
mTORC2 Is the Major Second Layer Kinase Negatively Regulating FOXO3 Activity

 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Results
2.1. FOXO3 Accumulates in the Nucleus upon the Inhibition of Regulatory Kinases
2.2. Lack of Effect of MEK and ERK Inhibitors Is Not Cell Type Specific
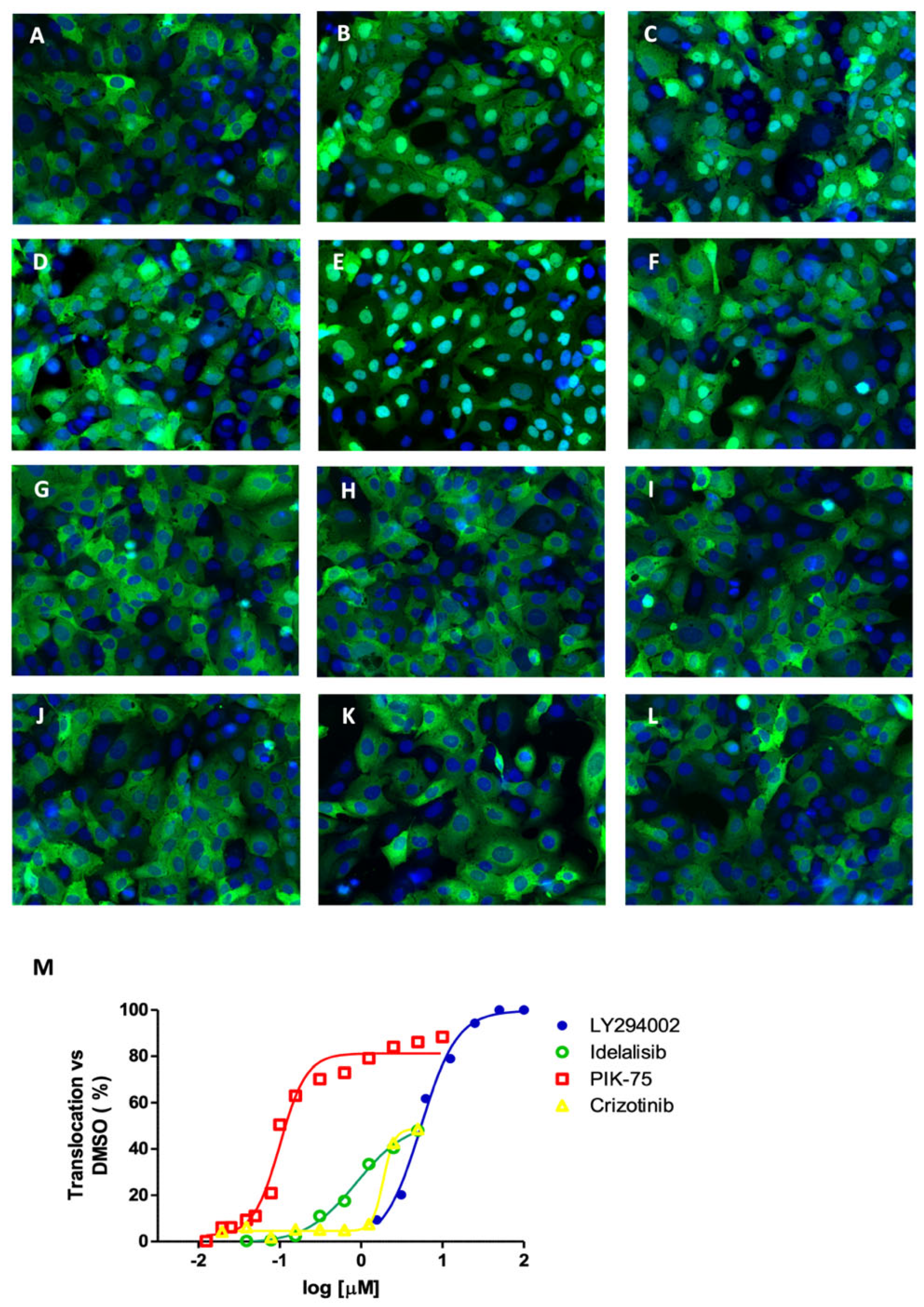
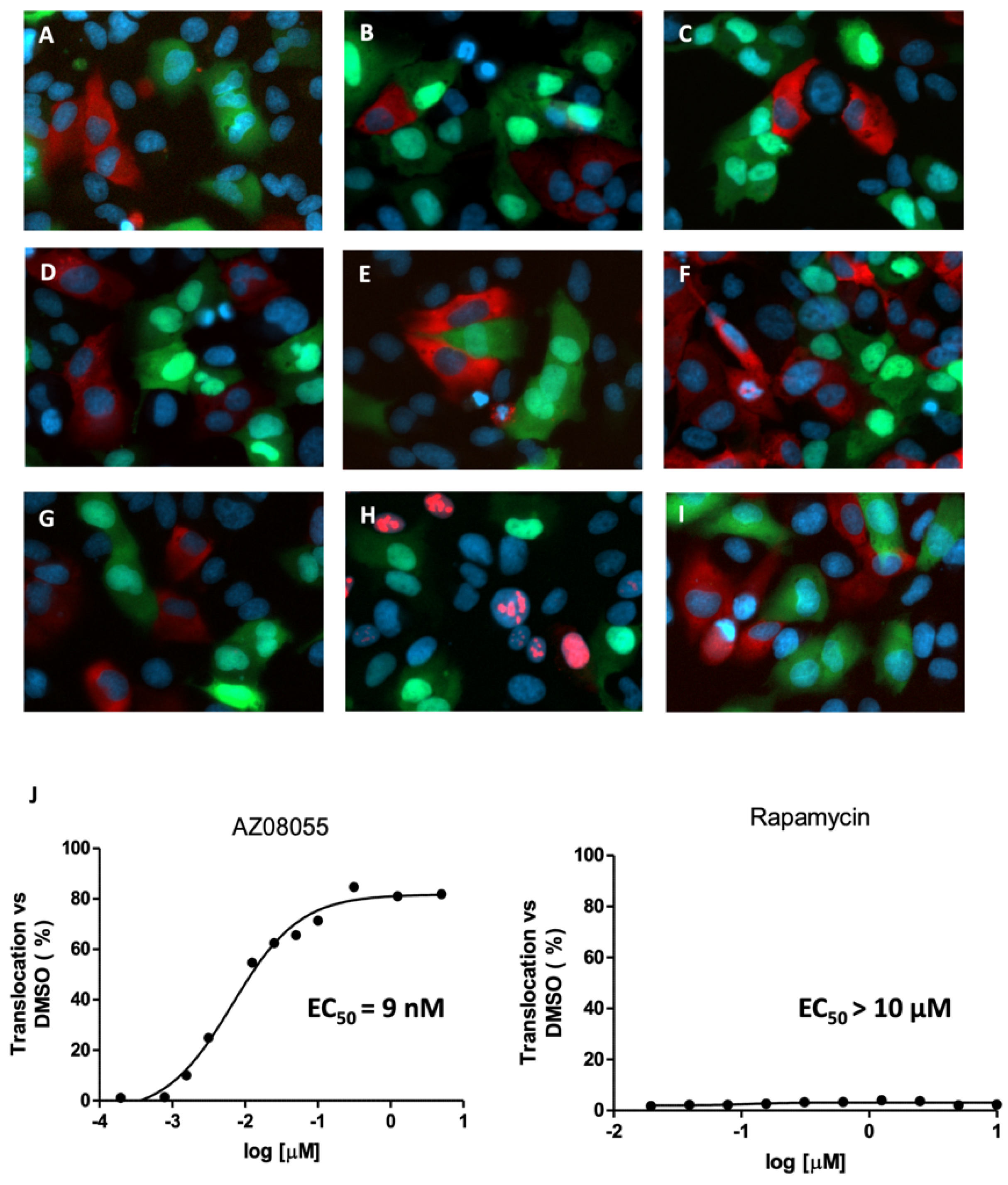
2.3. Inhibition of mTOR Kinase Induces Nuclear FOXO3 Accumulation
2.4. mTORC1 Inhibition Does Not Affect FOXO3 Localization
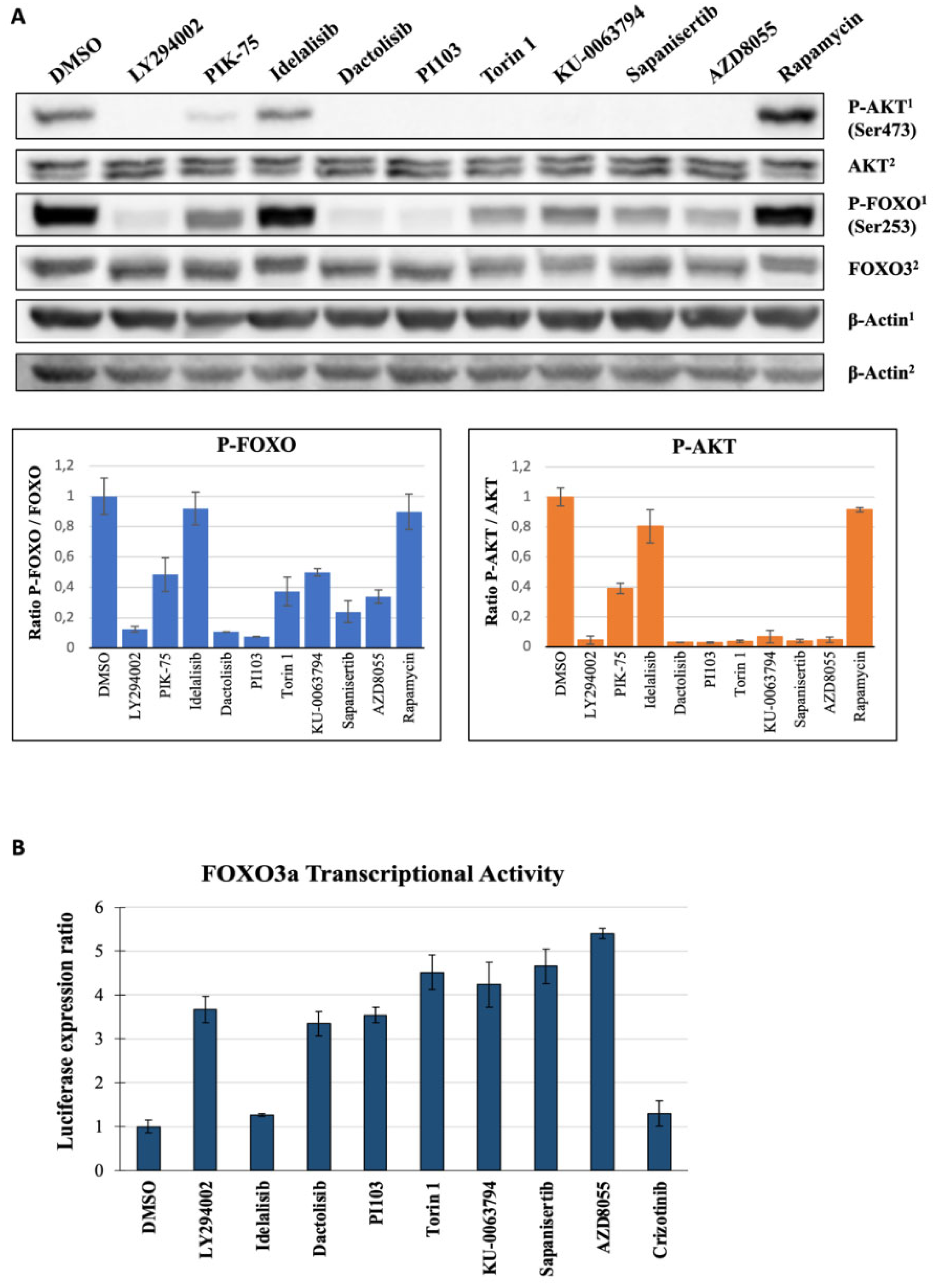
2.5. Second Layer Kinase Inhibitors Differentially Affect FOXO and AKT Phosphorylation
2.6. Inhibition of PI3K and mTOR Drive FOXO-Dependent Transcriptional Activity
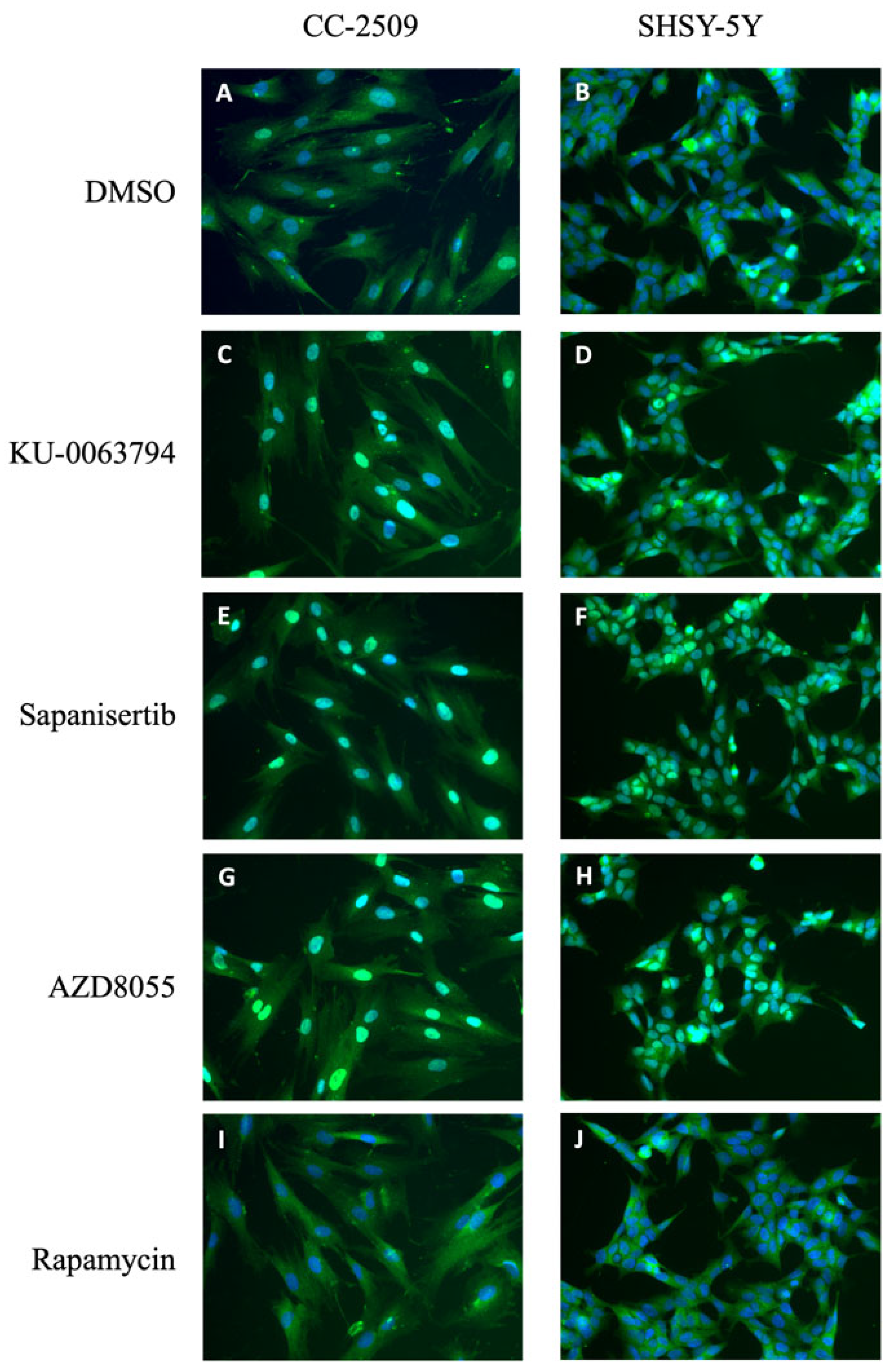
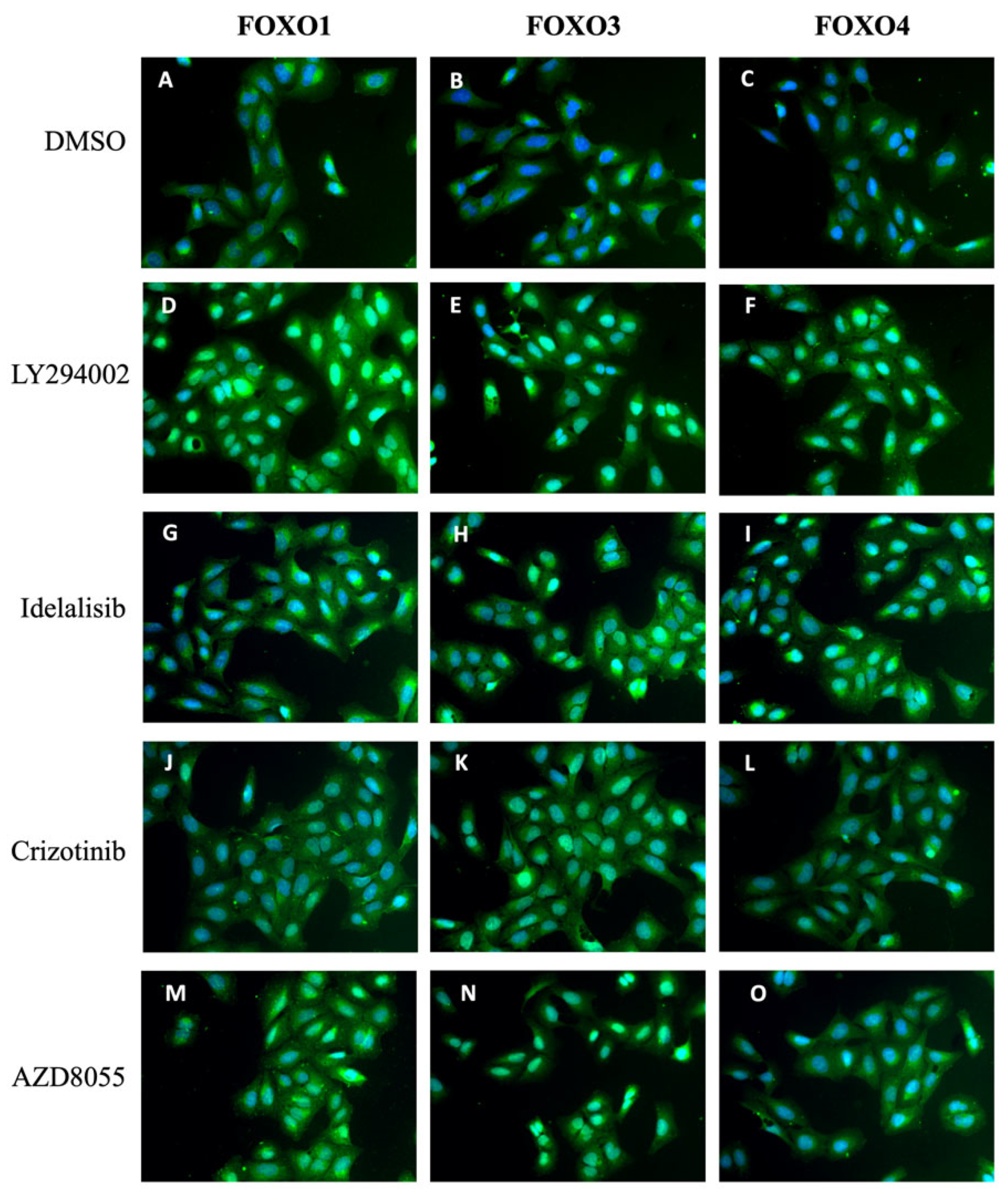
2.7. PI3K and mTOR Inhibition Affects the Localization of FOXO Isoforms FOXO1 and FOXO4
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. FOXO Translocation Assay in U2foxRELOC and U2redNES
4.3. EGF Induction
4.4. Dose-Response Assay in U2foxRELOC
4.5. Data Analysis
4.6. Immunofluorescence
4.7. Gene Reporter Assay
4.8. Western Blot Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Eijkelenboom, A.; Burgering, B.M. FOXOs: Signalling integrators for homeostasis maintenance. Nat. Rev. Mol. Cell Biol. 2013, 14, 83–97. [Google Scholar] [CrossRef]
