
Chemoenzymatic Synthesis of Select Intermediates and Natural Products of the Desferrioxamine E Siderophore Pathway
 , ,
, ,
Abstract
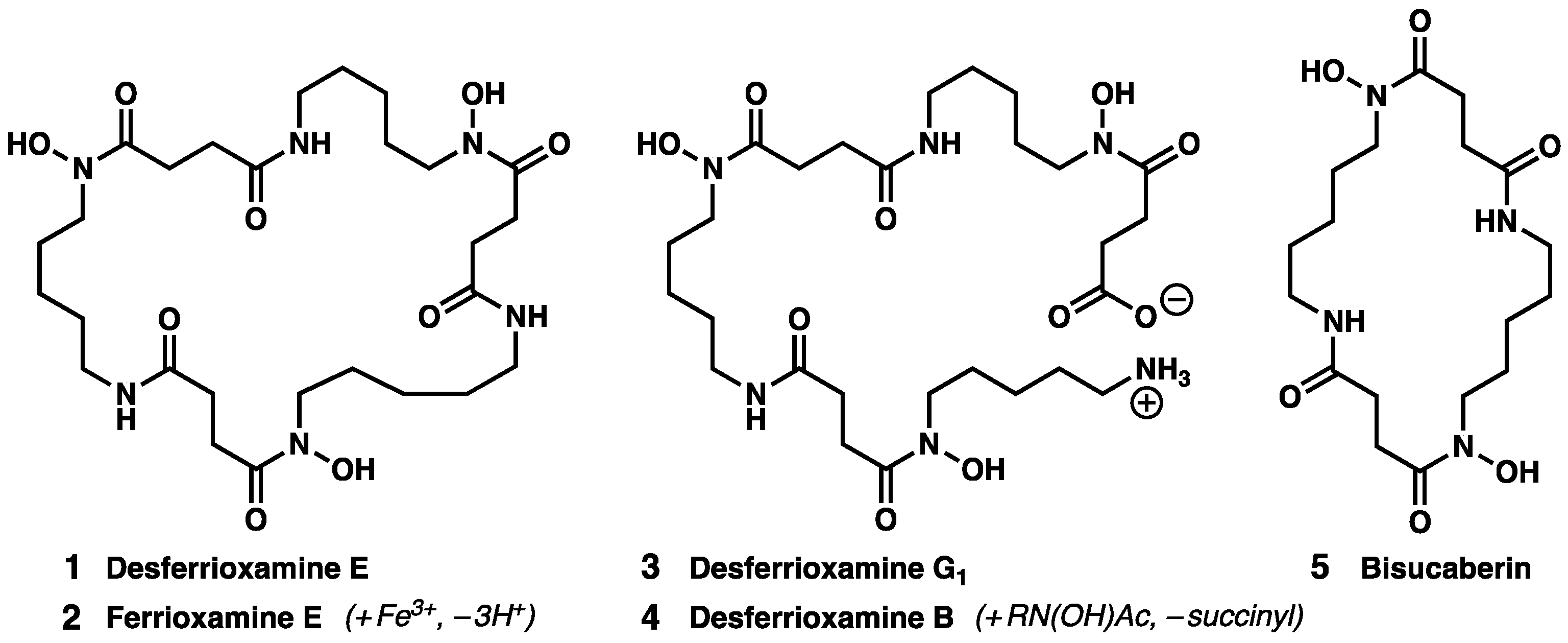
:1. Introduction
2. Results
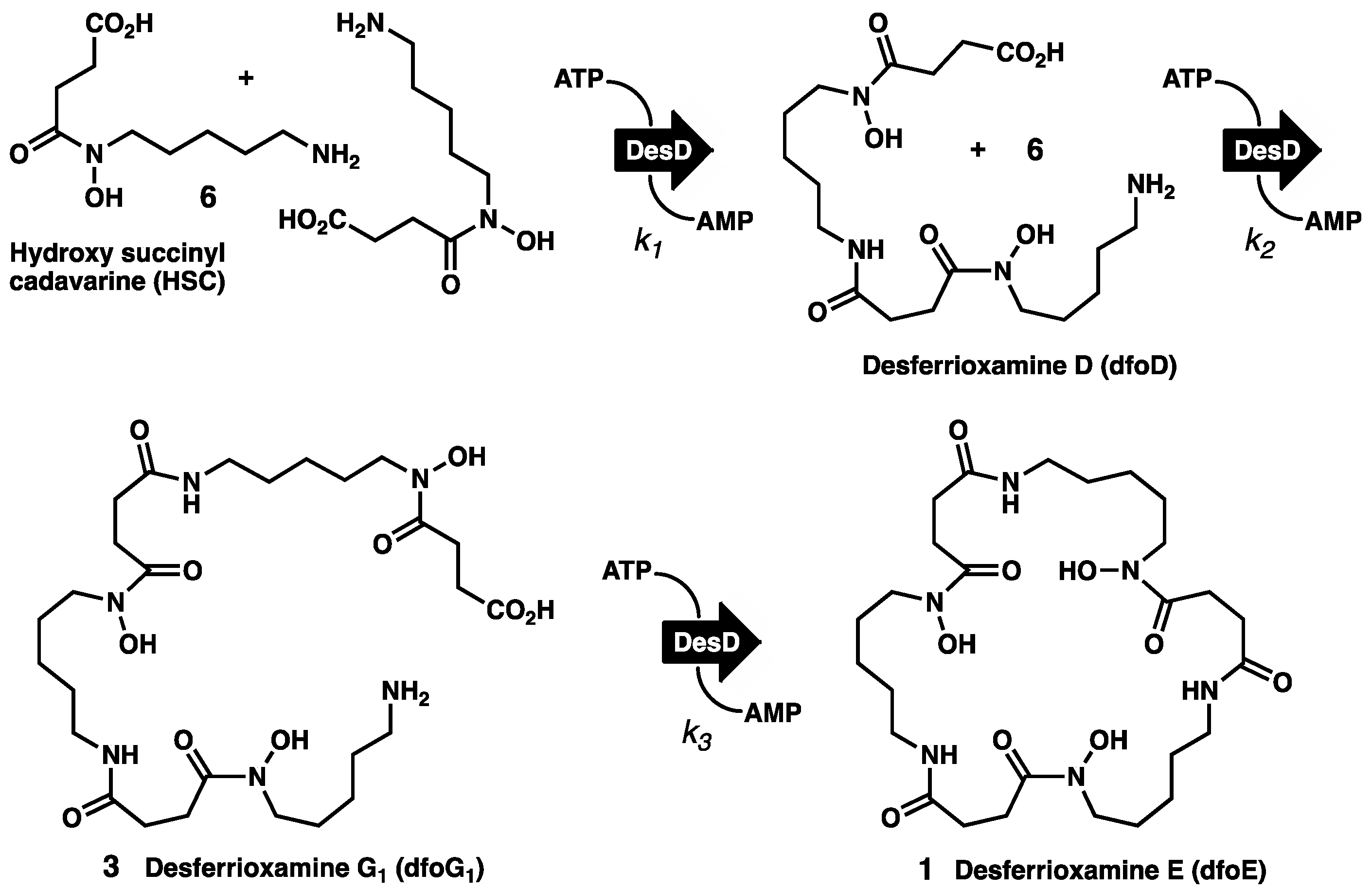
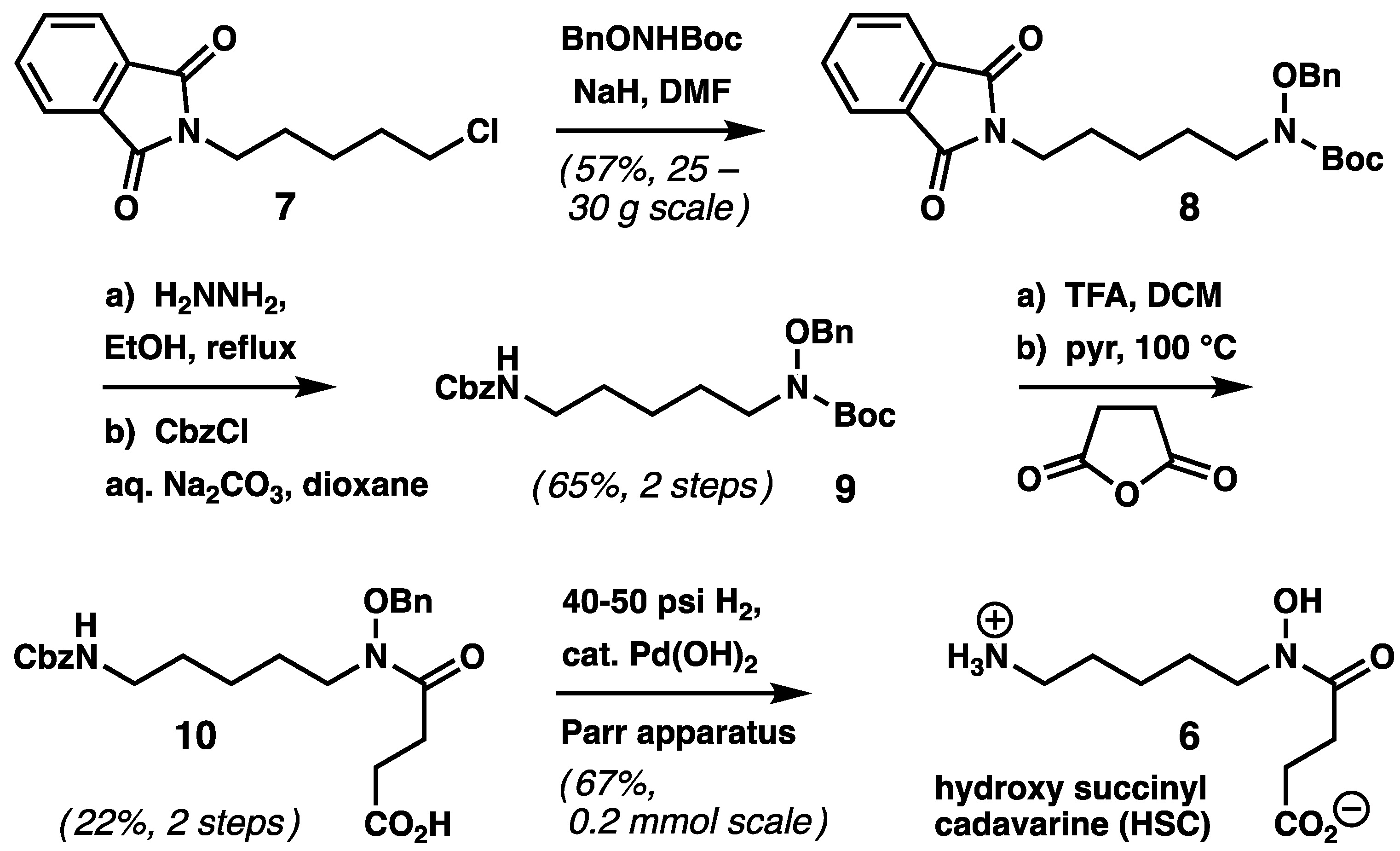
2.1. Chemical Synthesis of HSC
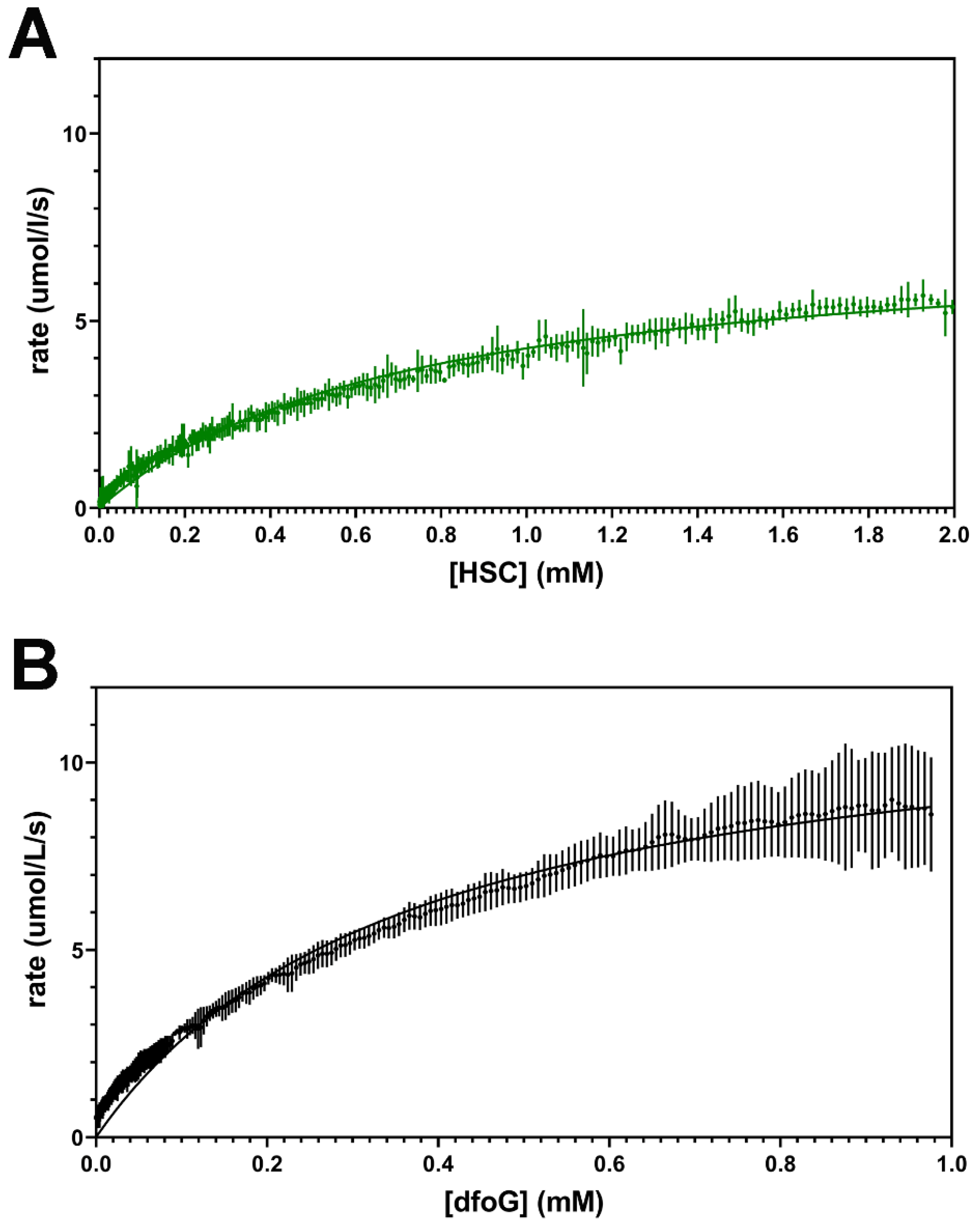
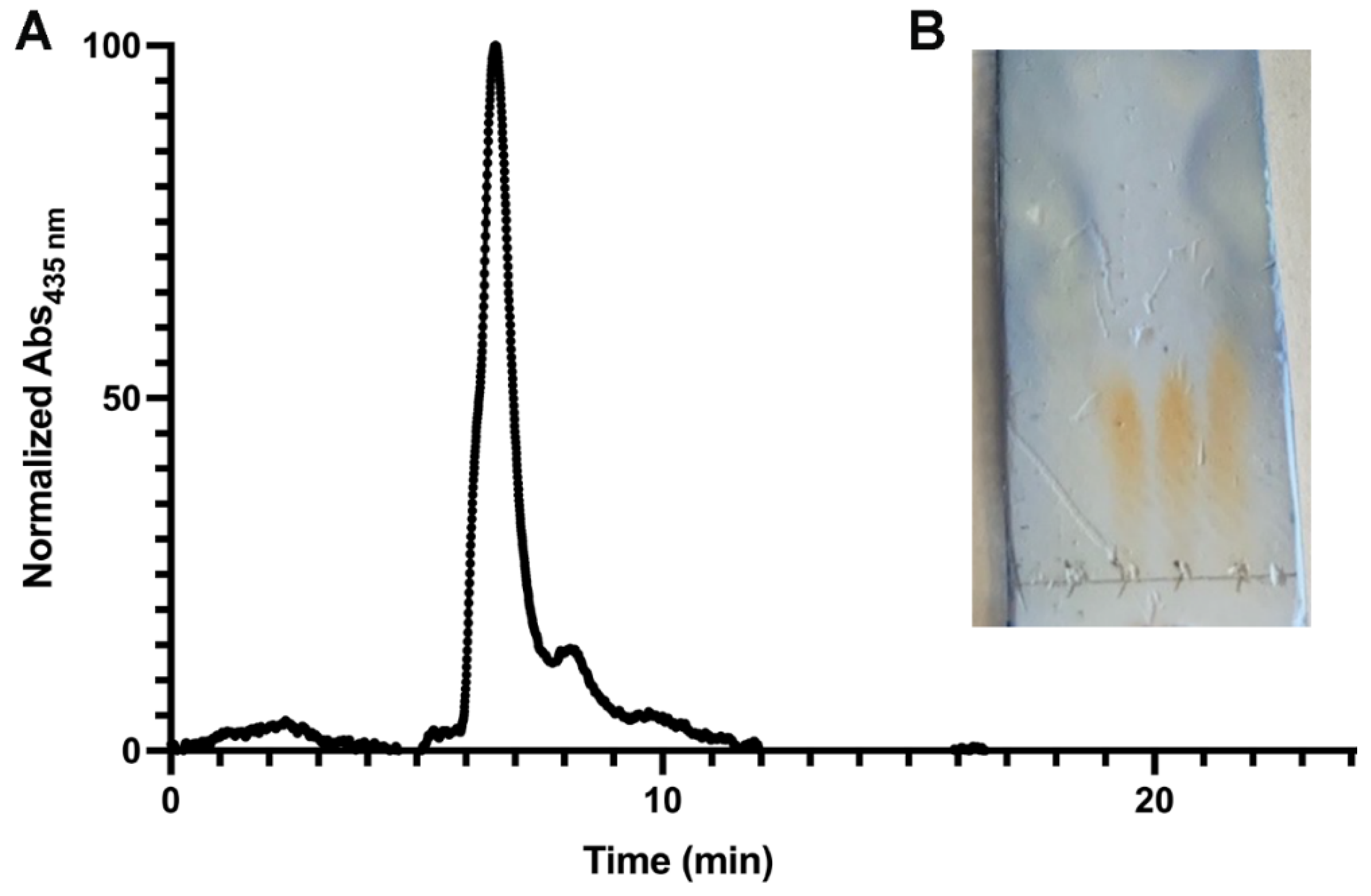
2.2. Enzymatic Synthesis of dfoE
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Neilands, J.B. Siderophores: Structure and function of microbial iron transport compounds. J. Biol. Chem. 1995, 270, 26723–26726. [Google Scholar] [CrossRef] [PubMed]
- Neilands, J.B. Siderophores. Arch. Biochem. Biophys. 1993, 302, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Braun, V.; Killman, H. Bacterial solutions to the iron-supply problem. Trends Biochem. Sci. 1999, 24, 104–109. [Google Scholar] [CrossRef]
- Barona-Gomez, F.; Lautru, S.; Francou, F.X.; Leblond, P.; Pernodet, J.L.; Challis, G.L. Multiple biosynthetic and uptake systems mediate siderophore-dependent iron acquisition in Streptomyces coelicolor A3(2) and Streptomyces ambofaciens ATCC 23877. Microbiology 2006, 152, 3355–3366. [Google Scholar] [CrossRef]
- Keller-Schierlein, W.; Prelog, V. Stoffwechselprodukte von Actinomyceten. Über das Ferrioxamine E; ein Beitrag zur Konstitution des Nocardamins. Helv. Chim. Acta 1961, 44, 244–245. [Google Scholar]
