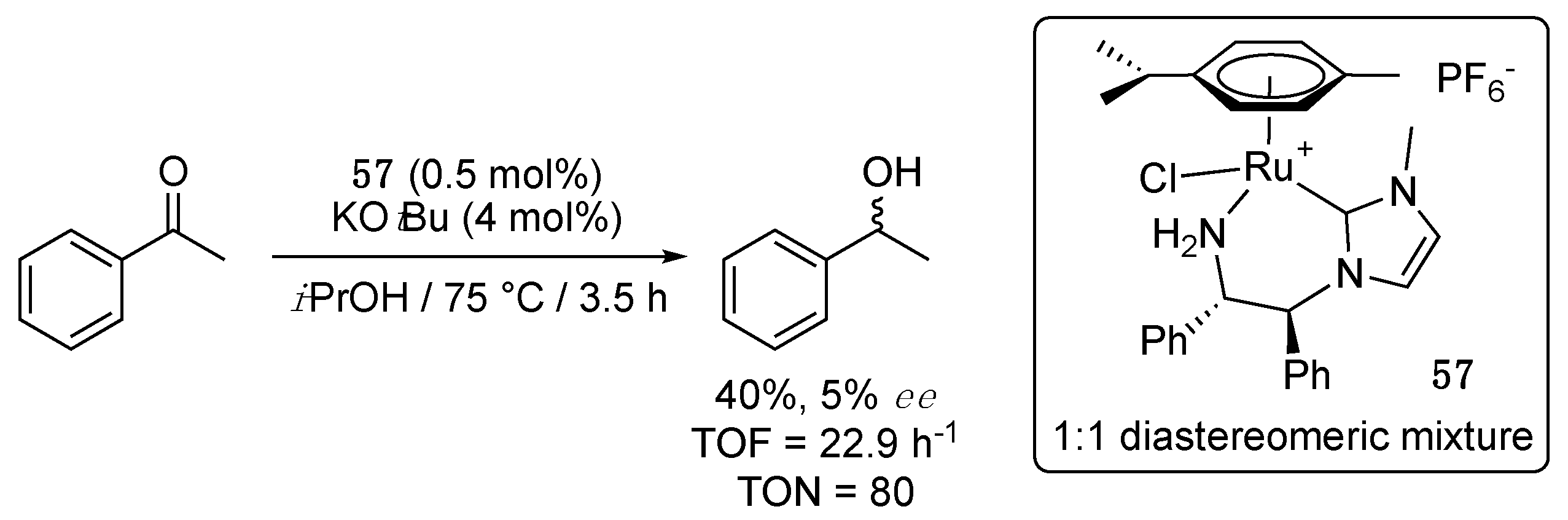
In 2016, Morris et al. disclosed an asymmetric version of their cationic amino-functionalized imidazol-2-ylidene complex 4a [14]. The chiral complex 57, bearing a (S,S)-1,2-diphenylethylamine-functionalized imidazol-2-ylidene, was obtained as a 1:1 diastereomeric mixture with opposite chirality at the ruthenium center by transmetalation from silver [71]. In isopropanol at 75 °C, 57 (0.5 mol%), along with 4 mol% KOtBu, afforded 40% conversion of acetophenone to 1-phenylethanol after 3.5 h of reaction (TOF = 22.9 h−1, TON = 80), with an enantioselectivity of only 5% (Scheme 29). The poor activity compared to 4a (TOF = 216 h−1, TON = 1080 in 5 h) was attributed to the low solubility of 57 in isopropanol, and the low enantioselectivity to its 1:1 diastereomeric composition or, tentatively, to an inner-sphere mechanism involving the decoordination of the amine side arm, as was proposed for 4a [14]. Of note, 57 (0.5 mol% with 4 mol% KOtBu) also showed moderate direct hydrogenation activity under 25 bar H2 at 50 °C in THF (TOF = 200 h−1, TON = 200), but no enantioselectivity at all in this case.
3.2. Ketones’ Asymmetric Transfer Hydrogenation with (κ2-CNHC,O)-Complexes
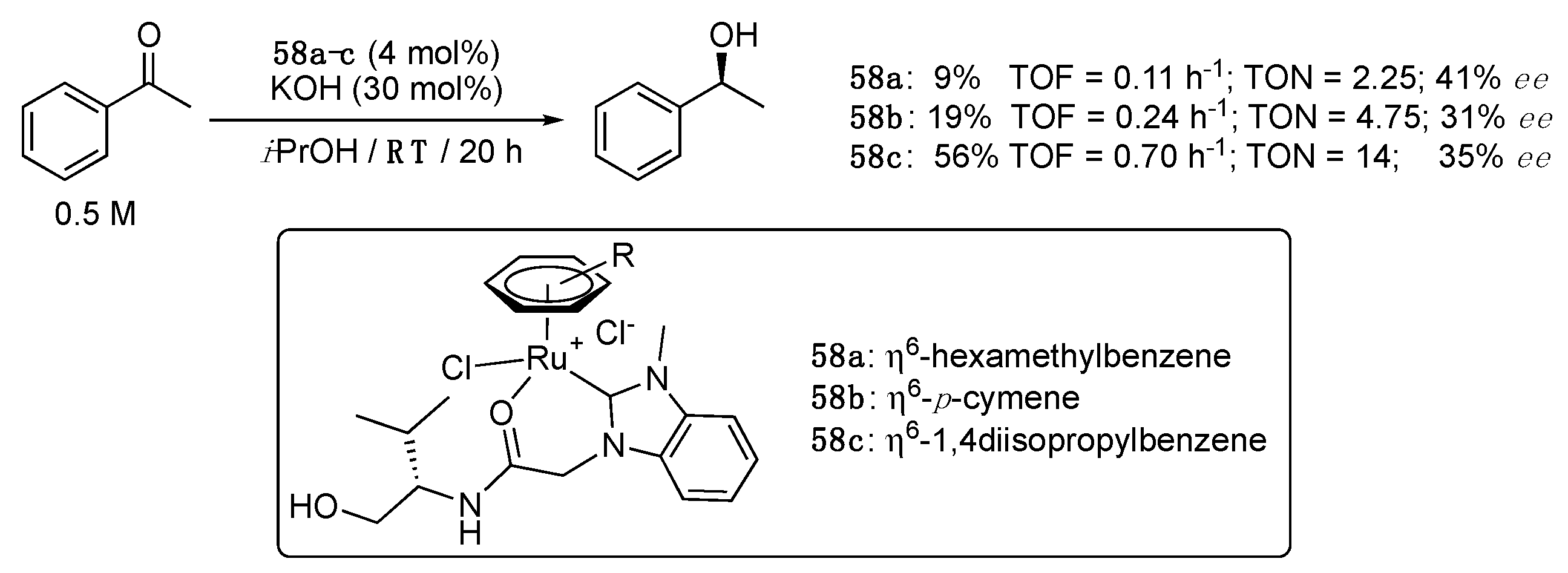
In 2013, Sakaguchi et al. prepared via transmetalation from silver to the corresponding [RuCl2(η6-arene)]2 precursor a series of benzimidazol-2-ylidene (η6-arene)Ru(II) complexes 58a–c bearing a hydroxy-amide chelating side arm, and applied it to the ATH of acetophenone [37]. All complexes acted as precatalysts at room temperature with low to moderate activity and relatively low enantioselectivity (Scheme 30). Interestingly, the nature of η6-arene ligand was shown to significantly alter the activity and selectivity of these complexes, with the η6-hexamethylbenzene derivative 58a giving the highest enantioselectivity (41% ee) and the η6–1,4-(diisopropyl)benzene complex 58c the highest activity (TOF = 0.70 h−1 after 20 h vs. 0.11 and 0.24 h−1 for 58a and 58b). Furthermore, the hydroxy functional group on the NHC side arm was found to be of critical importance, as its replacement by a methoxy group resulted in no activity. However, use of the dianionic alkoxy-amidate-NHC-Ru complex 59, obtained by treating the complex 58d with KOH at room temperature (Scheme 31), resulted in no activity in acetophenone TH, showing the probable importance of having a possible vacant site to which the reacting ketone can coordinate.
4. Transfer Hydrogenation of Aldehydes
The TH of aldehydes is rarely studied, and the catalytic efficiency is generally found to be low with limited substrate scope along with the formation of aldol condensation or carboxylic acid byproducts [5]. We are, thus, aware of only a small number of (κ2-CNHC,N)-complexes as precatalysts for this transformation.
Aldehydes TH with (κ2-CNHC,N)-Complexes
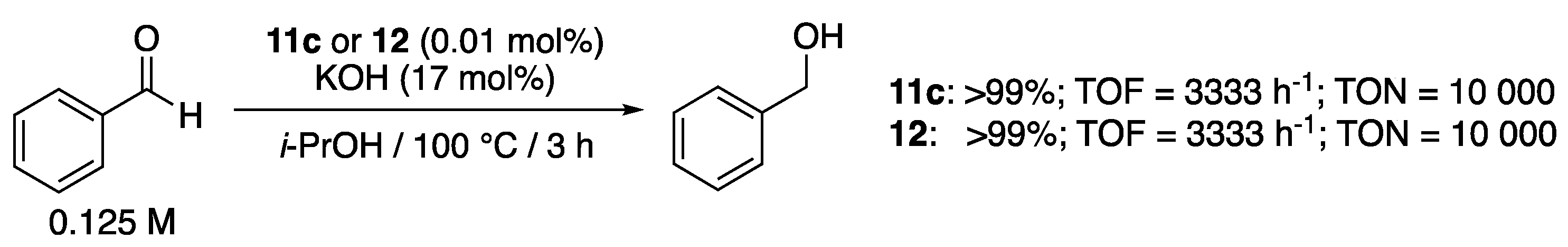
The pyridine- or pyrimidine-functionalized triazolylidene complexes 11c and 12 (0.01 mol%), described by Sarkar et al. [24,25], showed exceptional activities for the TH of benzaldehyde to benzylic alcohol in isopropanol at reflux in the presence of KOH (17 mol%), both achieving TOF of 3333 h−1 and TON of 10,000 (Scheme 32). Remarkably, no aldol-type condensation product was observed. This is in sharp contrast to what was observed by Albrecht et al. with the related pyridyl-functionalized triazolylidene complex 9a, that mainly differs from 11c and 12 by the bonding of the pyridyl unit to the triazole carbon instead of the triazole nitrogen, and with which double aldol condensation products were formed predominantly during the attempted TH of aldehydes such as benzaldehyde, p-chlorobenzaldehyde and thiophenyl aldehyde under similar conditions: 9a (0.5 mol%), KOH (10 mol%) in isopropanol at reflux [23]. Polysubstituted aryl aldehydes and aliphatic aldehydes could nevertheless be transfer hydrogenated by 9a to the corresponding primary alcohols, but with much lower efficiency (TOF = 39–72 h−1; TON = 78–144) than what was observed with 11c and 12 for benzaldehyde.
The pyridyl-functionalized imidazol-2-ylidene complex 21, bearing two propylsulfonate groups, that was shown by Voutchkova-Kostal et al. to be moderately active for the TH of acetophenone from glycerol under microwave heating at 140 °C (TOF = 7.5 h−1, TON = 15, Scheme 13), proved to be also active for the TH of benzaldehyde [32]. Thus, under the same reaction conditions, 79% yield to benzylic alcohol was achieved after 2 h reaction with 2 mol% 21 and 50 mol% KOH, giving TOF and TON values that are about 2.5 times higher than those obtained for the TH of acetophenone (Scheme 33). No aldol condensation product was reported.
5. Transfer Hydrogenation of Imines
Despite the importance of the resulting amines in the field of natural products, pharmaceuticals and agronomical compounds, the TH imines has rarely been studied with donor-functionalized NHC-ruthenium complexes, and has been limited to that of aldimines.
5.1. Aldimines TH with (κ2-CNHC,N)-Complexes
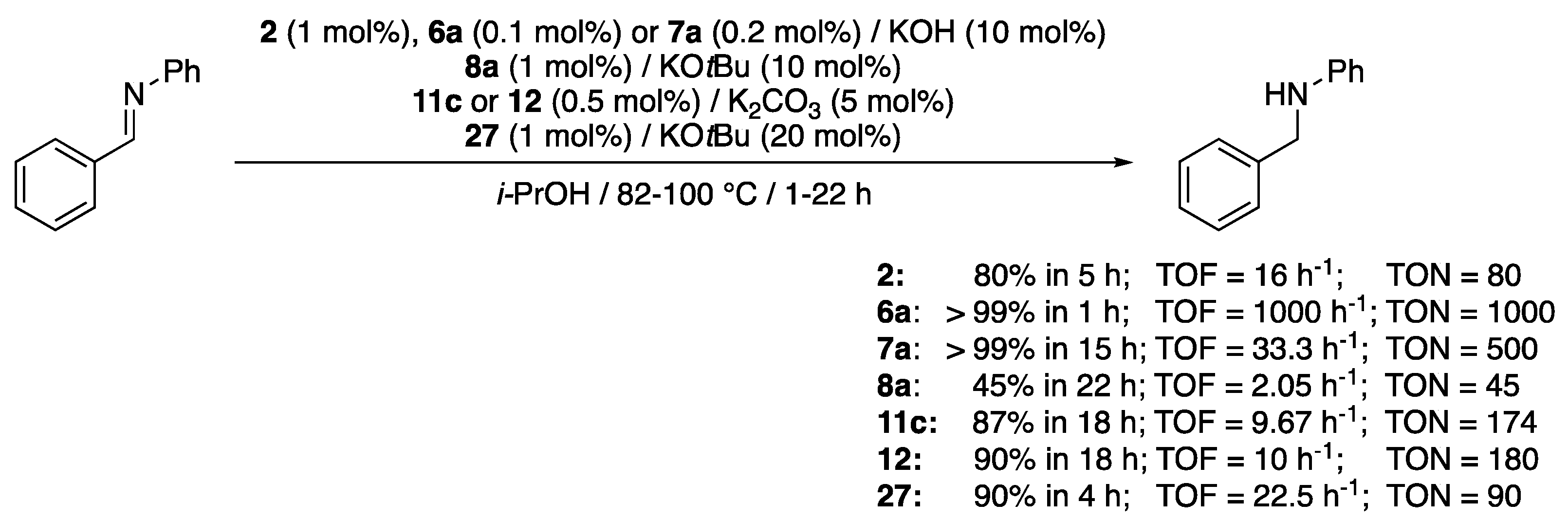