- Myatt, S.S.; Lam, E.W.F. The emerging roles of forkhead box (Fox) proteins in cancer. Nat. Rev. Cancer 2007, 7, 847–859. [Google Scholar] [CrossRef]
- Link, W. Introduction to FOXO Biology. In FOXO Transcription Factors; Methods in Molecular Biology; Humana: New York, NY, USA, 2019; Volume 1890, pp. 1–9. [Google Scholar] [CrossRef]
- Link, W.; Fernandez-Marcos, P.J. FOXO transcription factors at the interface of metabolism and cancer. Int. J. Cancer 2017, 141, 2379–2391. [Google Scholar] [CrossRef]
- Dansen, T.B.; Burgering, B.M. Unravelling the tumor-suppressive functions of FOXO proteins. Trends Cell Biol. 2008, 18, 421–429. [Google Scholar] [CrossRef]
- Beretta, G.L.; Corno, C.; Zaffaroni, N.; Perego, P.; Beretta, G.L.; Corno, C.; Zaffaroni, N.; Perego, P. Role of FoxO Proteins in Cellular Response to Antitumor Agents. Cancers 2019, 11, 90. [Google Scholar] [CrossRef]
- van Doeselaar, S.; Burgering, B.M.T. FOXOs Maintaining the Equilibrium for Better or for Worse. In Current Topics in Developmental Biology; Academic Press Inc.: Cambridge, MA, USA, 2018; Volume 127, pp. 49–103. ISBN 9780128042533. [Google Scholar]
- Martins, R.; Lithgow, G.J.; Link, W. Long live FOXO: Unraveling the role of FOXO proteins in aging and longevity. Aging Cell 2016, 15, 196–207. [Google Scholar] [CrossRef]