- Keller-Schierlein, W. Stoffwechselprodukte von Actinomyceten. Ferrioxamine G. Helv. Chim. Acta 1962, 45, 590–595. [Google Scholar] [CrossRef]
- Reissbrodt, R.; Rabsch, W.; Chapeaurouge, A.; Jung, G.; Winkelmann, G. Isolation and identification of ferrioxamine G and E in Hafnia alvei. Biol. Met. 1990, 3, 54–60. [Google Scholar] [CrossRef]
- Kameyama, T. Bisucaberin, a new siderophore, sensitizing tumor cells to macrophage-mediated cytolysis. I. Taxonomy of the producing organism, isolation, and biological properties. J. Antibiot. 1987, 40, 1664–1670. [Google Scholar] [CrossRef]
- Fujita, M.J.; Sakai, R. Production of avaroferrin and putrebactin by heterologous expression of a deep-sea metagenomic DNA. Mar. Drugs 2014, 12, 4799–4809. [Google Scholar] [CrossRef]
- Bellotti, D.; Remelli, M. Deferoxamine B: A natural, excellent, and versatile metal chelator. Molecules 2021, 26, 3255. [Google Scholar] [CrossRef]
- Kumar, A.; Yang, T.; Chakravorty, S.; Majumdar, A.; Nairn, B.M.; Six, D.A.; dos Santos, N.M.; Price, S.L.; Lawrenz, M.B.; Actis, L.A.; et al. Fluorescent sensors of siderophores produced by bacterial pathogens. J. Biol. Chem. 2022, 298, 101651–101667. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.; Gotsbacher, M.; Codd, R. Improved access to linear tetrameric hydroxamic acids with potential as radiochemical ligands for zirconium(IV)-89 PET imaging. Aust. J. Chem. 2020, 73, 969–978. [Google Scholar] [CrossRef]
