
New Histamine-Related Five-Membered N-Heterocycle Derivatives as Carbonic Anhydrase I Activators

 , , , , , , and
, , , , , , and
Abstract
:1. Introduction
2. Results
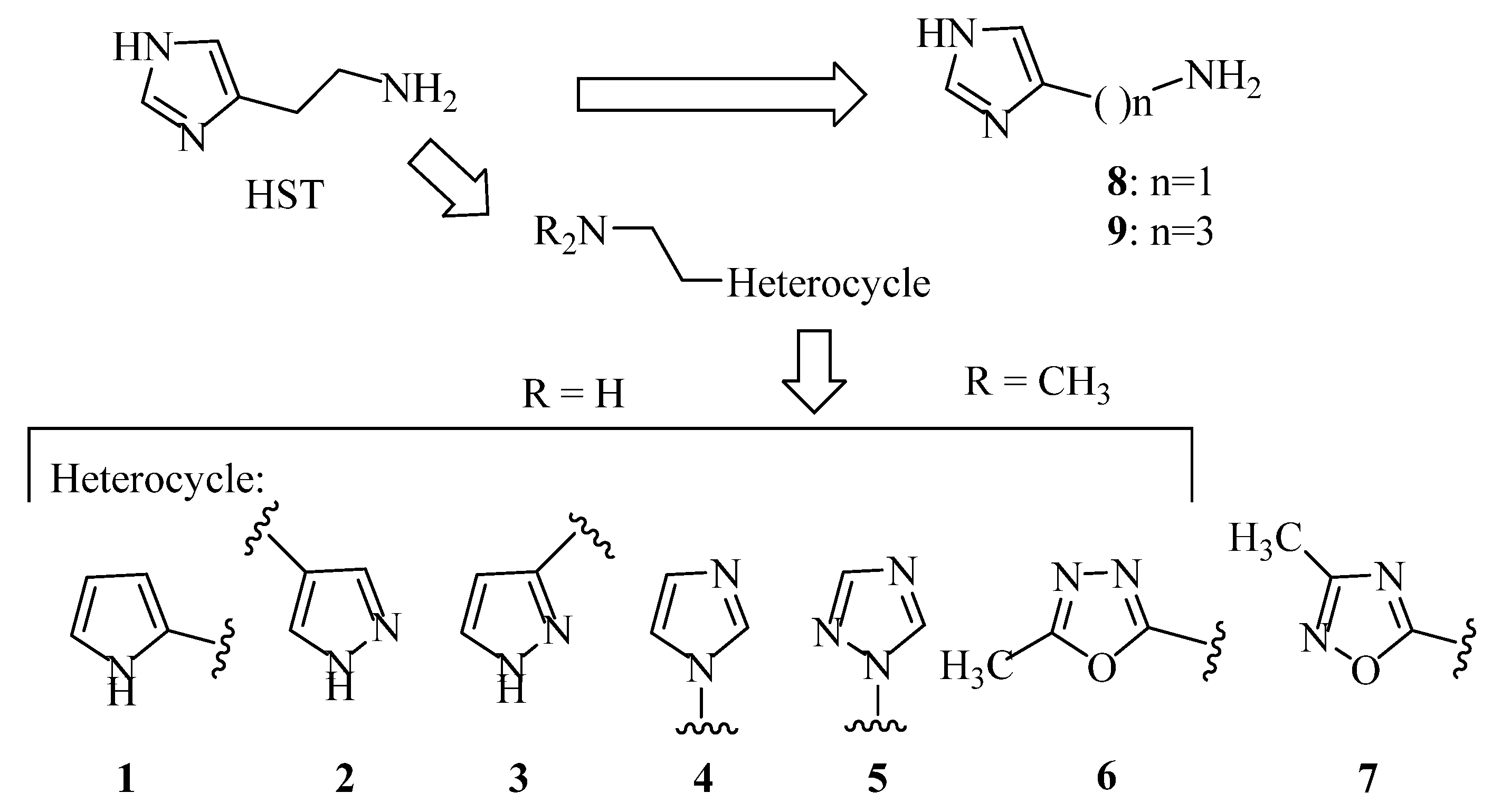
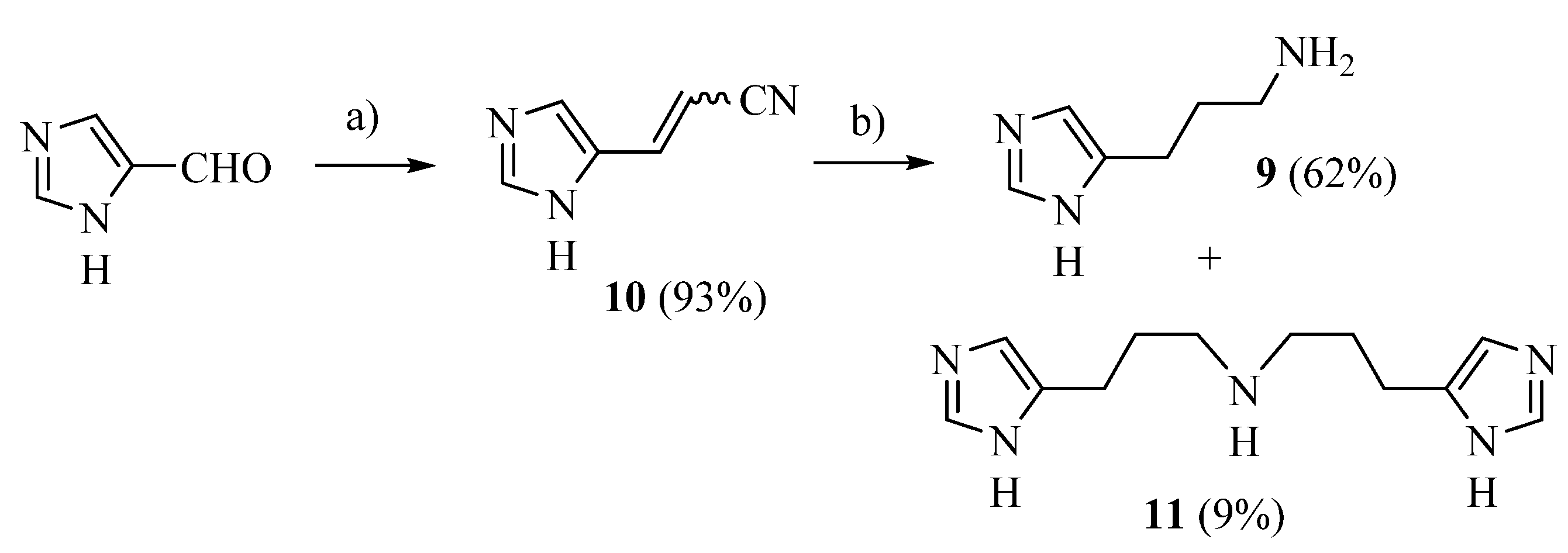
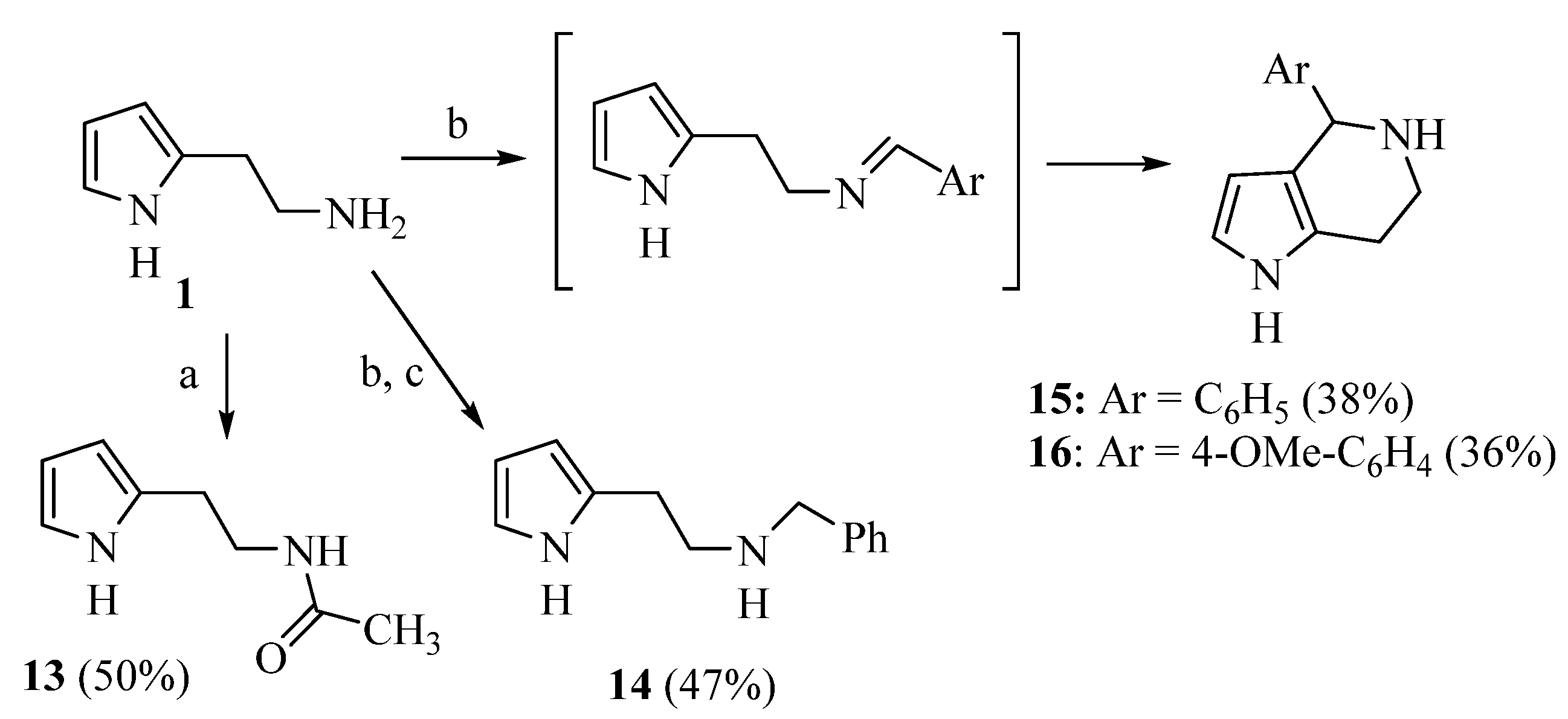
2.1. Chemistry
2.2. CA Activating Properties
2.2.1. Activity on hCA I
2.2.2. Activity on hCA VA, VII and XIII and Selectivity
3. Discussion
4. Materials and Methods
4.1. Chemistry
4.1.1. General Considerations
4.1.2. Compounds Prepared According to Literature Procedures
4.1.3. 3-(1H-imidazol-4-yl)propan-1-amine (9)
4.1.4. N-benzyl-2-(1H-pyrrol-2-yl)ethan-1-amine (14)
4.1.5. 4-phenyl-4,5,6,7-tetrahydro-1H-pyrrolo[3,2-c]pyridine (15)
4.1.6. 4-(4-methoxyphenyl)-4,5,6,7-tetrahydro-1H-pyrrolo[3,2-c]pyridine (16)
4.1.7. General Procedure for the Synthesis of Schiff Bases (17–19)
4.1.8. General Procedure for the Reduction of Schiff Bases to Amines 20–22
4.2. CA Activation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Supuran, C.T. Structure and function of carbonic anhydrases. Biochem. J. 2016, 473, 2023–2032. [Google Scholar] [CrossRef]
- Supuran, C.T. Carbonic anhydrase activators. Future Med. Chem. 2018, 10, 561–573. [Google Scholar] [CrossRef]
- Datta, R.; Shah, G.N.; Rubbelke, T.S.; Waheed, A.; Rauchman, M.; Goodman, A.G.; Katze, M.G.; Sly, W.S. Progressive renal injury from transgenic expression of human carbonic anhydrase IV folding mutants is enhanced by deficiency of p58IPK. Proc. Natl. Acad. Sci. USA 2010, 107, 6448–6452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, G.N.; Bonapace, G.; Hu, P.Y.; Strisciuglio, P.; Sly, W.S. Carbonic anhydrase II deficiency syndrome (osteopetrosis with renal tubular acidosis and brain calcification): Novel mutations in CA2 identified by direct sequencing expand the opportunity for genotype-phenotype correlation. Hum. Mutat. 2004, 24, 272. [Google Scholar] [CrossRef]
