
Prediction of Drug–Target Interaction Using Dual-Network Integrated Logistic Matrix Factorization and Knowledge Graph Embedding


Abstract
:1. Introduction
2. Related Work
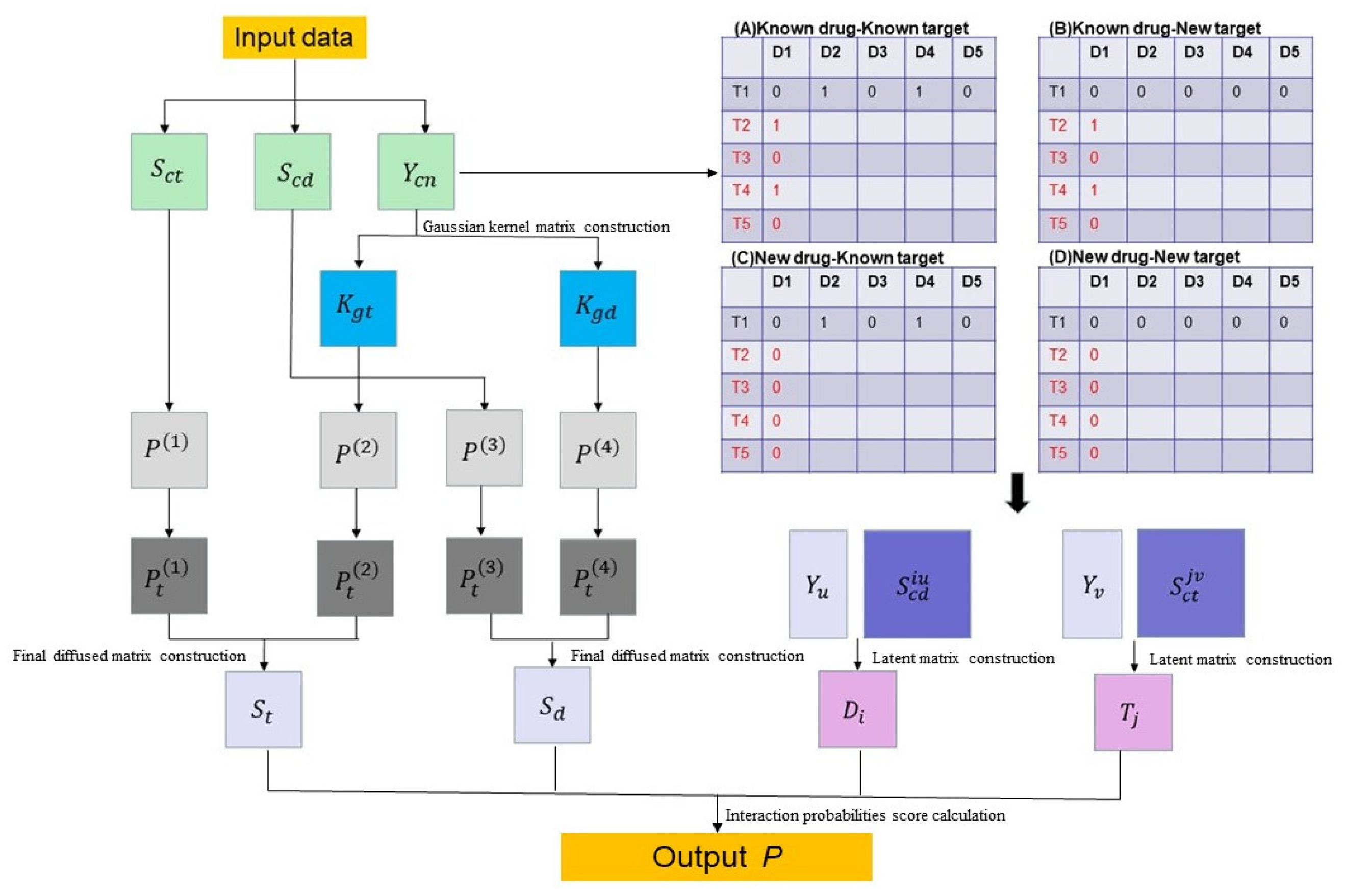
2.1. Principle of the DNILMF
2.1.1. Data Preparation
2.1.2. Definition
2.1.3. Latent Matrix and Gaussian Kernel Matrix Construction
2.1.4. Final Diffused Matrix Construction
2.1.5. Interaction Probabilities Score Calculation
2.2. RotatE
2.2.1. Three Relation Patterns Definition
2.2.2. Embeddings Optimization
2.2.3. Score Function Definition
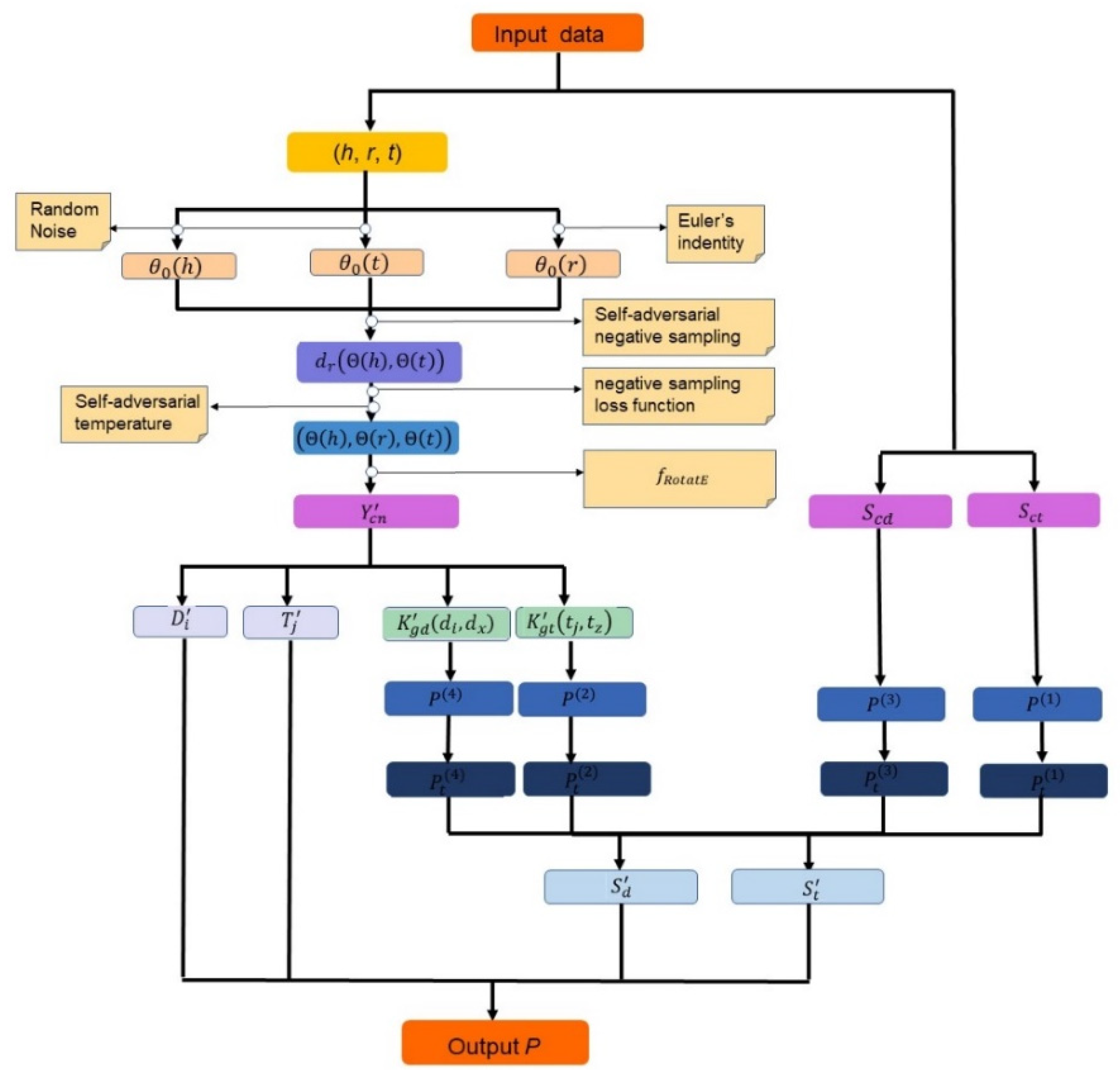
3. Our Proposed Prediction Scheme
3.1. Architecture
Data Preparation
3.2. Interaction Adjacency Matrix Construction with RotatE
3.2.1. Embedding Initialization
3.2.2. Embedding Optimization
3.2.3. Interaction Adjacency Matrix Construction
3.3. Predicting DTI with DNILMF
3.3.1. Latent Variable Matrix and Gaussian Kernel Matrix Construction
3.3.2. Final Diffused Matrix Construction
3.3.3. Interaction Probability Calculation
4. Experimental Results
4.1. Data Preparation and Experimental Settings
4.1.1. Dataset Preparation
4.1.2. Experimental Environment
4.2. Results and Discussion
4.2.1. Parameter Setting of Ro-DNILMF
4.2.2. The Optimization Function Determination of Ro-DNILMF
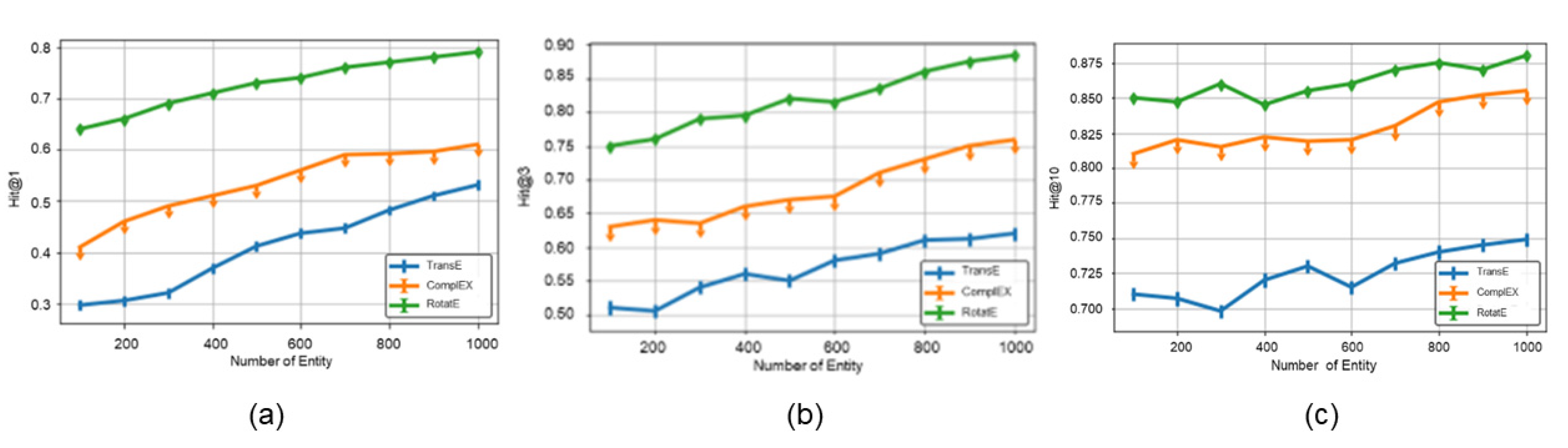
4.2.3. Performance of the Score Function in Ro-DNILMF
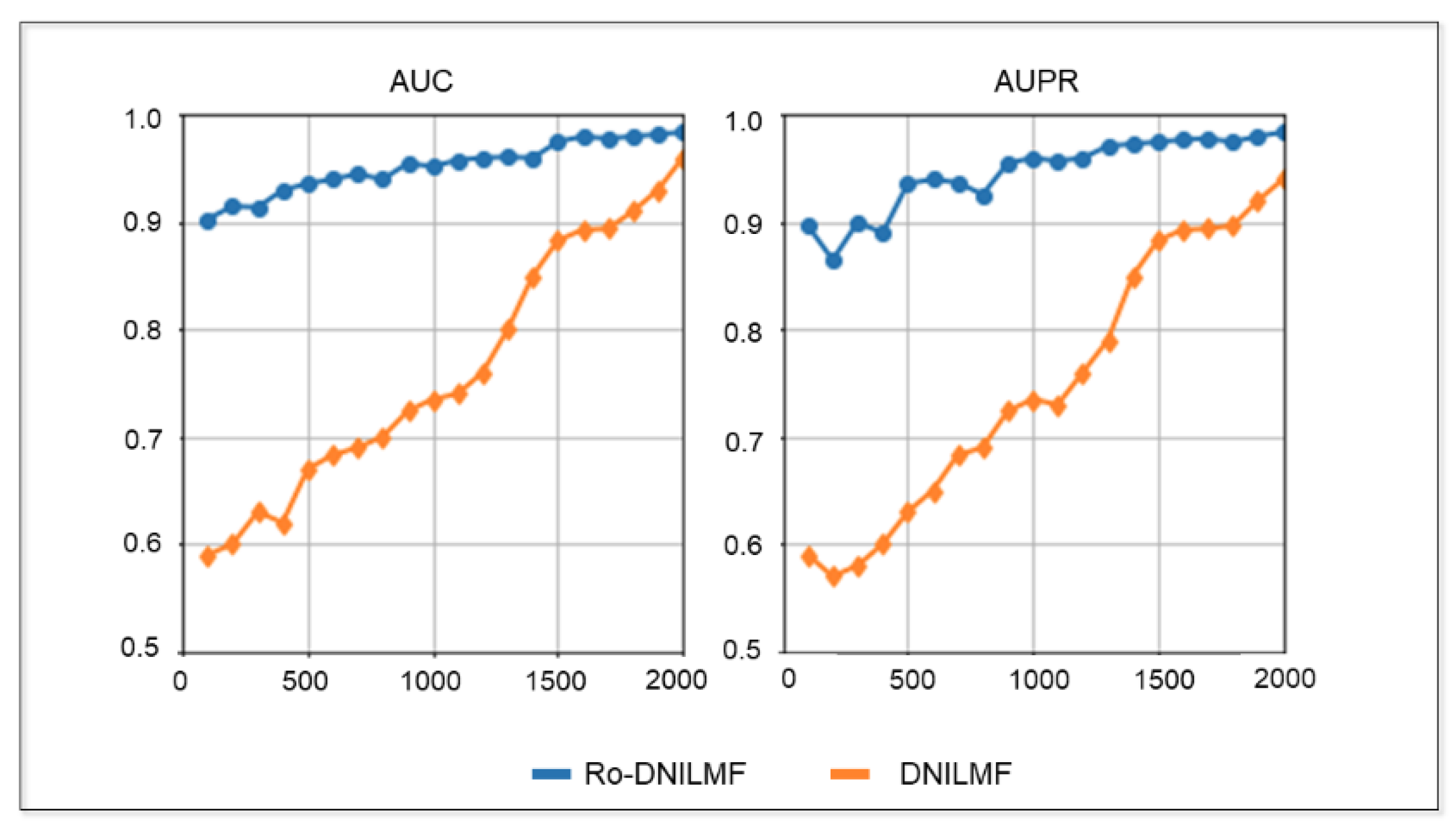
4.2.4. Performance of Ro-DNILMF under Different Samples
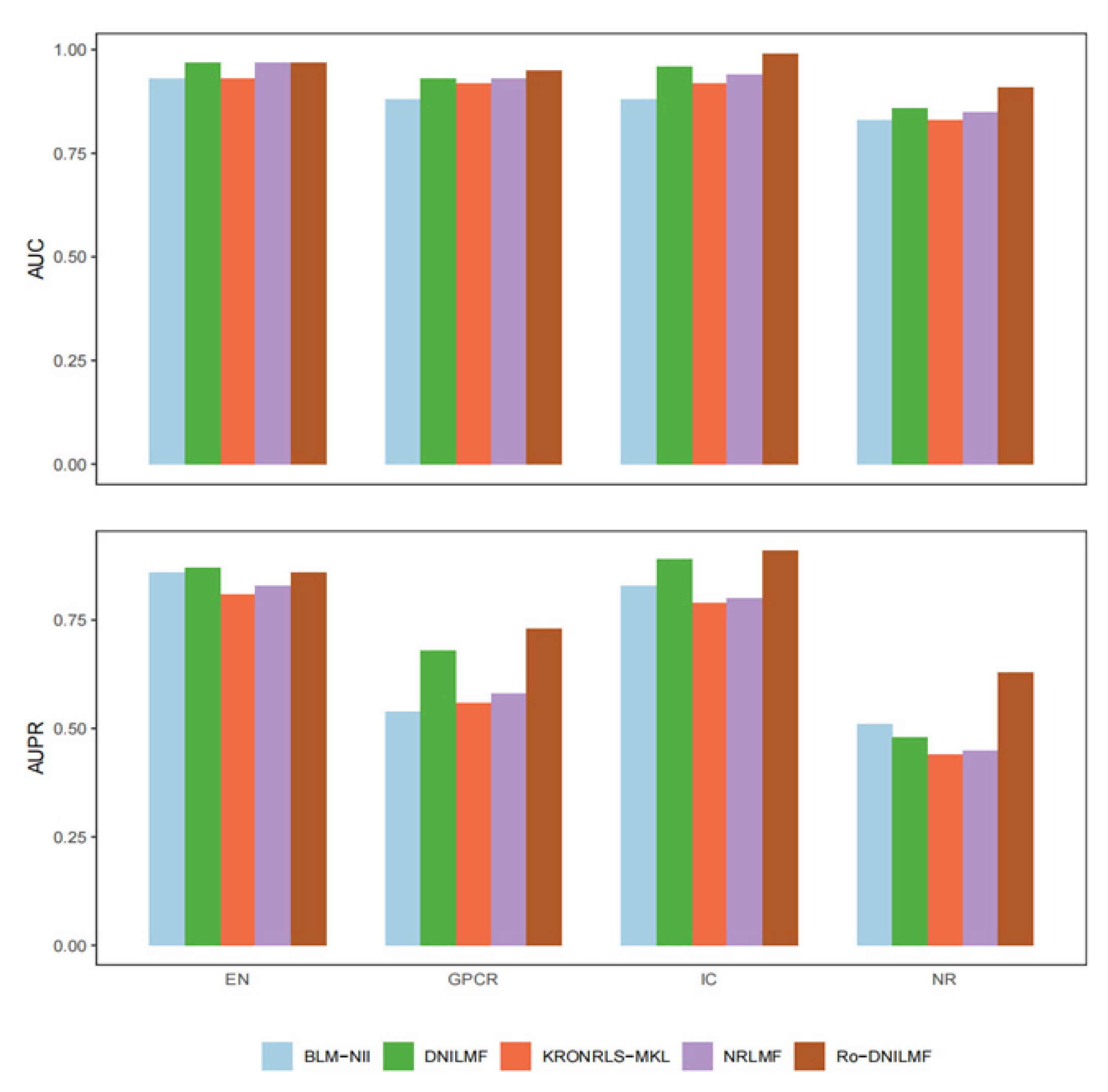
4.2.5. Comparison with Other Mainstream Methods
4.2.6. Comparison with Other Combination Models
5. Conclusions and Future Work