- Patel, P.; Song, L.; Challis, G.L. Distinct extracytoplasmic siderophore binding proteins recognize ferrioxamines and ferricoelichelin in Streptomyces coelicolor A3(2). Biochemistry 2010, 49, 8033–8042. [Google Scholar] [CrossRef] [PubMed]
- Challis, G.L. A widely distributed bacterial pathway for siderophore biosynthesis independent of nonribosomal peptide synthetases. Chembiochem 2005, 6, 601–611. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, J.T.; Jeffery, E.F.; Shannon, J.D.; Ramakrishnan, G. Characterization of the siderophore of Francisella tularensis and role of fslA in siderophore production. J. Bacteriol. 2006, 188, 3785–3795. [Google Scholar] [CrossRef]
- Allred, B.E.; Correnti, C.; Clifton, M.C.; Strong, R.K.; Raymond, K.N. Siderocalin outwits the coordination chemistry of vibriobactin, a siderophore of Vibrio cholerae. ACS Chem. Biol. 2013, 8, 1882–1887. [Google Scholar] [CrossRef]
- Cendrowski, S.; MacArthur, W.; Hanna, P. Bacillus anthracis requires siderophore biosynthesis for growth in macrophages and mouse virulence. Mol. Microbiol. 2004, 51, 407–417. [Google Scholar] [CrossRef]