- Ogilvie, J.; Ohlemiller, K.K.; Shah, G.N.; Ulmasov, B.; Becker, T.A.; Waheed, A.; Hennig, A.K.; Lukasiewicz, P.D.; Sly, W.S. Carbonic anhydrase XIV deficiency produces a functional defect in the retinal light response. Proc. Natl. Acad. Sci. USA 2007, 104, 8514–8519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Karnebeek, C.D.; Sly, W.S.; Ross, C.J.; Salvarinova, R.; Yaplito-Lee, J.; Santra, S.; Shyr, C.; Horvath, G.A.; Eydoux, P.; Lehman, A.M.; et al. Mitochondrial Carbonic Anhydrase VA Deficiency Resulting from CA5A Alterations Presents with Hyperammonemia in Early Childhood. Am. J. Hum. Genet. 2014, 94, 453–461. [Google Scholar] [CrossRef] [Green Version]
- Avital, D.; Hershkovitz, E.; Loewenthal, N. Exertional rhabdomyolysis in carbonic anhydrase 12 deficiency. J. Pediatr. Endocrinol. Metab. 2018, 31, 697–699. [Google Scholar] [CrossRef]
- Feinstein, Y.; Yerushalmi, B.; Loewenthal, N.; Alkrinawi, S.; Birk, O.S.; Parvari, R.; Hershkovitz, E. Natural History and Clinical Manifestations of Hyponatremia and Hyperchlorhidrosis due to Carbonic Anhydrase XII Deficiency. Horm. Res. Paediatr. 2014, 81, 336–342. [Google Scholar] [CrossRef] [PubMed]
- Boyne, K.; Corey, D.A.; Zhao, P.; Lu, B.; Boron, W.F.; Moss, F.J.; Kelley, T.J. Carbonic anhydrase and soluble adenylate cyclase regulation of cystic fibrosis cellular phenotypes. Am. J. Physiol. Cell. Mol. Physiol. 2022. [Google Scholar] [CrossRef]
- de Souza, L.C.; Provensi, G.; Vullo, D.; Carta, F.; Scozzafava, A.; Costa, A.; Schmidt, S.D.; Passani, M.B.; Supuran, C.T.; Blandina, P. Carbonic anhydrase activation enhances object recognition memory in mice through phosphorylation of the extracellular signal-regulated kinase in the cortex and the hippocampus. Neuropharmacology 2017, 118, 148–156. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, S.D.; Costa, A.; Rani, B.; Nachtigall, E.G.; Passani, M.B.; Carta, F.; Nocentini, A.; Myskiw, J.D.C.; Furini, C.R.G.; Supuran, C.T.; et al. The role of carbonic anhydrases in extinction of contextual fear memory. Proc. Natl. Acad. Sci. USA 2020, 117, 16000–16008. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Schröder, H.C.; Schlossmacher, U.; Neufurth, M.; Feng, Q.; Diehl-Seifert, B.; Müller, W.E.G. Modulation of the Initial Mineralization Process of SaOS-2 Cells by Carbonic Anhydrase Activators and Polyphosphate. Calcif. Tissue Int. 2013, 94, 495–509. [Google Scholar] [CrossRef]
- Briganti, F.; Mangani, S.; Orioli, P.; Scozzafava, A.; Vernaglione, G.; Supuran, C.T. Carbonic Anhydrase Activators: X-ray Crystallographic and Spectroscopic Investigations for the Interaction of Isozymes I and II with Histamine. Biochemistry 1997, 36, 10384–10392. [Google Scholar] [CrossRef]
- Akocak, S.; Lolak, N.; Vullo, D.; Durgun, M.; Supuran, C.T. Synthesis and biological evaluation of histamine Schiff bases as carbonic anhydrase I, II, IV, VII, and IX activators. J. Enzyme Inhib. Med. Chem. 2017, 32, 1305–1312. [Google Scholar] [CrossRef] [Green Version]
- Saada, M.-C.; Vullo, D.; Montero, J.-L.; Scozzafava, A.; Winum, J.-Y.; Supuran, C.T. Carbonic anhydrase I and II activation with mono- and dihalogenated histamine derivatives. Bioorg. Med. Chem. Lett. 2011, 21, 4884. [Google Scholar] [CrossRef] [PubMed]
- Dave, K.; Scozzafava, A.; Vullo, D.; Supuran, C.T.; Ilies, M.A. Pyridinium derivatives of histamine are potent activators of cytosolic carbonic anhydrase isoforms I, II and VII. Org. Biomol. Chem. 2011, 9, 2790–2800. [Google Scholar] [CrossRef]
- Supuran, C.; Barboiu, M.; Luca, C.; Pop, E.; Brewster, M.; Dinculescu, A. Carbonic anhydrase activators. Part 14. Syntheses of mono and bis pyridinium salt derivatives of 2-amino-5-(2-aminoethyl)- and 2-amino-5-(3-aminopropyl)-1,3,4-thiadiazole and their interaction with isozyme II. Eur. J. Med. Chem. 1996, 31, 597–606. [Google Scholar] [CrossRef]