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jourdan, J.-P.; Bureau, R.; Rochais, C.; Dallemagne, P. Drug repositioning: A brief overview. J. Pharm. Pharmacol. 2020, 72, 1145–1151. [Google Scholar] [CrossRef] [PubMed]
- Popovic, G.; Kirby, N.C.; Dement, T.C.; Peterson, K.M.; Daub, C.E.; Belcher, H.A.; Guthold, M.; Offenbacher, A.R.; Hudson, N.E. Development of Transient Recombinant Expression and Affinity Chromatography Systems for Human Fibrinogen. IJMS 2022, 23, 1054. [Google Scholar] [CrossRef] [PubMed]
- Riggs, A.D. Making, Cloning, and the Expression of Human Insulin Genes in Bacteria: The Path to Humulin. Endocr. Rev. 2021, 42, 374–380. [Google Scholar] [CrossRef]
- Zhang, P.; Wei, Z.; Che, C.; Jin, B. DeepMGT-DTI: Transformer network incorporating multilayer graph information for Drug–Target interaction prediction. Comput. Biol. Med. 2022, 142, 105214. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Zhong, W.; Zhao, L.; Chen, C.Y.-C. ML-DTI: Mutual Learning Mechanism for Interpretable Drug–Target Interaction Prediction. J. Phys. Chem. Lett. 2021, 12, 4247–4261. [Google Scholar] [CrossRef]
- Mei, J.-P.; Kwoh, C.-K.; Yang, P.; Li, X.-L.; Zheng, J. Drug–target interaction prediction by learning from local information and neighbors. Bioinformatics 2013, 29, 238–245. [Google Scholar] [CrossRef]
- Jung, Y.-S.; Kim, Y.; Cho, Y.-R. Comparative Analysis of Network-Based Approaches and Machine Learning Algorithms for Predicting Drug-Target Interactions. Methods 2022, 198, 19–31. [Google Scholar] [CrossRef]
- Hao, M.; Wang, Y.; Bryant, S.H. Improved prediction of drug-target interactions using regularized least squares integrating with kernel fusion technique. Anal. Chim. Acta 2016, 909, 41–50. [Google Scholar] [CrossRef]