- Calnan, D.R.; Brunet, A. The FoxO code. Oncogene 2008, 27, 2276–2288. [Google Scholar] [CrossRef]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 1999, 96, 857–868. [Google Scholar] [CrossRef]
- Essers, M.A.; Weijzen, S.; de Vries-Smits, A.M.; Saarloos, I.; de Ruiter, N.D.; Bos, J.L.; Burgering, B.M. FOXO transcription factor activation by oxidative stress mediated by the small GTPase Ral and JNK. EMBO J. 2004, 23, 4802–4812. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.Y.; Zong, C.S.; Xia, W.; Yamaguchi, H.; Ding, Q.; Xie, X.; Lang, J.Y.; Lai, C.C.; Chang, C.J.; Huang, W.C.; et al. ERK promotes tumorigenesis by inhibiting FOXO3a via MDM2-mediated degradation. Nat. Cell Biol. 2008, 10, 138–148. [Google Scholar] [CrossRef]
- Hu, M.C.T.; Lee, D.-F.; Xia, W.; Golfman, L.S.; Ou-Yang, F.; Yang, J.-Y.; Zou, Y.; Bao, S.; Hanada, N.; Saso, H.; et al. IkappaB kinase promotes tumorigenesis through inhibition of forkhead FOXO3a. Cell 2004, 117, 225–237. [Google Scholar] [CrossRef]
- Lehtinen, M.K.; Yuan, Z.; Boag, P.R.; Yang, Y.; Villé, J.; Becker, E.B.E.; Dibacco, S.; Ria De La Iglesia, N.; Gygi, S.; Keith Blackwell, T.; et al. A Conserved MST-FOXO Signaling Pathway Mediates Oxidative-Stress Responses and Extends Life Span. Cell 2006, 125, 987–1001. [Google Scholar] [CrossRef]