- Provensi, G.; Nocentini, A.; Passani, M.B.; Blandina, P.; Supuran, C.T. Activation of carbonic anhydrase isoforms involved in modulation of emotional memory and cognitive disorders with histamine agonists, antagonists and derivatives. J. Enzyme Inhib. Med. Chem. 2021, 36, 719–726. [Google Scholar] [CrossRef]
- Temperini, C.; Scozzafava, A.; Puccetti, L.; Supuran, C.T. Carbonic anhydrase activators: X-ray crystal structure of the adduct of human isozyme II with l-histidine as a platform for the design of stronger activators. Bioorg. Med. Chem. Lett. 2005, 15, 5136–5141. [Google Scholar] [CrossRef]
- Temperini, C.; Scozzafava, A.; Vullo, D.; Supuran, C.T. Carbonic Anhydrase Activators. Activation of Isoforms I, II, IV, VA, VII, and XIV with l- and d-Phenylalanine and Crystallographic Analysis of Their Adducts with Isozyme II: Stereospecific Recognition within the Active Site of an Enzyme and Its Consequences for the Drug Design. J. Med. Chem. 2006, 49, 3019–3027. [Google Scholar] [CrossRef]
- Temperini, C.; Innocenti, A.; Scozzafava, A.; Supuran, C.T. Carbonic anhydrase activators: Kinetic and X-ray crystallographic study for the interaction of d- and l-tryptophan with the mammalian isoforms I–XIV. Bioorganic Med. Chem. 2008, 16, 8373–8378. [Google Scholar] [CrossRef] [PubMed]
- Temperini, C.; Scozzafava, A.; Supuran, C.T. Carbonic anhydrase activators: The first X-ray crystallographic study of an adduct of isoform I. Bioorganic Med. Chem. Lett. 2006, 16, 5152–5156. [Google Scholar] [CrossRef] [PubMed]
- Bozdag, M.; Altamimi, A.S.A.; Vullo, D.; Supuran, C.T.; Carta, F. State of the Art on Carbonic Anhydrase Modulators for Biomedical Purposes. Curr. Med. Chem. 2019, 26, 2558–2573. [Google Scholar] [CrossRef] [PubMed]