- Liu, Y.; Wu, M.; Miao, C.; Zhao, P.; Li, X.-L. Neighborhood Regularized Logistic Matrix Factorization for Drug-Target Interaction Prediction. PLoS Comput. Biol. 2016, 12, e1004760. [Google Scholar] [CrossRef]
- Hao, M.; Bryant, S.H.; Wang, Y. Predicting drug-target interactions by dual-network integrated logistic matrix factorization. Sci. Rep. 2017, 7, 40376. [Google Scholar] [CrossRef]
- Chu, Y.-Y.; Zhang, Y.-F.; Wang, W.; Wang, X.-G.; Shan, X.-Q.; Xiong, Y.; Wei, D.-Q. DTI-CDF: A CDF model towards the prediction of DTIs based on hybrid features. Bioinformatics 2019, 198, 19–31. [Google Scholar] [CrossRef]
- Chu, Y.; Shan, X.; Chen, T.; Jiang, M.; Wang, Y.; Wang, Q.; Salahub, D.R.; Xiong, Y.; Wei, D.-Q. DTI-MLCD: Predicting drug-target interactions using multi-label learning with community detection method. Brief. Bioinform. 2021, 22, bbaa205. [Google Scholar] [CrossRef] [PubMed]
- Olayan, R.S.; Ashoor, H.; Bajic, V.B. DDR: Efficient computational method to predict drug–target interactions using graph mining and machine learning approaches. Bioinformatics 2018, 34, 3779. [Google Scholar] [CrossRef] [PubMed]
- Soh, J.; Park, S.; Lee, H. HIDTI: Integration of heterogeneous information to predict drug-target interactions. Sci. Rep. 2022, 12, 3793. [Google Scholar] [CrossRef]
- Mohamed, S.K.; Nováček, V.; Nounu, A. Discovering Protein Drug Targets Using Knowledge Graph Embeddings. Bioinformatics 2020, 36, 603–610. [Google Scholar] [CrossRef]
- Sun, Z.; Deng, Z.-H.; Nie, J.-Y.; Tang, J. RotatE: Knowledge Graph Embedding by Relational Rotation in Complex Space. In Proceedings of the International Conference on Learning Representations, New Orleans, LA, USA, 6–9 May 2019. [Google Scholar]