To our knowledge, the first example with (κ2-CNHC,N)-complexes only appeared in 2009 with the pyrimidine-functionalized Ru(II)-NHC complex 2 (Scheme 2) disclosed by Crabtree et al. [12]. It was shown to reduce N-benzylideneaniline in 80% yield after 5 h reaction in isopropanol at reflux with a 1 mol% catalyst loading in the presence of 10 mol% KOH (Scheme 34). A couple of years later, Valerga and Puerta’s picolyl-imidazol-2-ylidene complexes 6a and 7a demonstrated a better efficacy, in particular the Cp* derivative 6a, as it could fully reduce N-benzylideneaniline in 1 h with a catalyst loading of only 0.1 mol% under otherwise similar conditions, achieving a TOF of 1000 h−1 and a TON of 1000 (Scheme 34) [16,18]. The latter is still today the most efficient (κ2-CNHC,N)-precatalyst, as later reports from Hou [22] with his imino-imidazol-2-ylidene complex 8a, Sarkar [25] with his pyridine- or pyrimidine-mesoionic carbene complexes 11c and 12, and Rit [35] with his triazole-imidazol-2-ylidene complex 27 only gave low to moderate activities (Scheme 34).
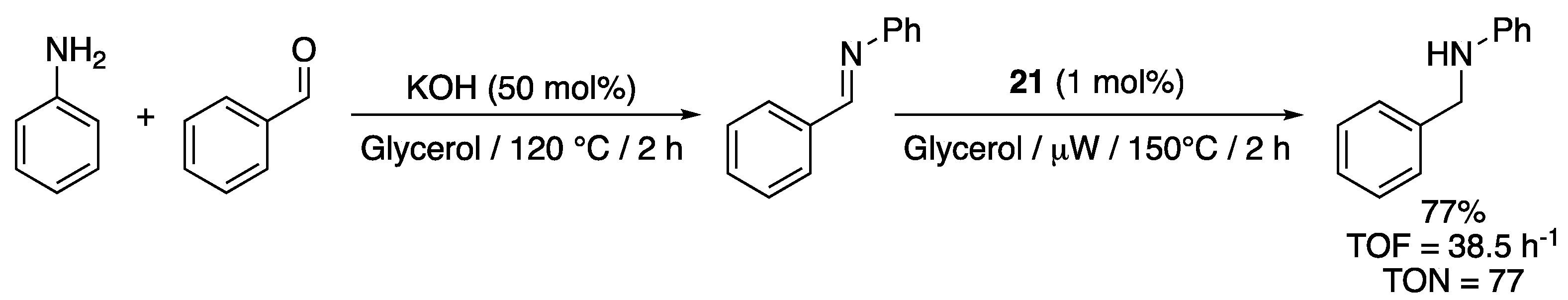
The versatile bis-sulfonated pyridyl-functionalized imidazole-2-ylidene complex 21 (1 mol%) catalyzed the TH of in situ prepared N-benzylideneaniline—using aniline and benzaldehyde—from glycerol under microwave irradiation at 150 °C giving N-benzylaniline in 77% yield after 2 h reaction (TOF = 38.5 h−1) (Scheme 35) [32]. To the best of our knowledge, this was the first report of TH of imines from glycerol. The reaction could also be carried out under conventional heating but only gave a poor 17% yield after 6 h reaction (TOF = 2.83 h−1). The important increase in activity brought by the microwave heating was tentatively attributed to an improved dispersion of the reactants and base in the polar and viscous glycerol solvent under these conditions, as well as to the highly polar zwitterionic character of 21, which likely enhances microwave irradiation’s absorbance. Surprisingly, a control experiment carried out with a preformed imine in the absence of a base gave the reduced product in 63% yield under microwave irradiation. The authors suggested that this observed activity in the absence of KOH may result from the sulfonate arms acting as an internal base. No mechanistic investigation was however conducted to confirm this supposition.
5.2. Aldimines TH with (κ2-CNHC,O)-Complexes
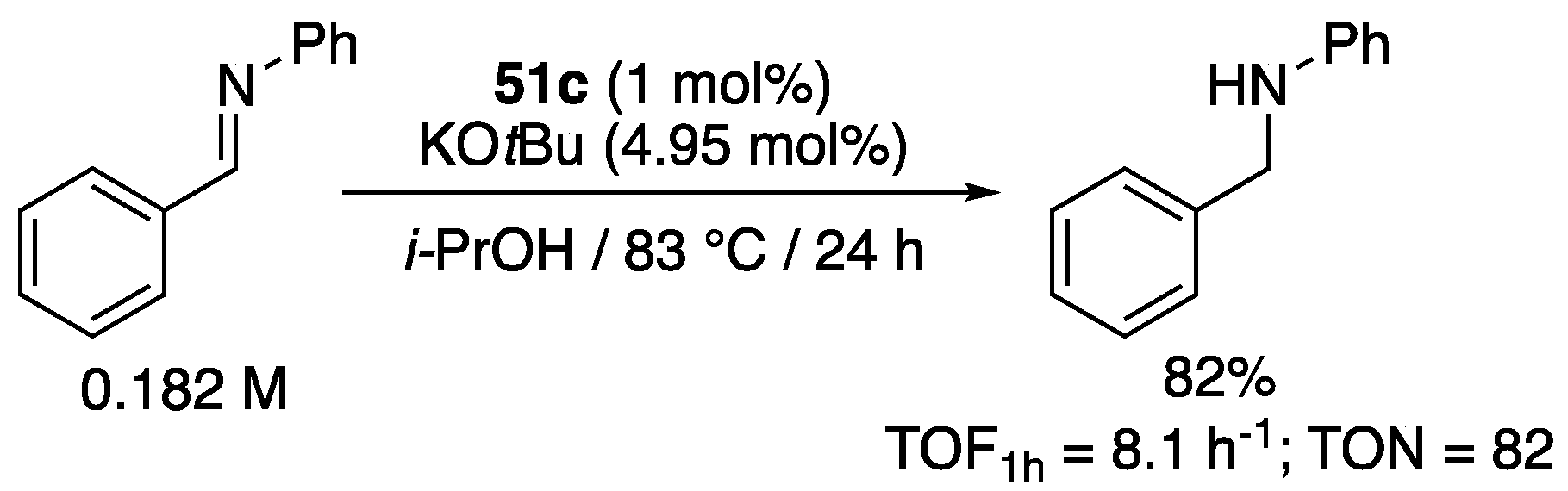
As seen in Section 2.3 and Section 3.2, (κ2-CNHC,O)-complexes are rare, and we are aware of only one example of TH of imines with such complexes, that was described with the carboxylate-functionalized benzimidazol-2-ylidene complex 51c (Figure 6) [68]. In the presence of 4.95 mol% KOtBu, the latter proved able, with a loading of 1 mol%, and an initial TOF1h after 1 h of 8.1 h−1, to reduce N-benzylidineaniline in 82% yield after 24 h reaction in isopropanol at reflux (Scheme 36), which is significantly slower and lower than what was observed with acetophenone (TOF1h = 31 h−1, TON = 96) under similar conditions. However, this reduced activity with aldimines is in line with what was observed with the majority of the κ2-CNHC,N-complexes that proved able to catalyze the TH of imines, i.e., complexes 2, 8a, 11c, and 12 (see Section 5.1).
6. Transfer Hydrogenation of Alkenes
Examples of ruthenium-catalyzed TH of C–C multiple bonds are exceedingly rare [7], and we are aware of only three examples with donor-functionalized N-heterocyclic carbene complexes, two that deal with the reduction in a unfunctionalized alkenes [25,72], and another one that deals with the TH of a polarized C=C double bond [68].
6.1. Alkenes TH with (κ2-C,N)-Complexes
The pyridyl- and pyrimidyl-functionalized mesoionic triazolylidene complexes 11c and 12 proved also active for the TH of a couple of alkenes [25]. Notably, cyclooctene could be fully reduced to cyclooctane with either 11c or 12 (1 mol%) in isopropanol at 100 °C for 24 h (TOF = 4.17 h−1) in the presence of a large amount of KOH (17 mol%) (Scheme 37). Excellent conversions were also observed with catalytic loadings of 0.5 mol%, in particular with the methoxy-functionalized pyridyl derivative 11c (TOF = 7.83 h−1 and TON = 188). Lower efficiencies were however observed with both pre-catalysts for the reductions of trans-β-methylstyrene and trans-stilbene, with maximum TON values of 78 and 41, respectively.
6.2. Alkenes TH with (κ2-C,O)-Complexes
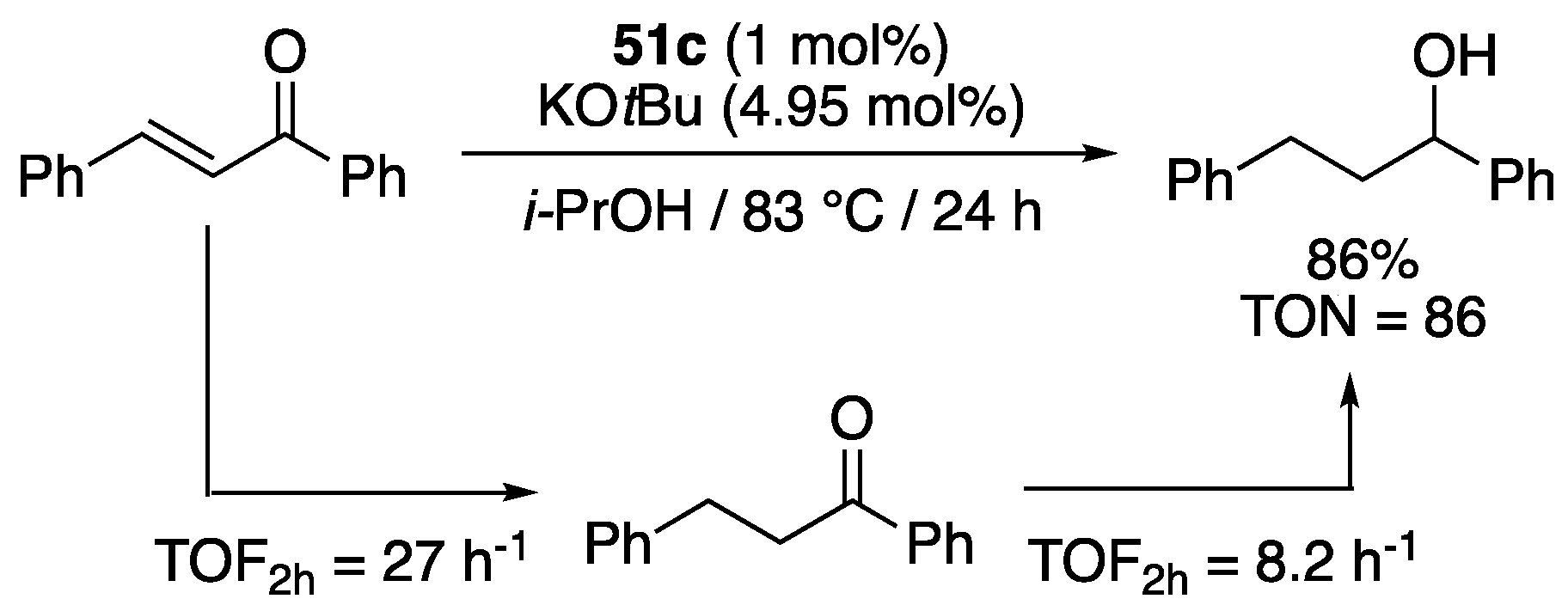
The carboxylate-functionalized benzimidazol-2-ylidene complex 51c (1 mol%) fully reduced the α,β-unsaturated ketone, chalcone, to 1,3-diphenyl-propan-1-ol in 86% yield in 24 h in isopropanol at reflux in the presence of KOtBu (4.95 mol%) (Scheme 38). 1H NMR studies revealed that hydrogenation initially occurred at the C=C double bond and that 1,3-diphenylpropan-1-one was formed as the sole product with a TOF of 27 h−1 for the first 2 h. After 2h, 1,3-diphenyl-propan-1-ol began to appear from hydrogenation of the C=O bond of 1,3-diphenylpropan-1-one with a TOF of 8.2 h−1 for the next 2 h. Despite this encouraging result for the TH of alkenes, 51c proved unable to reduce stilbene under similar conditions, which suggested that polarization of the C=C double bond was necessary to observe reactivity [68].
6.3. Alkenes TH with (κ2-C,S)-Complexes
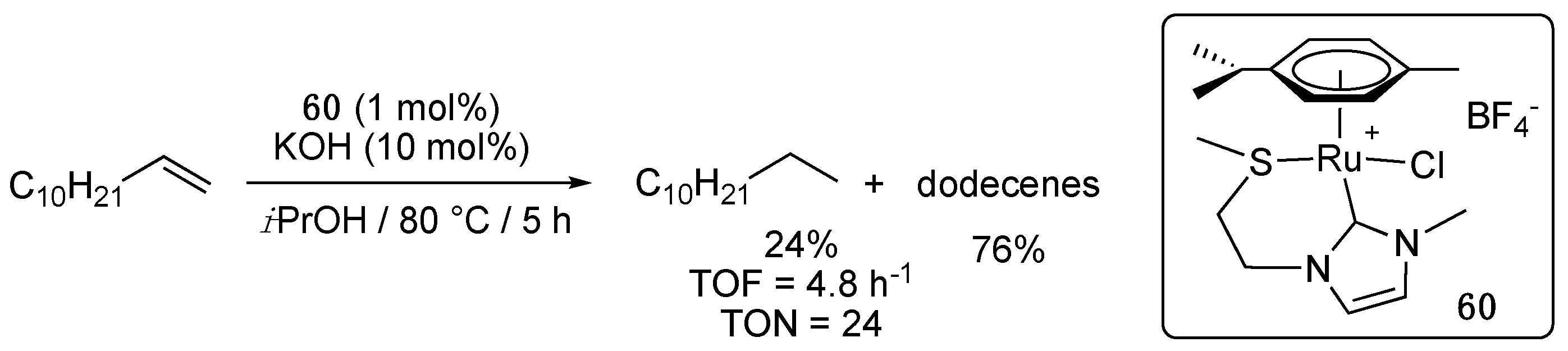
The carboxylate-functionalized imidazole-2-ylidene complex 51a and the thioether-functionalized imidazole-2-ylidene complex 60, which were both prepared via transmetalation from the in situ generated adequate silver carbene, were evaluated as catalyst precursors (1 mol%) for the TH of the non-activated alkene, 1-dodecene, under classical TH conditions, i.e., in isopropanol as hydrogen source and solvent at 80 °C in the presence of KOH (10 mol%) [72]. Whereas 51a was completely inactive under these conditions, 60 fully converted 1-dodecene to a mixture of dodecane (24%) and various dodecenes (76%) in 5 h (Scheme 39). However, prolonged heating to 24 h only allowed a slight increase in the hydrogenated product to 32%.