- Du, Z.; Lovly, C.M. Mechanisms of receptor tyrosine kinase activation in cancer. Mol. Cancer 2018, 17, 58. [Google Scholar] [CrossRef]
- Vanhaesebroeck, B.; Perry, M.W.D.; Brown, J.R.; André, F.; Okkenhaug, K. PI3K inhibitors are finally coming of age. Nat. Rev. Drug Discov. 2021, 20, 741–769. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef]
- Link, W.; Oyarzabal, J.; Serelde, B.G.; Albarran, M.I.; Rabal, O.; Cebriá, A.; Alfonso, P.; Fominaya, J.; Renner, O.; Peregrina, S.; et al. Chemical interrogation of FOXO3a nuclear translocation identifies potent and selective inhibitors of phosphoinositide 3-kinases. J. Biol. Chem. 2009, 284, 28392–28400. [Google Scholar] [CrossRef]
- Zanella, F.; Rosado, A.; Garcia, B.; Carnero, A.; Link, W. Chemical genetic analysis of FOXO nuclear-cytoplasmic shuttling by using image-based cell screening. Chembiochem 2008, 9, 2229–2237. [Google Scholar] [CrossRef] [PubMed]
- Chiabotto, G.; Grignani, G.; Todorovic, M.; Martin, V.; Centomo, M.L.; Prola, E.; Giordano, G.; Merlini, A.; Miglio, U.; Berrino, E.; et al. Pazopanib and Trametinib as a Synergistic Strategy against Osteosarcoma: Preclinical Activity and Molecular Insights. Cancers 2020, 12, 1519. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [Green Version]