- K., F.M.; Mohan, M. Ensemble Learning Models for Drug Target Interaction Prediction. In Proceedings of the 2022 International Conference on Applied Artificial Intelligence and Computing (ICAAIC), Salem, India, 9–11 May 2022; IEEE: Salem, India, 2022; pp. 138–143. [Google Scholar]
- Tang, C.; Zhong, C.; Chen, D.; Wang, J. Drug-target interactions prediction using marginalized denoising model on heterogeneous networks. BMC Bioinform. 2020, 21, 330. [Google Scholar] [CrossRef]
- Cheng, L.; YuanFei, T.; Gu, S.; Zheng, Y.; Yang, X.; Li, C.; Ke, Y.; Hu, J. Pattern matching of alarm sequences by using an improved Smith-Waterman algorithm. In Proceedings of the Third International Conference on Electronics and Communication; Network and Computer Technology (ECNCT 2021), Harbin, China, 7 March 2022; Mohiddin, M.K., Chen, S., EL-Zoghdy, S.F., Eds.; SPIE: Harbin, China, 2022; p. 106. [Google Scholar]
- Vila-Santa, A.; Islam, M.A.; Ferreira, F.C.; Prather, K.L.J.; Mira, N.P. Prospecting Biochemical Pathways to Implement Microbe-Based Production of the New-to-Nature Platform Chemical Levulinic Acid. ACS Synth. Biol. 2021, 10, 724–736. [Google Scholar] [CrossRef]
- Wang, B.; Mezlini, A.M.; Demir, F.; Fiume, M.; Tu, Z.; Brudno, M.; Haibe-Kains, B.; Goldenberg, A. Similarity network fusion for aggregating data types on a genomic scale. Nat. Methods 2014, 11, 333–337. [Google Scholar] [CrossRef]
- Denessen, E.J.S.; Heuts, S.; Daemen, J.H.T.; Vroemen, W.H.M.; Sels, J.W.; Segers, P.; Van ’T Hof, A.W.J.; Maessen, J.G.; Bekers, O.; Van Der Horst, I.C.C.; et al. High-sensitivity cardiac troponin I and T kinetics after coronary artery bypass grafting in relation to current definitions of myocardial infarction: A systematic review and meta-anal. Eur. Heart J. 2021, 42, ehab724.2251. [Google Scholar] [CrossRef]