7. Miscellaneous Transfer Hydrogenation
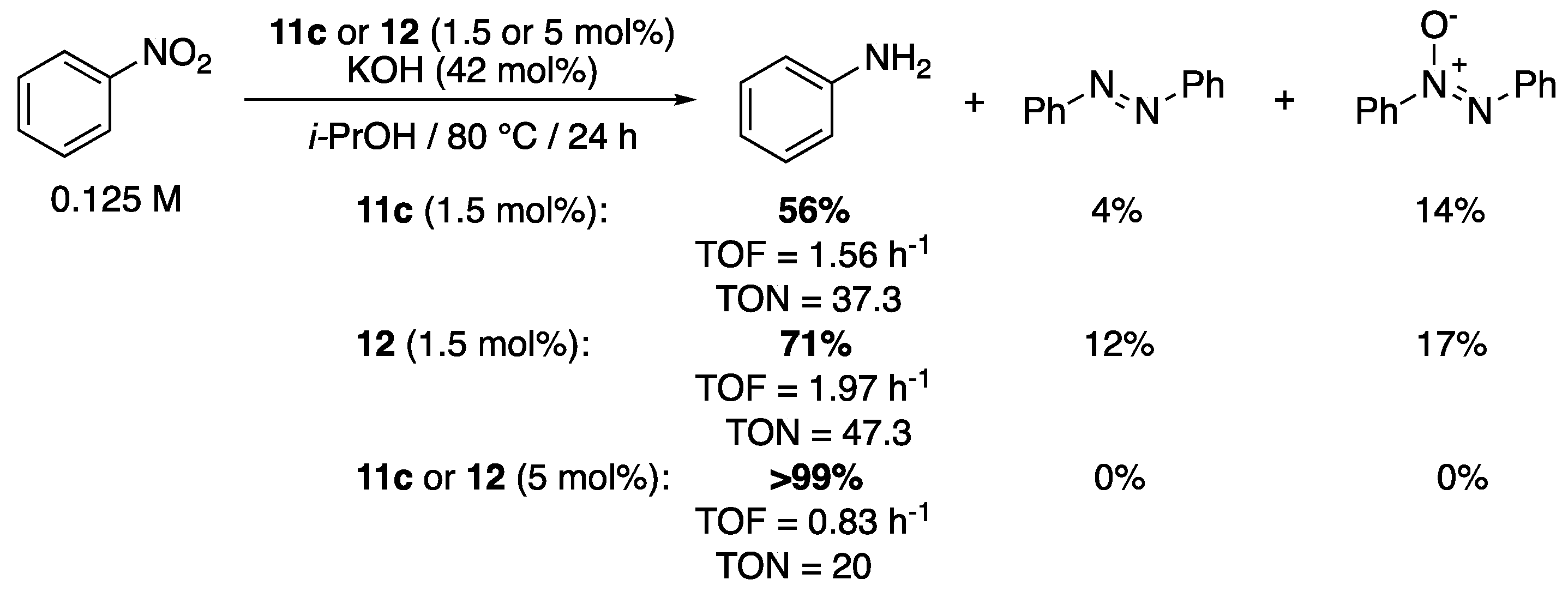
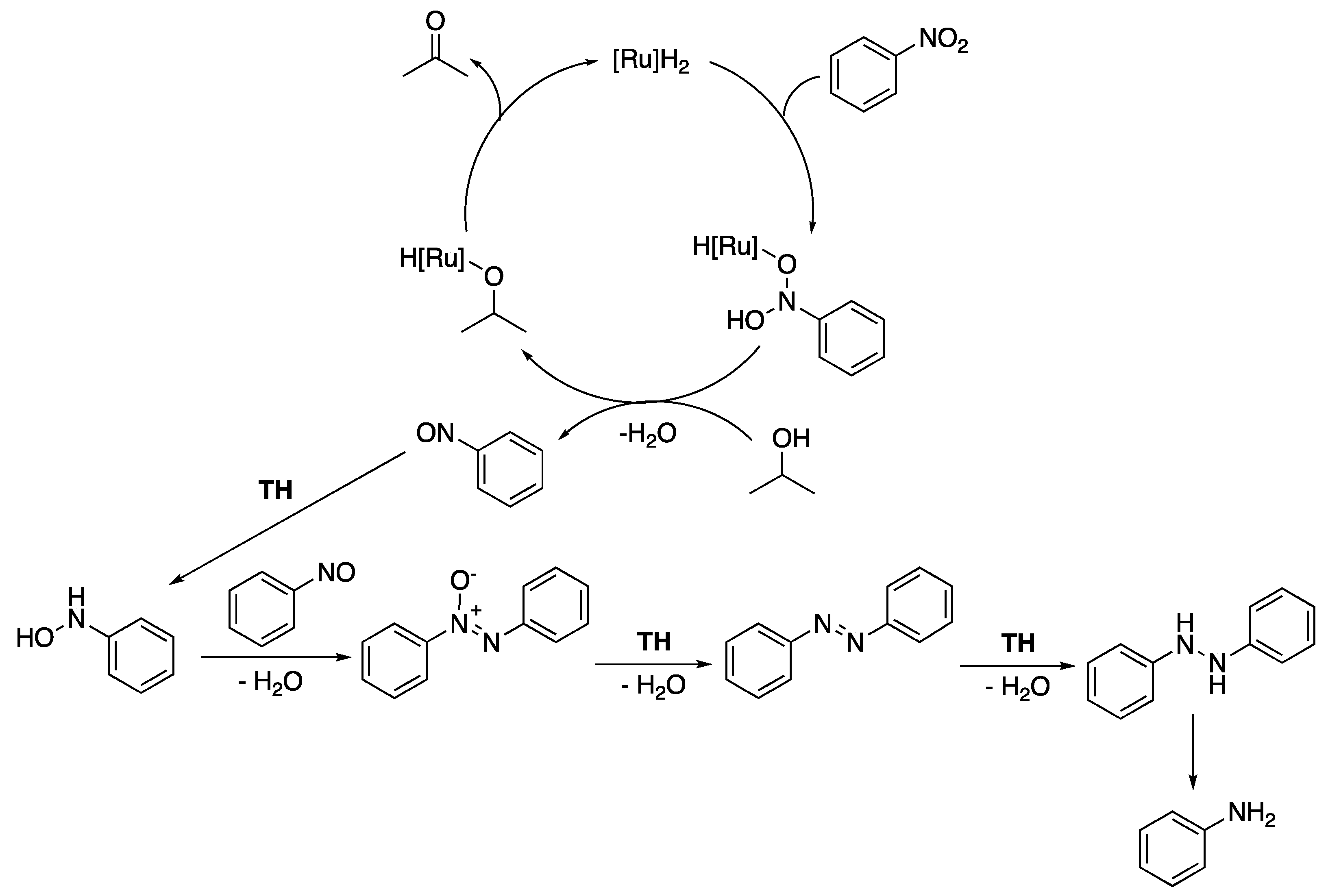
The versatile pyridyl- and pyrimidyl-functionalized mesoionic triazolylidene complexes 11c and 12 were also shown to be efficient precatalysts for the reduction in nitrobenzene to aniline [25]. In isopropanol at 80 °C, in the presence of 0.42 equiv. of KOH, the pyrimidyl derivative 12 (1.5 mol%), in particular, allowed the full conversion of nitrobenzene to aniline (71%), azobenzene (12%), and azoxybenzene (17%) in 24 h reaction, while 11c proved slightly less reactive with only 56% conversion to aniline and, respectively, 4% and 14% to azobenzene and azoxybenzene (Scheme 40). When 5 mol% of either precatalyst was used, a dramatic improvement to full conversion of nitrobenzene to aniline was observed.
Based on a previously reported mechanistic study conducted with related arene-Ru(II) and -Ir(III) complexes bearing pyridyl-triazole or bis-triazole ligands [73], the authors proposed the mechanism depicted in Scheme 41. In this mechanism, the precatalysts would be first converted to ruthenium hydrides “[Ru]H2” in basic isopropanol. Following the formation of nitrosobenzene through a first TH, the latter would then be successively transfer hydrogenated to azoxybenzene, azobenzene, and aniline.
8. Conclusions
Bidentate donor-functionalized NHC-ruthenium complexes have been successfully used as precatalysts for the TH of ketones, aldehydes, aldimines, alkenes, and nitrobenzene. The most widely studied reaction by far is the TH of ketones, and a large number of (κ2-CNHC,N)-ruthenium precatalysts have been investigated for this transformation, comprising amine, pyridine-, pycolyl-, pyrimidine-, imine-, oxazoline-, pyrazole, triazole-, and pyridylideneamide-functionalized NHC ligands. (κ2-CNHC,P)- and (κ2-CNHC,O)-complexes as ketone TH precatalysts are, in contrast, relatively rare, and (κ2-CNHC,S)-complexes have curiously not been investigated. Nevertheless, the (diphenylphosphino)methylene-functionalized bis-imidazol-4-ylidene ruthenium complex 49 is by far the most active donor-functionalized NHC-ruthenium complex giving TOF as high as 198,000 h−1. The TH of aldehydes, aldimines, alkenes, and nitrobenzene, have also been mainly studied with the (κ2-CNHC,N)-ruthenium species. It thus appears from this review that, despite the easy modulation of NHCs, donor-functionalized NHC ligands comprising nitrogen atoms have attracted most of the attention, while the other donors have been clearly under-exploited, leaving a broad avenue for further improvements, in particular for the enantioselective variants of these reactions, which have been barely studied.
Author Contributions
Review and editing, C.M.; writing—original draft preparation, review, and editing, V.R. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
VR and CM gratefully acknowledge the University of Strasbourg and the CNRS for their support.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Wang, D.; Astruc, D. The Golden Age of Transfer Hydrogenation. Chem. Rev. 2015, 115, 6621–6686. [Google Scholar] [CrossRef]
- Zassinovich, G.; Mestroni, G.; Gladiali, S. Asymmetric hydrogen transfer reactions promoted by homogeneous transition metal catalysts. Chem. Rev. 1992, 92, 1051–1069. [Google Scholar] [CrossRef]
- Noyori, R.; Hashiguchi, S. Asymmetric Transfer Hydrogenation Catalyzed by Chiral Ruthenium Complexes. Acc. Chem. Res. 1997, 30, 97–102. [Google Scholar] [CrossRef]
- Clapham, S.E.; Hadzovic, A.; Morris, R.H. Mechanisms of the H2-hydrogenation and transfer hydrogenation of polar bonds catalyzed by ruthenium hydride complexes. Coord. Chem. Rev. 2004, 248, 2201–2237. [Google Scholar] [CrossRef]
- Gladiali, S.; Alberico, E. Asymmetric transfer hydrogenation: Chiral ligands and applications. Chem. Soc. Rev. 2005, 35, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Hey, D.A.; Reich, R.M.; Baratta, W.; Kühn, F.E. Current advances on ruthenium(II) N-heterocyclic carbenes in hydrogenation reactions. Coord. Chem. Rev. 2018, 374, 114–132. [Google Scholar] [CrossRef] [Green Version]
- Ritleng, V.; de Vries, J. Ruthenacycles and Iridacycles as Transfer Hydrogenation Catalysts. Molecules 2021, 26, 4076. [Google Scholar] [CrossRef]
- Chernyshev, V.M.; Denisova, E.; Eremin, D.B.; Ananikov, V.P. The key role of R–NHC coupling (R = C, H, heteroatom) and M–NHC bond cleavage in the evolution of M/NHC complexes and formation of catalytically active species. Chem. Sci. 2020, 11, 6957–6977. [Google Scholar] [CrossRef]
- Normand, A.T.; Cavell, K.J. Donor-Functionalised N-Heterocyclic Carbene Complexes of Group 9 and 10 Metals in Catalysis: Trends and Directions. Eur. J. Inorg. Chem. 2008, 2008, 2781–2800. [Google Scholar] [CrossRef]
- Neshat, A.; Mastrorilli, P.; Mobarakeh, A.M. Recent Advances in Catalysis Involving Bidentate N-Heterocyclic Carbene Ligands. Molecules 2021, 27, 95. [Google Scholar] [CrossRef]
- Poyatos, M.; Maisse-François, A.; Bellemin-Laponnaz, S.; Peris, E.; Gade, L.H. Synthesis and structural chemistry of arene-ruthenium half-sandwich complexes bearing an oxazolinyl–carbene ligand. J. Organomet. Chem. 2006, 691, 2713–2720. [Google Scholar] [CrossRef]
- Gnanamgari, D.; Sauer, E.L.O.; Schley, N.; Butler, C.; Incarvito, C.D.; Crabtree, R.H. Iridium and Ruthenium Complexes with Chelating N-Heterocyclic Carbenes: Efficient Catalysts for Transfer Hydrogenation, β-Alkylation of Alcohols, and N-Alkylation of Amines. Organometallics 2008, 28, 321–325. [Google Scholar] [CrossRef]