- Chiaramonte, N.; Maach, S.; Biliotti, C.; Angeli, A.; Bartolucci, G.; Braconi, L.; Dei, S.; Teodori, E.; Supuran, C.T.; Romanelli, M.N. Synthesis and carbonic anhydrase activating properties of a series of 2-amino-imidazolines structurally related to clonidine1. J. Enzyme Inhib. Med. Chem. 2020, 35, 1003–1010. [Google Scholar] [CrossRef]
- Dudkin, V.Y. Bioisosteric equivalence of five-membered heterocycles. Chem. Heterocycl. Compd. 2012, 48, 27–32. [Google Scholar] [CrossRef]
- Dawande, S.G.; Kanchupalli, V.; Kalepu, J.; Chennamsetti, H.; Lad, B.S.; Katukojvala, S. Rhodium Enalcarbenoids: Direct Synthesis of Indoles by Rhodium(II)-Catalyzed [4+2] Benzannulation of Pyrroles. Angew. Chem. Int. Ed. 2014, 53, 4076–4080. [Google Scholar] [CrossRef]
- Jones, R.G.; Mann, M.J. New Methods of Synthesis of β-Aminoethylpyrazoles. J. Am. Chem. Soc. 1953, 75, 4048–4052. [Google Scholar] [CrossRef]
- Buchholz, M.; Heiser, U.; Schilling, S.; Niestroj, A.J.; Zunkel, K.; Demuth, H.-U. The First Potent Inhibitors for Human Glutaminyl Cyclase: Synthesis and Structure−Activity Relationship. J. Med. Chem. 2005, 49, 664–677. [Google Scholar] [CrossRef]
- Wright, W.B.; Press, J.B.; Chan, P.S.; Marsico, J.W.; Haug, M.F.; Lucas, J.; Tauber, J.; Tomcufcik, A.S. Thromboxane synthetase inhibitors and antihypertensive agents. 1. N-[(1H-imidazol-1-yl)alkyl]aryl amides and N-[(1H-1,2,4-triazol-1-yl)alkyl]aryl amides. J. Med. Chem. 1986, 29, 523–530. [Google Scholar] [CrossRef]
- Turner, W.W.; Arnold, L.D.; Maag, H.; Zlotnick, A. Hepatitis B Core Protein Allosteric Modulators. World patent WO2015138895, 17 September 2015. [Google Scholar]
- Macor, J.E.; Ordway, T.; Smith, R.L.; Verhoest, P.R.; Mack, R.A. Synthesis and Use of 5-Vinyl-1,2,4-oxadiazoles as Michael Acceptors. A Rapid Synthesis of the Potent Muscarinic Agonist L-670,548. J. Org. Chem. 1996, 61, 3228–3229. [Google Scholar] [CrossRef]