- Aradnia, A.; Haeri, M.A.; Ebadzadeh, M.M. Adaptive Explicit Kernel Minkowski Weighted K-means. Inf. Sci. 2022, 584, 503–518. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Kawashima, M. KEGG mapping tools for uncovering hidden features in biological data. Protein Sci. 2022, 31, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Xian, W.; Li, X.; Chen, Y.; Geng, J.; Wang, Q.; Gao, Q.; Tang, B.; Wang, H.; Kang, P. Mechanisms of Quercetin against atrial fibrillation explored by network pharmacology combined with molecular docking and experimental validation. Sci. Rep. 2022, 12, 9777. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Hsieh, C.-Y.; Yang, Z.; Kang, Y.; Chen, J.; Cao, D.; He, S.; Hou, T. A unified drug–target interaction prediction framework based on knowledge graph and recommendation system. Nat. Commun. 2021, 12, 6775. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, J.; Bian, N. DNILMF-LDA: Prediction of lncRNA-Disease Associations by Dual-Network Integrated Logistic Matrix Factorization and Bayesian Optimization. Genes 2019, 10, 608. [Google Scholar] [CrossRef] [PubMed]
- Bordes, A.; Usunier, N.; Garcia-Duran, A.; Weston, J.; Yakhnenko, O. Translating embeddings for modeling multi-relational data. Adv. Neural Inf. Process. Syst. 2013, 2, 2787–2795. [Google Scholar]
- Trouillon, T.; Welbl, J.; Riedel, S.; Gaussier, É.; Bouchard, G. Complex embeddings for simple link prediction. In Proceedings of the International Conference on Machine Learning, PMLR, New York, NY, USA, 20–22 June 2016; pp. 2071–2080. [Google Scholar]
- Liu, S.; An, J.; Zhao, J.; Zhao, S.; Lv, H.; Wang, S. Drug-Target Interaction Prediction Based on Multisource Information Weighted Fusion. Contrast Media Mol. Imaging 2021, 2021, 1–10. [Google Scholar] [CrossRef]