- Van Der Heide, L.P.; Jacobs, F.M.J.; Burbach, J.P.H.; Hoekman, M.F.M.; Smidt, M.P. FoxO6 transcriptional activity is regulated by Thr26 and Ser184, independent of nucleo-cytoplasmic shuttling. Biochem. J. 2005, 391, 623–629. [Google Scholar] [CrossRef]
- Calissi, G.; Lam, E.W.F.; Link, W. Therapeutic strategies targeting FOXO transcription factors. Nat. Rev. Drug Discov. 2021, 20, 21–38. [Google Scholar] [CrossRef]
- Patel, M.N.; Halling-Brown, M.D.; Tym, J.E.; Workman, P.; Al-Lazikani, B. Objective assessment of cancer genes for drug discovery. Nat. Rev. Drug Discov. 2013, 12, 35–50. [Google Scholar] [CrossRef]
- Henley, M.J.; Koehler, A.N. Advances in targeting ‘undruggable’ transcription factors with small molecules. Nat. Rev. Drug Discov. 2021, 20, 669–688. [Google Scholar] [CrossRef] [PubMed]
- Hagenbuchner, J.; Obsilova, V.; Kaserer, T.; Kaiser, N.; Rass, B.; Psenakova, K.; Docekal, V.; Alblova, M.; Kohoutova, K.; Schuster, D.; et al. Modulating FOXO3 transcriptional activity by small, DBD-binding molecules. Elife 2019, 8, e48876. [Google Scholar] [CrossRef]
- Martini, M.; De Santis, M.C.; Braccini, L.; Gulluni, F.; Hirsch, E. PI3K/AKT signaling pathway and cancer: An updated review. Ann. Med. 2014, 46, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Chresta, C.M.; Davies, B.R.; Hickson, I.; Harding, T.; Cosulich, S.; Critchlow, S.E.; Vincent, J.P.; Ellston, R.; Jones, D.; Sini, P.; et