- Wylie, W.N.; Lough, A.J.; Morris, R.H. Transmetalation of a Primary Amino-Functionalized N-Heterocyclic Carbene Ligand from an Axially Chiral Square-Planar Nickel(II) Complex to a Ruthenium(II) Precatalyst for the Transfer Hydrogenation of Ketones. Organometallics 2009, 28, 6755–6761. [Google Scholar] [CrossRef]
- Ohara, H.; Wylie, W.N.; Lough, A.J.; Morris, R.H. Effect of chelating ring size in catalytic ketone hydrogenation: Facile synthesis of ruthenium(ii) precatalysts containing an N-heterocyclic carbene with a primary amine donor for ketone hydrogenation and a DFT study of mechanisms. Dalton Trans. 2012, 41, 8797–8808. [Google Scholar] [CrossRef] [PubMed]
- Cross, W.B.; Daly, C.G.; Boutadla, Y.; Singh, K. Variable coordination of amine functionalised N-heterocyclic carbene ligands to Ru, Rh and Ir: C–H and N–H activation and catalytic transfer hydrogenation. Dalton Trans. 2011, 40, 9722–9730. [Google Scholar] [CrossRef] [PubMed]
- Fernández, F.E.; Puerta, M.C.; Valerga, P. Half-Sandwich Ruthenium(II) Picolyl-NHC Complexes: Synthesis, Characterization, and Catalytic Activity in Transfer Hydrogenation Reactions. Organometallics 2011, 30, 5793–5802. [Google Scholar] [CrossRef]
- Ning, J.; Shang, Z.; Xu, X. How Does the Hemilabile Group in Ruthenium-Cp* Picolyl-NHC Complexes Affect the Mechanism of Transfer Hydrogenation Reaction? A DFT Study. Catal. Lett. 2015, 145, 1331–1343. [Google Scholar] [CrossRef]
- Fernández, F.E.; Puerta, M.C.; Valerga, P. Ruthenium(II) Picolyl-NHC Complexes: Synthesis, Characterization, and Catalytic Activity in Amine N-alkylation and Transfer Hydrogenation Reactions. Organometallics 2012, 31, 6868–6879. [Google Scholar] [CrossRef]
- Piyasaengthong, A.; Williams, L.J.; Yufit, D.S.; Walton, J.W. Novel ruthenium complexes bearing bipyridine-based and N-heterocyclic carbene-supported pyridine (NCN) ligands: The influence of ligands for catalytic transfer hydrogenation of ketones. Dalton Trans. 2022, 51, 340–351. [Google Scholar] [CrossRef]
- Pámies, O.; Bäckvall, J.-E. Studies on the Mechanism of Metal-Catalyzed Hydrogen Transfer from Alcohols to Ketones. Chem. Eur. J. 2001, 7, 5052–5058. [Google Scholar] [CrossRef]
- Albrecht, M.; Miecznikowski, J.; Samuel, A.; Faller, J.; Crabtree, R.H. Chelated Iridium(III) Bis-carbene Complexes as Air-Stable Catalysts for Transfer Hydrogenation. Organometallics 2002, 21, 3596–3604. [Google Scholar] [CrossRef]
- Guo, X.-Q.; Wang, Y.-N.; Wang, D.; Cai, L.-H.; Chen, Z.-X.; Hou, X.-F. Palladium, iridium and ruthenium complexes with acyclic imino-N-heterocyclic carbenes and their application in aqua-phase Suzuki–Miyaura cross-coupling reaction and transfer hydrogenation. Dalton Trans. 2012, 41, 14557–14567. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Rebollo, M.; Canseco-Gonzalez, D.; Hollering, M.; Mueller-Bunz, H.; Albrecht, M. Synthesis and catalytic alcohol oxidation and ketone transfer hydrogenation activity of donor-functionalized mesoionic triazolylidene ruthenium(ii) complexes. Dalton Trans. 2013, 43, 4462–4473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolje, A.; Hohloch, S.; van der Meer, M.; Košmrlj, J.; Sarkar, B. RuII, OsII, and IrIIIComplexes with Chelating Pyridyl-Mesoionic Carbene Ligands: Structural Characterization and Applications in Transfer Hydrogenation Catalysis. Chem.-A Eur. J. 2015, 21, 6756–6764. [Google Scholar] [CrossRef]
- Bolje, A.; Hohloch, S.; Košmrlj, J.; Sarkar, B. RuII, IrIII and OsII mesoionic carbene complexes: Efficient catalysts for transfer hydrogenation of selected functionalities. Dalton Trans. 2016, 45, 15983–15993. [Google Scholar] [CrossRef] [Green Version]
- Olguín, J.; Paz-Sandoval, M. Synthesis and transfer hydrogenation catalysis of chelating triazolylidene ruthenium(II) complexes: Effect of the pendant arm, p-cymene, acetonitrile and butadienesulfonyl co-ligands. J. Organomet. Chem. 2017, 848, 309–317. [Google Scholar] [CrossRef]
- Sabater, S.; Müller-Bunz, H.; Albrecht, M. Carboxylate-Functionalized Mesoionic Carbene Precursors: Decarboxylation, Ruthenium Bonding, and Catalytic Activity in Hydrogen Transfer Reactions. Organometallics 2016, 35, 2256–2266. [Google Scholar] [CrossRef]
- Olguín, J.; Díaz-Fernández, M.; de la Cruz-Cruz, J.I.; Paz-Sandoval, M.A. Mixed heteropentadienyl and N-heterocyclic carbene ruthenium(II) complexes: Synthesis and transfer hydrogenation catalysis. J. Organomet. Chem. 2016, 824, 33–41. [Google Scholar] [CrossRef]
- Smith, I.G.; Zgrabik, J.C.; Gutauskas, A.C.; Gray, D.L.; Domski, G.J. Synthesis, characterization, and catalytic behavior of mono- and bimetallic ruthenium(II) and iridium(III) complexes supported by pyridine-functionalized N -heterocyclic carbene ligands. Inorg. Chem. Commun. 2017, 81, 27–32. [Google Scholar] [CrossRef]
- Viji, M.; Tyagi, N.; Naithani, N.; Ramaiah, D. Aryl appended neutral and cationic half-sandwich ruthenium(ii)–NHC complexes: Synthesis, characterisation and catalytic applications. New J. Chem. 2017, 41, 12736–12745. [Google Scholar] [CrossRef]
- Castañón, E.B.; Kaposi, M.; Reich, R.M.; Kühn, F.E. Water-soluble transition metal complexes of ruthenium(ii), osmium(ii), rhodium(iii) and iridium(iii) with chelating N-heterocyclic carbene ligands in hydrogenation and transfer hydrogenation catalysis. Dalton Trans. 2018, 47, 2318–2329. [Google Scholar] [CrossRef]