- Dale, S.E.; Doherty-Kirby, A.; Lajoie, G.; Heinrichs, D.E. Role of siderophore biosynthesis in virulence of Staphylococcus aureus: Identification and characterization of genes involved in production of a siderophore. Infect. Immun. 2004, 72, 29–37. [Google Scholar] [CrossRef]
- Franza, T.; Mahé, B.; Expert, D. Erwinia chrysanthemi requires a second iron transport route dependent of the siderophore achromobactin for extracellular growth and plant infection. Mol. Microbiol. 2005, 55, 261–275. [Google Scholar] [CrossRef]
- Koppisch, A.T.; Browder, C.C.; Moe, A.L.; Shelley, J.T.; Kinkel, B.A.; Hersman, L.E.; Iyer, S.; Ruggiers, C.E. Petrobactin is the primary siderophore synthesized by Bacillus anthracis str. Sterne under conditions of iron starvation. Biometals 2005, 18, 577–585. [Google Scholar] [CrossRef]
- Oves-Costales, D.; Kadi, N.; Challis, G.L. The long-overlooked enzymology of a nonribosomal peptide synthetase-independent pathway for virulence-conferring siderophore biosynthesis. Chem. Commun. 2009, 6530–6541. [Google Scholar] [CrossRef] [PubMed]
- Kadi, N.; Oves-Costales, D.; Barona-Gomez, F.; Challis, G.L. A new family of ATP-dependent oligomerization-macrocyclization biocatalysts. Nat. Chem. Biol. 2007, 3, 652–656. [Google Scholar] [CrossRef] [PubMed]