al. AZD8055 is a potent, selective, and orally bioavailable ATP-competitive mammalian target of rapamycin kinase inhibitor with in vitro and in vivo antitumor activity. Cancer Res. 2010, 70, 288–298. [Google Scholar] [CrossRef]
- Serra, V.; Markman, B.; Scaltriti, M.; Eichhorn, P.J.A.; Valero, V.; Guzman, M.; Botero, M.L.; Llonch, E.; Atzori, F.; Di Cosimo, S.; et al. NVP-BEZ235, a Dual PI3K/mTOR Inhibitor, Prevents PI3K Signaling and Inhibits the Growth of Cancer Cells with Activating PI3K Mutations. Cancer Res. 2008, 68, 8022–8030. [Google Scholar] [CrossRef]
- Feehan, R.P.; Shantz, L.M. Negative regulation of the FOXO3a transcription factor by mTORC2 induces a pro-survival response following exposure to ultraviolet-B irradiation. Cell. Signal. 2016, 28, 798–809. [Google Scholar] [CrossRef]
- Zanella, F.; Rosado, A.; Blanco, F.; Henderson, B.R.; Carnero, A.; Link, W. An HTS approach to screen for antagonists of the nuclear export machinery using high content cell-based assays. Assay Drug Dev. Technol. 2007, 5, 333–341. [Google Scholar] [CrossRef]
- Zanella, F.; Rosado, A.; Garcia, B.; Carnero, A.; Link, W. Using multiplexed regulation of luciferase activity and GFP translocation to screen for FOXO modulators. BMC Cell Biol. 2009, 10, 14. [Google Scholar] [CrossRef]