- Azua, A.; Finn, M.; Yi, H.; Dantas, A.B.; Voutchkova-Kostal, A. Transfer Hydrogenation from Glycerol: Activity and Recyclability of Iridium and Ruthenium Sulfonate-Functionalized N-Heterocyclic Carbene Catalysts. ACS Sustain. Chem. Eng. 2017, 5, 3963–3972. [Google Scholar] [CrossRef]
- Nair, A.G.; McBurney, R.T.; Walker, D.B.; Page, M.J.; Gatus, M.R.D.; Bhadbhade, M.; Messerle, B.A. Ruthenium(ii) complexes of hemilabile pincer ligands: Synthesis and catalysing the transfer hydrogenation of ketones. Dalton Trans. 2016, 45, 14335–14342. [Google Scholar] [CrossRef] [PubMed]
- Hollering, M.; Albrecht, M.; Kühn, F.E. Bonding and Catalytic Application of Ruthenium N-Heterocyclic Carbene Complexes Featuring Triazole, Triazolylidene, and Imidazolylidene Ligands. Organometallics 2016, 35, 2980–2986. [Google Scholar] [CrossRef]
- Illam, P.M.; Donthireddy, S.N.R.; Chakrabartty, S.; Rit, A. Heteroditopic Ru(II)– and Ir(III)–NHC Complexes with Pendant 1,2,3-Triazole/Triazolylidene Groups: Stereoelectronic Impact on Transfer Hydrogenation of Unsaturated Compounds. Organometallics 2019, 38, 2610–2623. [Google Scholar] [CrossRef]
- Sinha, A.; Daw, P.; Rahaman, S.W.; Saha, B.; Bera, J.K. A RuII–N-heterocyclic carbene (NHC) complex from metal–metal singly bonded diruthenium(I) precursor: Synthesis, structure and catalytic evaluation. J. Organomet. Chem. 2011, 696, 1248–1257. [Google Scholar] [CrossRef]
- Yoshimura, M.; Kamisue, R.; Sakaguchi, S. Synthesis of Ru(II) complexes containing N-heterocyclic carbenes functionalized with secondary donor groups: Catalytic activity toward enantioselective transfer hydrogenation. J. Organomet. Chem. 2013, 740, 26–32. [Google Scholar] [CrossRef]
- Sortais, J.-B.; Ritleng, V.; Voelklin, A.; Holuigue, A.; Smail, H.; Barloy, L.; Sirlin, C.; Verzijl, G.K.M.; Boogers, J.A.F.; de Vries, A.H.M.; et al. Cycloruthenated Primary and Secondary Amines as Efficient Catalyst Precursors for Asymmetric Transfer Hydrogenation. Org. Lett. 2005, 7, 1247–1250. [Google Scholar] [CrossRef] [Green Version]
- Jerphagnon, T.; Haak, R.; Berthiol, F.; Gayet, A.J.A.; Ritleng, V.; Holuigue, A.; Pannetier, N.; Pfeffer, M.; Voelklin, A.; Lefort, L.; et al. Ruthenacycles and Iridacycles as Catalysts for Asymmetric Transfer Hydrogenation and Racemisation. Top. Catal. 2010, 53, 1002–1008. [Google Scholar] [CrossRef] [Green Version]
- Kilic, A.; Kayan, C.; Aydemir, M.; Durap, F.; Durgun, M.; Baysal, A.; Tas, E.; Gümgüm, B. Synthesis of new boron complexes: Application to transfer hydrogenation of acetophenone derivatives. Appl. Organomet. Chem. 2011, 25, 390–394. [Google Scholar] [CrossRef]
- Kilic, A.; Aydemir, M.; Durgun, M.; Meriç, N.; Ocak, Y.S.; Keles, A.; Temel, H. Fluorine/phenyl chelated boron complexes: Synthesis, fluorescence properties and catalyst for transfer hydrogenation of aromatic ketones. J. Fluor. Chem. 2014, 162, 9–16. [Google Scholar] [CrossRef]
- Leigh, V.; Carleton, D.J.; Olguin, J.; Mueller-Bunz, H.; Wright, L.J.; Albrecht, M. Solvent-Dependent Switch of Ligand Donor Ability and Catalytic Activity of Ruthenium(II) Complexes Containing Pyridinylidene Amide (PYA) N-Heterocyclic Carbene Hybrid Ligands. Inorg. Chem. 2014, 53, 8054–8060. [Google Scholar] [CrossRef] [Green Version]
- Donnelly, K.F.; Segarra, C.; Shao, L.-X.; Suen, R.; Müller-Bunz, H.; Albrecht, M. Adaptive N-Mesoionic Ligands Anchored to a Triazolylidene for Ruthenium-Mediated (De)Hydrogenation Catalysis. Organometallics 2015, 34, 4076–4084. [Google Scholar] [CrossRef]
- Navarro, M.; Segarra, C.; Pfister, T.; Albrecht, M. Structural, Electronic, and Catalytic Modulation of Chelating Pyridylideneamide Ruthenium(II) Complexes. Organometallics 2020, 39, 2383–2391. [Google Scholar] [CrossRef]
- Malan, F.P.; Singleton, E.; van Rooyen, P.H.; Albrecht, M.; Landman, M. Synthesis, Stability, and (De)hydrogenation Catalysis by Normal and Abnormal Alkene- and Picolyl-Tethered NHC Ruthenium Complexes. Organometallics 2019, 38, 2624–2635. [Google Scholar] [CrossRef]
- Albrecht, M. C4-bound imidazolylidenes: From curiosities to high-impact carbene ligands. Chem. Commun. 2008, 3601–3610. [Google Scholar] [CrossRef] [Green Version]
- Crabtree, R.H. Abnormal, mesoionic and remote N-heterocyclic carbene complexes. Coord. Chem. Rev. 2013, 257, 755–766. [Google Scholar] [CrossRef]
- Juzgado, A.; Lorenzo-García, M.M.; Barrejón, M.; Rodríguez, A.M.; Rodríguez-López, J.; Merino, S.; Tejeda, J. Chelation assistance as a tool for the selective preparation of an imidazole-based mesoionic palladium carbene complex. Chem. Commun. 2014, 50, 15313–15315. [Google Scholar] [CrossRef]
- Cheng, Y.; Sun, J.-F.; Yang, H.-L.; Xu, H.-J.; Li, Y.-Z.; Chen, X.-T.; Xue, Z.-L. Syntheses, Structures, and Catalytic Properties of Ruthenium(II) Nitrosyl Complexes with Pyridine-Functionalized N-Heterocyclic Carbenes. Organometallics 2009, 28, 819–823. [Google Scholar] [CrossRef]
- Cheng, Y.; Xu, H.-J.; Sun, J.-F.; Li, Y.-Z.; Chen, X.-T.; Xue, Z.-L. Synthesis, structures and catalytic activities of ruthenium(ii) carbonyl chloride complexes containing pyridine-functionalised N-heterocyclic carbenes. Dalton Trans. 2009, 7132–7140. [Google Scholar] [CrossRef]
- Li, X.-W.; Wang, G.-F.; Chen, F.; Li, Y.-Z.; Chen, X.-T.; Xue, Z.-L. Ruthenium(II) carbonyl chloride complexes containing pyridine-functionalised bidentate N-heterocyclic carbenes: Synthesis, structures, and impact of the carbene ligands on catalytic activities. Inorg. Chim. Acta 2011, 378, 280–287. [Google Scholar] [CrossRef]