- Oves-Costales, D.; Kadi, N.; Fogg, M.J.; Song, L.; Wilson, K.S.; Challis, G.L. Petrobactin biosynthesis: AsbB catalyzes condensation of spermidine with N8-citryl-spermidine and its N1-(3,4-dihydroxybenzoyl) derivative. Chem. Commun. 2008, 4034–4036. [Google Scholar] [CrossRef] [PubMed]
- Schmelz, S.; Kadi, N.; McMahon, S.A.; Song, L.; Oves-Costales, D.; Oke, M.; Liu, H.; Johnson, K.A.; Carter, L.G.; Botting, C.H.; et al. AcsD catalyzes enantioselective citrate desymmetrization in siderophore biosynthesis. Nat. Chem. Biol. 2009, 5, 174–182. [Google Scholar] [CrossRef]
- Schmelz, S.; Botting, C.H.; Song, L.; Kadi, N.F.; Challis, G.L.; Naismith, J.H. Structural basis for acyl acceptor specificity in the achromobactin biosynthetic enzyme AcsD. J. Mol. Biol. 2011, 412, 495–504. [Google Scholar] [CrossRef]
- Nusca, T.D.; Kim, Y.; Maltseva, N.; Lee, J.Y.; Eschenfeldt, W.; Stols, L.; Schofield, M.M.; Scaglione, J.B.; Dixon, S.D.; Oves-Costales, D.; et al. Functional and structural analysis of the siderophore synthetase AsbB through reconstitution of the petrobactin biosynthetic pathway from Bacillus anthracis. J. Biol. Chem. 2012, 287, 16058–16072. [Google Scholar] [CrossRef]
- Bailey, D.C.; Drake, E.J.; Grant, T.D.; Gulick, A.M. Structural and functional characterization of aerobactin synthetase IucA from a hypervirulent pathotype of Klebsiella pneumonia. Biochemistry 2016, 55, 3559–3570. [Google Scholar] [CrossRef]