- Bagherian, M.; Kim, R.B.; Jiang, C.; Sartor, M.A.; Derksen, H.; Najarian, K. Coupled matrix–matrix and coupled tensor–matrix completion methods for predicting drug–target interactions. Brief. Bioinform. 2021, 22, 2161–2171. [Google Scholar] [CrossRef]
- Alshahrani, M.; Almansour, A.; Alkhaldi, A.; Thafar, M.A.; Uludag, M.; Essack, M.; Hoehndorf, R. Combining biomedical knowledge graphs and text to improve predictions for drug-target interactions and drug-indications. PeerJ 2022, 10, e13061. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, P.; Qiu, N.; Yang, D. A Knowledge Representation Method of Joint Entity and Neighbor Information. In Proceedings of the 2021 International Conference on Electronic Information Engineering and Computer Science (EIECS), Changchun, China, 23–26 September 2021; IEEE: Changchun, China, 2021; pp. 168–171. [Google Scholar]
- Zhang, S.; Tay, Y.; Yao, L.; Liu, Q. Quaternion Knowledge Graph Embeddings. In Proceedings of the 33rd International Conference on Neural Information Processing Systems, Vancouver, BC, Canada, 8–14 December 2019; Curran Associates Inc.: Red Hook, NY, USA, 2019. [Google Scholar]
- Li, J.; Li, A.; Liu, T. Feature Interaction Convolutional Network for Knowledge Graph Embedding. In Knowledge Science, Engineering and Management; Qiu, H., Zhang, C., Fei, Z., Qiu, M., Kung, S.-Y., Eds.; Lecture Notes in Computer Science; Springer International Publishing: Cham, Switzerland, 2021; Volume 12815, pp. 369–380. ISBN 978-3-030-82135-7. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset | Group | Drugs | Proteins | DTIs |
|---|---|---|---|---|
| Yamanishi_08 | EN | 445 | 664 | 2926 |
| IC | 210 | 204 | 1476 | |
| GPCR | 223 | 95 | 635 | |