- Chen, C.; Lu, C.; Zheng, Q.; Ni, S.; Zhang, M.; Chen, W. Synthesis and structures of ruthenium–NHC complexes and their catalysis in hydrogen transfer reaction. Beilstein J. Org. Chem. 2015, 11, 1786–1795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miranda-Soto, V.; Grotjahn, D.B.; Cooksy, A.L.; Golen, J.A.; Moore, C.E.; Rheingold, A.L. A Labile and Catalytically Active Imidazol-2-yl Fragment System. Angew. Chem. Int. Ed. 2010, 50, 631–635. [Google Scholar] [CrossRef]
- Kuwata, S.; Ikariya, T. Metal-ligand bifunctional reactivity and catalysis of protic N-heterocyclic carbene and pyra-zole complexes featuring β-NH units. Chem. Commun. 2014, 50, 14290–14300. [Google Scholar] [CrossRef] [PubMed]
- Humphries, M.E.; Pecak, W.H.; Hohenboken, S.A.; Alvarado, S.R.; Swenson, D.C.; Domski, G.J. Ruthenium(II) supported by phosphine-functionalized N-heterocyclic carbene ligands as catalysts for the transfer hydrogenation of ketones. Inorg. Chem. Commun. 2013, 37, 138–143. [Google Scholar] [CrossRef]
- Witt, J.; Pöthig, A.; Kühn, F.E.; Baratta, W. Abnormal N-Heterocyclic Carbene-Phosphine Ruthenium(II) Complexes as Active Catalysts for Transfer Hydrogenation. Organometallics 2013, 32, 4042–4045. [Google Scholar] [CrossRef]
- Yamakawa, M.; Ito, H.; Noyori, R. The Metal−Ligand Bifunctional Catalysis: A Theoretical Study on the Ruthenium(II)-Catalyzed Hydrogen Transfer between Alcohols and Carbonyl Compounds. J. Am. Chem. Soc. 2000, 122, 1466–1478. [Google Scholar] [CrossRef]
- Bitzer, M.J.; Pöthig, A.; Jandl, C.; Kühn, F.E.; Baratta, W. Ru–Ag and Ru–Au dicarbene complexes from an abnormal carbene ruthenium system. Dalton Trans. 2015, 44, 11686–11689. [Google Scholar] [CrossRef] [Green Version]
- Pardatscher, L.; Bitzer, M.J.; Jandl, C.; Kück, J.W.; Reich, R.M.; Kühn, F.E.; Baratta, W. Cationic abnormal N-heterocyclic carbene ruthenium complexes as suitable precursors for the synthesis of heterobimetallic compounds. Dalton Trans. 2018, 48, 79–89. [Google Scholar] [CrossRef]
- Pardatscher, L.; Hofmann, B.J.; Fischer, P.J.; Hölzl, S.M.; Reich, R.M.; Kühn, F.E.; Baratta, W. Highly Efficient Abnormal NHC Ruthenium Catalyst for Oppenauer-Type Oxidation and Transfer Hydrogenation Reactions. ACS Catal. 2019, 9, 11302–11306. [Google Scholar] [CrossRef]
- Chelucci, G.; Baldino, S.; Baratta, W. Ruthenium and osmium complexes containing 2-(aminomethyl)pyridine (Ampy)-based ligands in catalysis. Coord. Chem. Rev. 2015, 300, 29–85. [Google Scholar] [CrossRef]
- Baratta, W.; Schütz, J.; Herdtweck, E.; Herrmann, W.A.; Rigo, P. Fast transfer hydrogenation using a highly active orthometalated heterocyclic carbene ruthenium catalyst. J. Organomet. Chem. 2005, 690, 5570–5575. [Google Scholar] [CrossRef]
- Poyatos, M.; Mata, J.A.; Falomir, E.; Crabtree, R.H.; Peris, E. New Ruthenium(II) CNC-Pincer Bis(carbene) Complexes: Synthesis and Catalytic Activity. Organometallics 2003, 22, 1110–1114. [Google Scholar] [CrossRef]
- Hollering, M.; Weiss, D.T.; Bitzer, M.J.; Jandl, C.; Kühn, F.E. Controlling Coordination Geometries: Ru–Carbene Complexes with Tetra-NHC Ligands. Inorg. Chem. 2016, 55, 6010–6017. [Google Scholar] [CrossRef] [PubMed]
- Horn, S.; Gandolfi, C.; Albrecht, M. Transfer Hydrogenation of Ketones and Activated Olefins Using Chelating NHC Ruthenium Complexes. Eur. J. Inorg. Chem. 2011, 2011, 2863–2868. [Google Scholar] [CrossRef] [Green Version]
- Gandolfi, C.; Heckenroth, M.; Neels, A.; Laurenczy, G.; Albrecht, M. Chelating NHC Ruthenium(II) Complexes as Robust Homogeneous Hydrogenation Catalysts. Organometallics 2009, 28, 5112–5121. [Google Scholar] [CrossRef] [Green Version]
- DePasquale, J.; White, N.J.; Ennis, E.J.; Zeller, M.; Foley, J.P.; Papish, E.T. Synthesis of chiral N-heterocyclic carbene (NHC) ligand precursors and formation of ruthenium(II) complexes for transfer hydrogenation catalysts. Polyhedron 2012, 58, 162–170. [Google Scholar] [CrossRef]
- Mangalum, A.; McMillen, C.D.; Tennyson, A. Synthesis, coordination chemistry and reactivity of transition metal complexes supported by a chelating benzimidazolylidene carboxylate ligand. Inorg. Chim. Acta 2015, 426, 29–38. [Google Scholar] [CrossRef]
- Akkoç, M.; Öz, E.; Demirel, S.; Dorcet, V.; Roisnel, T.; Bayri, A.; Bruneau, C.; Altin, S.; Yaşar, S.; Özdemir, I. Investigation of potential hybrid capacitor property of chelated N-Heterocyclic carbene Ruthenium(II) complex. J. Organomet. Chem. 2018, 866, 214–222. [Google Scholar] [CrossRef]
- Pretorius, R.; Mazloomi, Z.; Albrecht, M. Synthesis, hemilability, and catalytic transfer hydrogenation activity of iridium(III) and ruthenium(II) complexes containing oxygen-functionalised triazolylidene ligands. J. Organomet. Chem. 2017, 845, 196–205. [Google Scholar] [CrossRef]
- Wan, K.Y.; Lough, A.J.; Morris, R.H. Transition Metal Complexes of an (S,S)-1,2-Diphenylethylamine-Functionalized N-Heterocyclic Carbene: A New Member of the Asymmetric NHC Ligand Family. Organometallics 2016, 35, 1604–1612. [Google Scholar] [CrossRef] [Green Version]
- Horn, S.; Albrecht, M. Transfer hydrogenation of unfunctionalised alkenes using N-heterocyclic carbene ruthenium catalyst precursors. Chem. Commun. 2011, 47, 8802–8804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hohloch, S.; Suntrup, L.; Sarkar, B. Arene–Ruthenium(II) and −Iridium(III) Complexes with “Click”-Based Pyridyl-triazoles, Bis-triazoles, and Chelating Abnormal Carbenes: Applications in Catalytic Transfer Hydrogenation of Nitrobenzene. Organometallics 2013, 32, 7376–7385. [Google Scholar] [CrossRef]
Scheme 1.
TH of acetophenone and 2-octanone catalyzed by the oxazolinyl-functionalized complex 1a.