- Turner, R.A.; Huebner, C.F.; Scholz, C.R. Studies on Imidazole Compounds. I. 4-Methylimidazole and Related Compounds. J. Am. Chem. Soc. 1949, 71, 2801–2803. [Google Scholar] [CrossRef]
- Sellier, C.; Buschauer, A.; Elz, S.; Schunack, W. Zur Synthese von (Z)- und (E)-3-(1H-Imidazol-4-yl)-2-propenamin und einigen 3-(1H-Imidazol-4-yl)propanaminen. Liebigs Ann. Chem. 1992, 1992, 317–323. [Google Scholar] [CrossRef]
- Komsani, J.R.; Koppireddi, S.; Avula, S.; Koochana, P.K.; Yadla, R. Demonic axe-like conjugated alkynes in combating microbes. Eur. J. Med. Chem. 2013, 68, 132–138. [Google Scholar] [CrossRef]
- Tait, B.; Cullen, M. Methods of Modulating CFTR Activity. World patent WO2014210159, 31 December 2014. [Google Scholar]
- Khorana, N.; Smith, C.; Herrick-Davis, K.; Purohit, A.; Teitler, M.; Grella, B.; Dukat, M.; Glennon, R.A. Binding of Tetrahydrocarboline Derivatives at Human 5-HT5A Receptors. J. Med. Chem. 2003, 46, 3930–3937. [Google Scholar] [CrossRef] [PubMed]
- Herz, W.; Tocker, S. Pyrrolo [3,2-c]pyridines1. J. Am. Chem. Soc. 1955, 77, 6353–6355. [Google Scholar] [CrossRef]
- Cox, E.D.; Cook, J.M. The Pictet-Spengler condensation: A new direction for an old reaction. Chem. Rev. 1995, 95, 1797–1842. [Google Scholar] [CrossRef]
- Galli, U.; Hysenlika, R.; Meneghetti, F.; Del Grosso, E.; Pelliccia, S.; Novellino, E.; Giustiniano, M.; Tron, G.C. Exploiting the Nucleophilicity of the Nitrogen Atom of Imidazoles: One-Pot Three-Component Synthesis of Imidazo-Pyrazines. Molecules 2019, 24, 1959. [Google Scholar] [CrossRef] [Green Version]
- National Center for Biotechnology Information. PubChem Compound Summary for CID 63001082, 2-[(1H-imidazol-5-ylmethylamino)methyl]phenol. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/2-_1H-imidazol-5-ylmethylamino_methyl_phenol (accessed on 21 December 2021).