- Kudo, N.; Matsumori, N.; Taoka, H.; Fujiwara, D.; Schreiner, E.P.; Wolff, B.; Yoshida, M.; Horinouchi, S. Leptomycin B inactivates CRM1/exportin 1 by covalent modification at a cysteine residue in the central conserved region. Proc. Natl. Acad. Sci. USA 1999, 96, 9112–9117. [Google Scholar] [CrossRef] [PubMed]
- Bouck, D.C.; Shu, P.; Cui, J.; Shelat, A.; Chen, T. A High-Content Screen Identifies Inhibitors of Nuclear Export of Forkhead Transcription Factors. J. Biomol. Screen. 2011, 16, 394–404. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, L.; Silva, A.; Calissi, G.; Grenho, I.; Monteiro, R.; Mayoral-Varo, V.; Blanco-Aparicio, C.; Pastor, J.; Bustos, V.; Bracher, F.; et al. Screening health-promoting compounds for their capacity to induce the activity of FOXO3. J. Gerontol. Ser. A 2022, 77, 1485–1493. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Inhibition Target | PubChem ID |
|---|---|---|
| Afatinib | EGFR, HER2 | 10184653 |
| AZD8055 | mTORC1/2 | 25262965 |
| Crizotinib | ALK, MET, ROS1 | 11626560 |
| Dactolisib (BEZ235) | PI3Kα/β/γ/δ, mTOR | 11977753 |
| Erlotinib | EGFR | 176870 |
| Idelalisib | PI3Kδ | 11625818 |
| KU-0063794 | mTORC1/2 | 16736978 |
| Lapatinib | EGFR, HER2 | 208908 |
| Lenvatinib | VEGFR1/2/3 | 9823820 |
| Leptomycin B | CRM1 | 6917907 |