Scheme 2.
TH of acetophenone and cyclohexanone catalyzed by the pyrimidine-functionalized complex 2.
Scheme 2.
TH of acetophenone and cyclohexanone catalyzed by the pyrimidine-functionalized complex 2.

Scheme 3.
TH of acetophenone by the amino-functionalized NHC-Ru(II) complex 3.

Scheme 4.
TH of ketones by the amino-functionalized imidazol-2-ylidene complex 4a.

Scheme 5.
TH of acetophenone catalyzed by the amino-functionalized NHC-Ru(II) complex 5.

Scheme 6.
TH of ketones catalyzed by the picolyl-functionalized NHC-Ru(II) complex 6a.

Figure 1.
(η6-p-cymene)Ru(II) complexes 7a–e bearing picolyl-NHC ligands.

Scheme 7.
Deuterium labeling experiment with 7a establishing a monohydride mechanism.

Scheme 8.
TH of acetophenone catalyzed by the imino-functionalized imidazol-2-ylidene-Ru(II) complexes 8a–c.
Scheme 8.
TH of acetophenone catalyzed by the imino-functionalized imidazol-2-ylidene-Ru(II) complexes 8a–c.

Scheme 9.
TH of benzophenone catalyzed by the pyridyl-functionalized mesoionic triazolylidene complexes 9a,b and 10.
Scheme 9.
TH of benzophenone catalyzed by the pyridyl-functionalized mesoionic triazolylidene complexes 9a,b and 10.

Scheme 10.
TH of various ketones catalyzed by the pyridyl- or pyrimidyl-functionalized mesoionic triazolylidene complexes 11a–g and 12.
Scheme 10.
TH of various ketones catalyzed by the pyridyl- or pyrimidyl-functionalized mesoionic triazolylidene complexes 11a–g and 12.

Scheme 11.
TH of benzophenone catalyzed by the picolyl-triazolylidene complexes 13a–c.