- Olszewski, T.K.; Boduszek, B. Application of Bis(trimethylsilyl) Phosphonite in the Efficient Preparation of New Heterocyclic α-Aminomethyl-H-phosphinic Acids. Synthesis 2010, 2011, 437–442. [Google Scholar] [CrossRef]
- Khalifah, R.G. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J. Biol. Chem. 1971, 246, 2561–2573. [Google Scholar] [CrossRef]
- Vullo, D.; Innocenti, A.; Nishimori, I.; Scozzafava, A.; Kaila, K.; Supuran, C.T. Carbonic anhydrase activators: Activation of the human isoforms VII (cytosolic) and XIV (transmembrane) with amino acids and amines. Bioorganic Med. Chem. Lett. 2007, 17, 4107–4112. [Google Scholar] [CrossRef] [PubMed]
- Ruusuvuori, E.; Huebner, A.K.; Kirilkin, I.; Yukin, A.Y.; Blaesse, P.; Helmy, M.; Kang, H.J.; El Muayed, M.; Hennings, J.C.; Voipio, J.; et al. Neuronal carbonic anhydrase VII provides GABAergic excitatory drive to exacerbate febrile seizures. EMBO J. 2013, 32, 2275–2286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karjalainen, S.L.; Haapasalo, H.K.; Aspatwar, A.; Barker, H.; Parkkila, S.; Haapasalo, J.A. Carbonic anhydrase related protein expression in astrocytomas and oligodendroglial tumors. BMC Cancer 2018, 18, 584. [Google Scholar] [CrossRef]
- Lehtonen, J.; Shen, B.; Vihinen, M.; Casini, A.; Scozzafava, A.; Supuran, C.T.; Parkkila, A.-K.; Saarnio, J.; Kivelä, A.J.; Waheed, A.; et al. Characterization of CA XIII, a Novel Member of the Carbonic Anhydrase Isozyme Family. J. Biol. Chem. 2004, 279, 2719–2727. [Google Scholar] [CrossRef] [Green Version]
- Lacruz, R.S.; Hilvo, M.; Kurtz, I.; Paine, M.L. A survey of carbonic anhydrase mRNA expression in enamel cells. Biochem. Biophys. Res. Commun. 2010, 393, 883–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maccallini, C.; Di Matteo, M.; Vullo, D.; Ammazzalorso, A.; Carradori, S.; De Filippis, B.; Fantacuzzi, M.; Giampietro, L.; Pandolfi, A.; Supuran, C.T.; et al. Indazole, Pyrazole, and Oxazole Derivatives Targeting Nitric Oxide Synthases and Carbonic Anhydrases. ChemMedChem 2016, 11, 1695–1699. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, M.; Kondeti, B.; Tu, C.; Maupin, C.M.; Silverman, D.N.; McKenna, R. Structural insight into activity enhancement and inhibition of H64A carbonic anhydrase II by imidazoles. IUCrJ 2014, 1, 129–135. [Google Scholar] [CrossRef]
- Scozzafava, A.; Supuran, C.T. Carbonic anhydrase activators–Part 21. Novel activators of isozymes I, II and IV incorporating carboxamido and ureido histamine moieties. Eur. J. Med. Chem. 2000, 35, 31–39. [Google Scholar] [CrossRef]
- Pala, N.; Cadoni, R.; Sechi, M. Carbonic Anhydrase I. In Carbonic Anhydrases as Biocatalysts. From Theory to Medical and Industrial Applications; De Simone, G., Supuran, C.T., Eds.; Elsevier: Amsterdam, The Netherlands, 2015; pp. 31–49. [Google Scholar]
- Gambhir, K.K.; Ornasir, J.; Headings, V.; Bonar, A. Decreased total carbonic anhydrase esterase activity and decreased levels of carbonic anhydrase 1 isozyme in erythrocytes of type II diabetic patients. Biochem. Genet. 2007, 45, 431–439. [Google Scholar] [CrossRef]