| LY294002 | PI3K (pan-inhibitor) | 3973 |
| PI103 | PI3Kα/β/γ/δ, mTOR | 9884685 |
| PIK-75 | PI3Kα | 10275789 |
| Rapamycin | mTORC1 | 5284616 |
| Sapanisertib | mTORC1/2 | 45375953 |
| SCH772984 | ERK1/2 | 24866313 |
| Torin 1 | mTORC1/2 | 49836027 |
| Trametinib | MEK1/2 | 11707110 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jimenez, L.; Amenabar, C.; Mayoral-Varo, V.; Mackenzie, T.A.; Ramos, M.C.; Silva, A.; Calissi, G.; Grenho, I.; Blanco-Aparicio, C.; Pastor, J.; et al. mTORC2 Is the Major Second Layer Kinase Negatively Regulating FOXO3 Activity. Molecules 2022, 27, 5414. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27175414
Jimenez L, Amenabar C, Mayoral-Varo V, Mackenzie TA, Ramos MC, Silva A, Calissi G, Grenho I, Blanco-Aparicio C, Pastor J, et al. mTORC2 Is the Major Second Layer Kinase Negatively Regulating FOXO3 Activity. Molecules. 2022; 27(17):5414. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27175414
Chicago/Turabian StyleJimenez, Lucia, Carlos Amenabar, Victor Mayoral-Varo, Thomas A. Mackenzie, Maria C. Ramos, Andreia Silva, Giampaolo Calissi, Inês Grenho, Carmen Blanco-Aparicio, Joaquin Pastor, and et al. 2022. "mTORC2 Is the Major Second Layer Kinase Negatively Regulating FOXO3 Activity" Molecules 27, no. 17: 5414. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27175414