- Bailey, D.C.; Alexander, E.; Rice, M.R.; Drake, E.J.; Mydy, L.S.; Aldrich, C.C.; Gulick, A.M. Structural and functional delineation of aerobactin biosynthesis in hypervirulent Klebsiella pneumoniae. J. Biol. Chem. 2018, 293, 7841–7852. [Google Scholar] [CrossRef]
- Salomone-Stagni, M.; Bartho, J.D.; Polsinelli, I.; Bellini, D.; Walsh, M.A.; Demitri, N.; Benini, S. A complete structural characterization of the desferrioxamine E biosynthetic pathway from the fire blight pathogen Erwinia amylovora. J. Struct. Biol. 2018, 202, 236–249. [Google Scholar] [CrossRef]
- Hoffmann, K.M.; Goncuian, E.S.; Karimi, K.L.; Amendola, C.R.; Mojab, Y.; Wood, K.M.; Prussia, G.A.; Nix, J.; Yamamoto, M.; Lathan, K.; et al. Cofactor complexes of DesD, a model enzyme in the virulence-related NIS synthetase family. Biochemistry 2020, 59, 3427–3437. [Google Scholar] [CrossRef]
- Yang, J.; Banas, V.S.; Patel, K.D.; Rivera, G.S.M.; Mydy, L.S.; Gulick, A.M.; Wencewicz, T.A. An acyl adenylate mimic reveals the structural basis for substrate recognition by the iterative siderophore synthetase DesD. J. Biol. Chem. 2022, 298, 102166. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Ju, Y.; Zhou, J.; Guo, J.; Gu, Q.; Xu, J.; Zhou, H. Structural and biochemical characterization of SbnC as a representative type B siderophore synthetase. ACS Chem. Bio. 2020, 15, 2731–2740. [Google Scholar] [CrossRef] [PubMed]