| NR | 54 | 26 | 90 | |
| ALL | 932 | 989 | 5127 | |
| KEGG | - | 10,979 | 13,959 | 12,112 |
| DrugBank | - | 1482 | 1408 | 9881 |
| Parameter | Value | |||||
|---|---|---|---|---|---|---|
| k | 125 | 250 | 500 | 750 | 1000 | - |
| b | 128 | 256 | 512 | 1024 | 2048 | - |
| 3 | 6 | 9 | 12 | 24 | 30 | |
| 0.5 | 0.6 | 0.7 | 0.8 | 0.9 | 1 | |
| 0.25 | 0.2 | 0.15 | 0.1 | 0.05 | 0 | |
| 0.25 | 0.2 | 0.15 | 0.1 | 0.05 | 0 | |
| K | 5 | 6 | 7 | 8 | 9 | 10 |
| Sigmoid | Tanh | |||||||
|---|---|---|---|---|---|---|---|---|
| MRR | H@1 | H@3 | H@10 | MRR | H@1 | H@3 | H@10 | |
| EN | 0.683 | 0.490 | 0.524 | 0.741 | 0.692 | 0.537 | 0.547 | 0.732 |
| IC | 0.359 | 0.512 | 0.537 | 0.817 | 0.467 | 0.564 | 0.582 | 0.861 |
| GPCR | 0.723 | 0.509 | 0.518 | 0.803 | 0.743 | 0.521 | 0.539 | 0.884 |
| NR | 0.436 | 0.427 | 0.469 | 0.654 | 0.584 | 0.433 | 0.486 | 0.747 |
| Metrics | Embedding Model | PredictingModel | EN | IC | GPCR | NR |
|---|---|---|---|---|---|---|
| AUPR | - - | NRLMF | 0.812 | 0.785 | 0.556 | 0.449 |
| DNILMF | 0.869 | 0.887 | 0.684 | 0.483 | ||
| Ro-DNILMF | 0.863 | 0.912 | 0.726 | 0.625 | ||
| TransE | NRLMF | 0.816 | 0.789 | 0.560 | 0.478 | |
| DNILMF | 0.873 | 0.889 | 0.684 | 0.535 | ||
| DisMult | NRLMF | 0.815 | 0.786 | 0.574 | 0.497 | |
| DNILMF | 0.872 | 0.893 | 0.687 | 0.530 | ||
| HolE | NRLMF | 0.813 | 0.795 | 0.587 | 0.513 | |
| DNILMF | 0.889 | 0.903 | 0.695 | 0.573 | ||
| ComplEx | NRLMF | 0.824 | 0.793 | 0.593 | 0.510 | |
| DNILMF | 0.886 | 0.904 | 0.703 | 0.542 | ||
| ConvE | NRLMF | 0.818 | 0.814 | 0.609 | 0.526 | |
| DNILMF | 0.873 | 0.903 | 0.721 | 0.568 | ||
| pRotatE | NRLMF | 0.820 | 0.817 | 0.614 | 0.523 | |
| DNILMF | 0.886 | 0.905 | 0.715 | 0.567 | ||
| RotatE | NRLMF | 0.823 | 0.826 | 0.627 | 0.527 | |