Figure 2.
Pyridyl-functionalized triazolylidene complexes 14a–c and 16 bearing a pendant carboxylic group and unfunctionalized complex 15.
Figure 2.
Pyridyl-functionalized triazolylidene complexes 14a–c and 16 bearing a pendant carboxylic group and unfunctionalized complex 15.

Figure 3.
Pyridyl-functionalized imidazol-2-ylidene complexes 17–20.

Scheme 12.
TH of acetophenone in aqueous solution catalyzed by the water soluble complex 17f.

Scheme 13.
TH of acetophenone from glycerol catalyzed by the bis-sulfonated complex 21.

Figure 4.
Triazole- and pyrazole-functionalized imidazole-2-ylidene complexes 22–27.

Scheme 14.
Low-temperature TH of ketones catalyzed by the anion-mixed pyrazole-imidazol-2-ylidene complex 22b.
Scheme 14.
Low-temperature TH of ketones catalyzed by the anion-mixed pyrazole-imidazol-2-ylidene complex 22b.

Scheme 15.
Solvent-dependent resonance forms of the pyridylideneamide-imidazol-2-ylidene-Ru(II) complexes 28a,b.
Scheme 15.
Solvent-dependent resonance forms of the pyridylideneamide-imidazol-2-ylidene-Ru(II) complexes 28a,b.

Scheme 16.
Solvent dependence of the TH of benzophenone catalyzed by the adaptive pyridylideneamide-triazolylidene complex 29.
Scheme 16.
Solvent dependence of the TH of benzophenone catalyzed by the adaptive pyridylideneamide-triazolylidene complex 29.

Scheme 17.
TH of benzophenone catalyzed by the pycolyl-functionalized imidazol-4-ylidene complexes 30 and 31/32.
Scheme 17.
TH of benzophenone catalyzed by the pycolyl-functionalized imidazol-4-ylidene complexes 30 and 31/32.

Figure 5.
Pyridyl- or picolyl-functionalized imidazol-, benzimidazol- or triazol-2-ylidene octahedral Ru(II) complexes 33–39.
Figure 5.
Pyridyl- or picolyl-functionalized imidazol-, benzimidazol- or triazol-2-ylidene octahedral Ru(II) complexes 33–39.

Scheme 18.
Low-temperature TH of aromatic ketones catalyzed by the aqua-Ru(II) complex 40 bearing a 1,8-naphtyrid-2-yl-imidazol-2-ylidene chelate.
Scheme 18.
Low-temperature TH of aromatic ketones catalyzed by the aqua-Ru(II) complex 40 bearing a 1,8-naphtyrid-2-yl-imidazol-2-ylidene chelate.

Scheme 19.
TH of ketones catalyzed by the pyrimidin-2-yl-functionalized imidazol-2-ylidene complexes 41 and 42.
Scheme 19.
TH of ketones catalyzed by the pyrimidin-2-yl-functionalized imidazol-2-ylidene complexes 41 and 42.

Scheme 20.
Synthesis of the hydride complex 44 from the protic NHC complex 43 (A); TH of acetophenone catalyzed by 43 and 44 (B); Proposed TH mechanism (C).
Scheme 20.
Synthesis of the hydride complex 44 from the protic NHC complex 43 (A); TH of acetophenone catalyzed by 43 and 44 (B); Proposed TH mechanism (C).

Scheme 21.
TH of ketones catalyzed by the ortho-(diphenylphosphino)benzyl-functionalized imidazol-2-ylidene complex 45.
Scheme 21.
TH of ketones catalyzed by the ortho-(diphenylphosphino)benzyl-functionalized imidazol-2-ylidene complex 45.

Scheme 22.
TH of acetophenone catalyzed by the (diphenylphosphino)ethylene-functionalized imidazol-4-ylidene complexes 46 and 47.
Scheme 22.
TH of acetophenone catalyzed by the (diphenylphosphino)ethylene-functionalized imidazol-4-ylidene complexes 46 and 47.

Scheme 23.
TH of ketones catalyzed by the (diphenylphosphino)methylene-functionalized imidazol-2-ylidene and imidazol-4-ylidene complex 48.
Scheme 23.
TH of ketones catalyzed by the (diphenylphosphino)methylene-functionalized imidazol-2-ylidene and imidazol-4-ylidene complex 48.

Scheme 24.
TH of ketones catalyzed by the (diphenylphosphino)methylene-functionalized bis-imidazol-4-ylidene complex 49.
Scheme 24.
TH of ketones catalyzed by the (diphenylphosphino)methylene-functionalized bis-imidazol-4-ylidene complex 49.

Scheme 25.
Formation of the dihydride bis-imidazol-2-ylidene complex 50 from 49.

Scheme 26.
TH of benzophenone catalyzed by the carboxylate-functionalized imidazol-2-ylidene and 1,2,3-triazolylidene complexes 51a, 52 and 53.
Scheme 26.
TH of benzophenone catalyzed by the carboxylate-functionalized imidazol-2-ylidene and 1,2,3-triazolylidene complexes 51a, 52 and 53.

Figure 6.
Carboxylate-functionalized NHC-Ru(II) complexes 51b–d.

Scheme 27.
TH of ketones catalyzed by the carboxylate-functionalized benzimidazolylidene complex 51d.
Scheme 27.
TH of ketones catalyzed by the carboxylate-functionalized benzimidazolylidene complex 51d.

Scheme 28.
TH of benzophenone catalyzed by the hydroxy-functionalized 1,2,3-triazolylidene complexes 54 and 55 and the 1,4-dibutyl-triazolylidene complex 56.
Scheme 28.
TH of benzophenone catalyzed by the hydroxy-functionalized 1,2,3-triazolylidene complexes 54 and 55 and the 1,4-dibutyl-triazolylidene complex 56.

Scheme 29.
ATH attempt of acetophenone with the (S,S)-1,2-diphenylethylamine-functionalized imidazol-2-ylidene complex 57.
Scheme 29.
ATH attempt of acetophenone with the (S,S)-1,2-diphenylethylamine-functionalized imidazol-2-ylidene complex 57.

Scheme 30.
ATH of acetophenone with the hydroxy-amide-functionalized benzimidazol-2-ylidene (η6-arene)Ru(II) complexes 58a–c.
Scheme 30.
ATH of acetophenone with the hydroxy-amide-functionalized benzimidazol-2-ylidene (η6-arene)Ru(II) complexes 58a–c.

Scheme 31.
Synthesis of the catalytically inactive dianionic alkoxy-amidate-NHC-Ru complex 59.

Scheme 32.
TH of benzaldehyde catalyzed by the pyridine- or pyrimidine-functionalized triazolylidene complexes 11c and 12.
Scheme 32.
TH of benzaldehyde catalyzed by the pyridine- or pyrimidine-functionalized triazolylidene complexes 11c and 12.

Scheme 33.
TH of benzaldehyde from glycerol catalyzed by the bis-sulfonated complex 21.

Scheme 34.
TH of N-benzylidineaniline catalyzed by complexes 2, 6a, 7a, 8a, 11c, 12 and 27.

Scheme 35.
TH of N-benzylidineaniline from glycerol catalyzed by the bis-sulfonated complex 21.

Scheme 36.
TH of N-benzylidineaniline catalyzed by the carboxylate-functionalized benzimidazol-2-ylidene complex 51c.
Scheme 36.
TH of N-benzylidineaniline catalyzed by the carboxylate-functionalized benzimidazol-2-ylidene complex 51c.

Scheme 37.
TH of alkenes by the pyridyl- and pyrimidyl-functionalized triazolylidene complexes 11c and 12.
Scheme 37.
TH of alkenes by the pyridyl- and pyrimidyl-functionalized triazolylidene complexes 11c and 12.

Scheme 38.
TH of chalcone catalyzed by the carboxylate-functionalized benzimidazol-2-ylidene complex 51c.
Scheme 38.
TH of chalcone catalyzed by the carboxylate-functionalized benzimidazol-2-ylidene complex 51c.

Scheme 39.
TH of 1-dodecene catalyzed by the thioether-functionalized imidazol-2-ylidene complex 60.
Scheme 39.
TH of 1-dodecene catalyzed by the thioether-functionalized imidazol-2-ylidene complex 60.

Scheme 40.
TH of nitrobenzene to aniline catalyzed by the pyridyl- and pyrimidyl-functionalized triazolylidene complexes 11c and 12.
Scheme 40.
TH of nitrobenzene to aniline catalyzed by the pyridyl- and pyrimidyl-functionalized triazolylidene complexes 11c and 12.

Scheme 41.
Proposed mechanism for the TH of nitrobenzene to aniline catalyzed by 11c and 12.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ritleng, V.; Michon, C. Bidentate Donor-Functionalized N-Heterocyclic Carbenes: Valuable Ligands for Ruthenium-Catalyzed Transfer Hydrogenation. Molecules 2022, 27, 4703. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27154703
AMA Style
Ritleng V, Michon C. Bidentate Donor-Functionalized N-Heterocyclic Carbenes: Valuable Ligands for Ruthenium-Catalyzed Transfer Hydrogenation. Molecules. 2022; 27(15):4703. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27154703
Chicago/Turabian StyleRitleng, Vincent, and Christophe Michon. 2022. "Bidentate Donor-Functionalized N-Heterocyclic Carbenes: Valuable Ligands for Ruthenium-Catalyzed Transfer Hydrogenation" Molecules 27, no. 15: 4703. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27154703