- Chiang, W.-L.; Chu, S.-C.; Yang, S.-S.; Li, M.-C.; Lai, J.-C.; Yang, S.-F.; Chiou, H.-L.; Hsieh, Y.-S. The aberrant expression of cytosolic carbonic anhydrase and its clinical significance in human non-small cell lung cancer. Cancer Lett. 2002, 188, 199–205. [Google Scholar] [CrossRef]
- Lee, W.-C.; Chou, H.-S.; Wu, T.-J.; Lee, C.-F.; Hsu, P.-Y.; Hsu, H.-Y.; Wu, T.-H.; Chan, K.-M. Down-regulation of metabolic proteins in hepatocellular carcinoma with portal vein thrombosis. Clin. Proteom. 2017, 14, 29. [Google Scholar] [CrossRef]
- Marshall, A.G.; Hendrickson, C.L. High-Resolution Mass Spectrometers. Ann. Rev. Anal. Chem. 2008, 1, 579–599. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cmp (Salt) a | Structure | KA (μM) b | ||||
|---|---|---|---|---|---|---|
| hCA I | hCA II | hCA VA | hCA VII | hCA XIII | ||
| HST |  | 2.10 | 125 | 0.010 | 37.5 | 4.6 |
| 1 (oxalate) |  | 2.16 | >150 | 29.8 | 44.6 | >100 |
| 2 (oxalate) |  | 11.6 | >150 | 37.9 | 32.8 | >100 |
| 3 |  | 28.4 | >150 | 51.0 | 23.7 | >100 |
| 3a |  | 2.19 | >150 | 78.5 | 120 | >100 |
| 4 |  | 13.5 | >150 | 42.7 | 25.4 | >100 |
| 5 |  | 9.84 | >150 | 24.6 | 35.5 | >100 |
| 6 (oxalate) |  | >150 | >150 | 21.7 | 23.0 | >100 |
| 7 (HCl) |  | >150 | >150 | 28.6 | 12.1 | >100 |
| 8.2HCl |  | 2.9 | >100 | 12.5 | 13.2 | 29.0 |
| 9.2HCl |  | 2.4 | >100 | 12.5 | 10.8 | 28.2 |
| 11.3HCl |  | 7.0 | >100 | 12.9 | 11.5 | 84.8 |
| 12 |  | 11.6 | >100 | 34.9 | 13.8 | 96.4 |
| 13 |  | 5.6 | >100 | 24.8 | 10.8 | >100 |
| 14 |  | 3.0 | >100 | 14.0 | 12.5 | 91.1 |
| 15 |  | 3.7 | >100 | 20.6 | 11.1 | >100 |
| 16 |  | 5.7 | >100 | 19.1 | 14.3 | >100 |
| 17 |  | 59.1 | >100 | 15.2 | 38.6 | 64.4 |
| 18 |  | 56.1 | >100 | 16.0 | 22.1 | 33.0 |
| 19 |  | 93.5 | >100 | 13.3 | 35.4 | 57.3 |
| 20 |  | 5.8 | >100 | 27.2 | 9.7 | 46.0 |
| 21 |  | 4.9 | >100 | 11.9 | 18.3 | 73.0 |
| 22 |  | 0.9 | >100 | 11.2 | 13.2 | >100 |
| 23 |  | 86.0 | >100 | 18.8 | 74.6 | >100 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiaramonte, N.; Gabellini, A.; Angeli, A.; Bartolucci, G.; Braconi, L.; Dei, S.; Teodori, E.; Supuran, C.T.; Romanelli, M.N. New Histamine-Related Five-Membered N-Heterocycle Derivatives as Carbonic Anhydrase I Activators. Molecules 2022, 27, 545. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27020545
Chiaramonte N, Gabellini A, Angeli A, Bartolucci G, Braconi L, Dei S, Teodori E, Supuran CT, Romanelli MN. New Histamine-Related Five-Membered N-Heterocycle Derivatives as Carbonic Anhydrase I Activators. Molecules. 2022; 27(2):545. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27020545
Chicago/Turabian StyleChiaramonte, Niccolò, Alessio Gabellini, Andrea Angeli, Gianluca Bartolucci, Laura Braconi, Silvia Dei, Elisabetta Teodori, Claudiu T. Supuran, and Maria Novella Romanelli. 2022. "New Histamine-Related Five-Membered N-Heterocycle Derivatives as Carbonic Anhydrase I Activators" Molecules 27, no. 2: 545. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27020545