| DNILMF | 0.860 | 0.908 | 0.724 | 0.590 | ||
| AUC | - | NRLMF | 0.966 | 0.943 | 0.930 | 0.851 |
| DNILMF | 0.971 | 0.962 | 0.933 | 0.856 | ||
| Ro-DNILMF | 0.967 | 0.986 | 0.945 | 0.913 | ||
| TransE | NRLMF | 0.966 | 0.944 | 0.930 | 0.859 | |
| DNILMF | 0.968 | 0.964 | 0.933 | 0.854 | ||
| DisMult | NRLMF | 0.965 | 0.947 | 0.932 | 0.864 | |
| DNILMF | 0.968 | 0.963 | 0.934 | 0.867 | ||
| HolE | NRLMF | 0.966 | 0.946 | 0.931 | 0.873 | |
| DNILMF | 0.971 | 0.975 | 0.936 | 0.875 | ||
| ComplEx | NRLMF | 0.967 | 0.969 | 0.940 | 0.884 | |
| DNILMF | 0.972 | 0.982 | 0.938 | 0.891 | ||
| ConvE | NRLMF | 0.965 | 0.974 | 0.939 | 0.873 | |
| DNILMF | 0.970 | 0.979 | 0.937 | 0.886 | ||
| pRotatE | NRLMF | 0.964 | 0.981 | 0.938 | 0.896 | |
| DNILMF | 0.971 | 0.982 | 0.939 | 0.893 | ||
| RotatE | NRLMF | 0.970 | 0.982 | 0.939 | 0.901 | |
| DNILMF | 0.969 | 0.984 | 0.940 | 0.903 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Yang, X.; Guan, Y.; Pan, Z. Prediction of Drug–Target Interaction Using Dual-Network Integrated Logistic Matrix Factorization and Knowledge Graph Embedding. Molecules 2022, 27, 5131. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27165131
Li J, Yang X, Guan Y, Pan Z. Prediction of Drug–Target Interaction Using Dual-Network Integrated Logistic Matrix Factorization and Knowledge Graph Embedding. Molecules. 2022; 27(16):5131. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27165131
Chicago/Turabian StyleLi, Jiaxin, Xixin Yang, Yuanlin Guan, and Zhenkuan Pan. 2022. "Prediction of Drug–Target Interaction Using Dual-Network Integrated Logistic Matrix Factorization and Knowledge Graph Embedding" Molecules 27, no. 16: 5131. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27165131