- Kadi, N.; Song, L.; Challis, G.L. Bisucaberin biosynthesis: An adenylating domain of the BibC multi-enzyme catalyzes cyclodimerization of N-hydroxy-N-succinylcadaverine. Chem. Commun. 2008, 5119–5121. [Google Scholar] [CrossRef] [PubMed]
- Bentley, S.D.; Chater, K.F.; Cerdeño-Tárraga, A.M.; Challis, G.L.; Thomson, N.R.; James, K.D.; Harris, D.E.; Quail, M.A.; Kieser, H.; Harper, D.; et al. Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2). Nature 2002, 417, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Dellagi, A.; Brisset, M.-N.; Paulin, J.-P.; Expert, D. Dual role of desferrioxamine in Erwinia amylovora pathogenicity. Mol. Plant Microbe Interact. 1998, 11, 734–742. [Google Scholar] [CrossRef]
- Schupp, T.; Toupet, C.; Divers, M. Cloning and expression of two genes of Streptomyces pilosus involved in the biosynthesis of the siderophore desferrioxamine B. Gene 1988, 64, 179–188. [Google Scholar] [CrossRef]
- Lifa, T.; Tieu, W.; Hocking, R.K.; Codd, R. Forward and reverse (retro) iron(III) or gallium(III) desferrioxamine E and ring-expanded analogues prepared using metal-templated synthesis from endo-hydroxamic acid monomers. Inorg. Chem. 2015, 54, 3573–3583. [Google Scholar] [CrossRef]
- Bergeron, R.J.; Liu, Z.-R.; McManis, J.S.; Wiegand, J. Structural alterations in desferrioxamine compatible with iron clearance in animals. J. Med. Chem. 1992, 35, 4739–4744. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| kcat (s−1) | KM (mM) | kcat/KM (M−1 s−1) | ΔH (kJ/mol) | |
|---|---|---|---|---|
| DesD (HSC) | 7.35 +/− 0.04 | 0.72 +/− 0.01 | 10,200 +/− 200 | −0.4 +/− 0.1 |
| DesD (dfoG1) | 11.1 +/− 0.2 | 0.35 +/− 0.01 | 32,000 +/− 1000 | −0.9 +/− 0.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hoffmann, K.M.; Kingsbury, J.S.; March, N.L.; Jang, Y.; Nguyen, J.H.; Hutt, M.M. Chemoenzymatic Synthesis of Select Intermediates and Natural Products of the Desferrioxamine E Siderophore Pathway. Molecules 2022, 27, 6144. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27196144
Hoffmann KM, Kingsbury JS, March NL, Jang Y, Nguyen JH, Hutt MM. Chemoenzymatic Synthesis of Select Intermediates and Natural Products of the Desferrioxamine E Siderophore Pathway. Molecules. 2022; 27(19):6144. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27196144
Chicago/Turabian StyleHoffmann, Katherine M., Jason S. Kingsbury, Nathan L. March, Yoojin Jang, James H. Nguyen, and Miranda M. Hutt. 2022. "Chemoenzymatic Synthesis of Select Intermediates and Natural Products of the Desferrioxamine E Siderophore Pathway" Molecules 27, no. 19: 6144. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27196144