
Functionalization of Rhodamine Platforms with 3-Hydroxy-4-pyridinone Chelating Units and Its Fluorescence Behavior towards Fe(III)

 , , and
, , and
Abstract
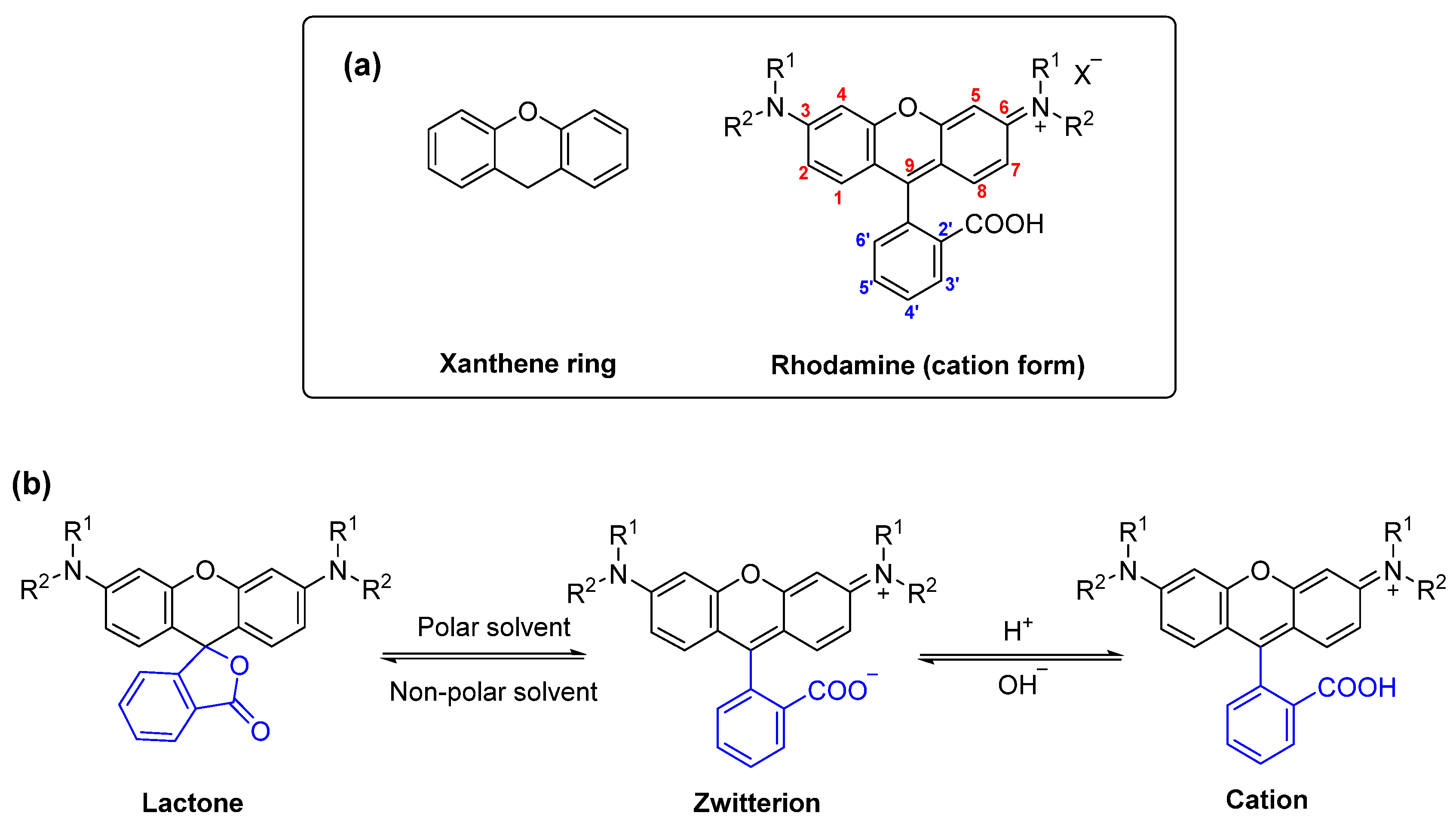
:1. Introduction
2. Results and Discussion
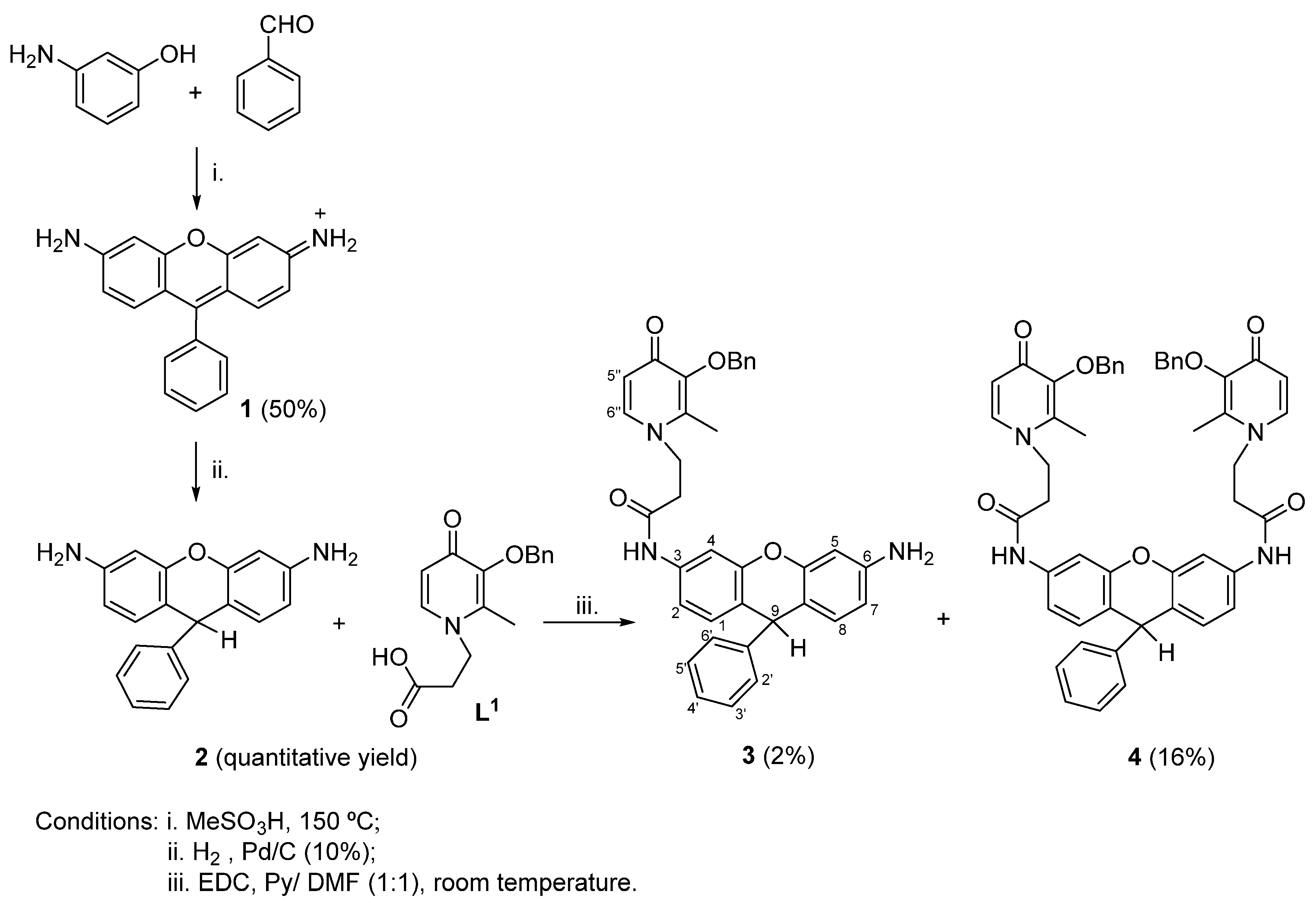
2.1. Synthesis
2.2. NMR Characterization
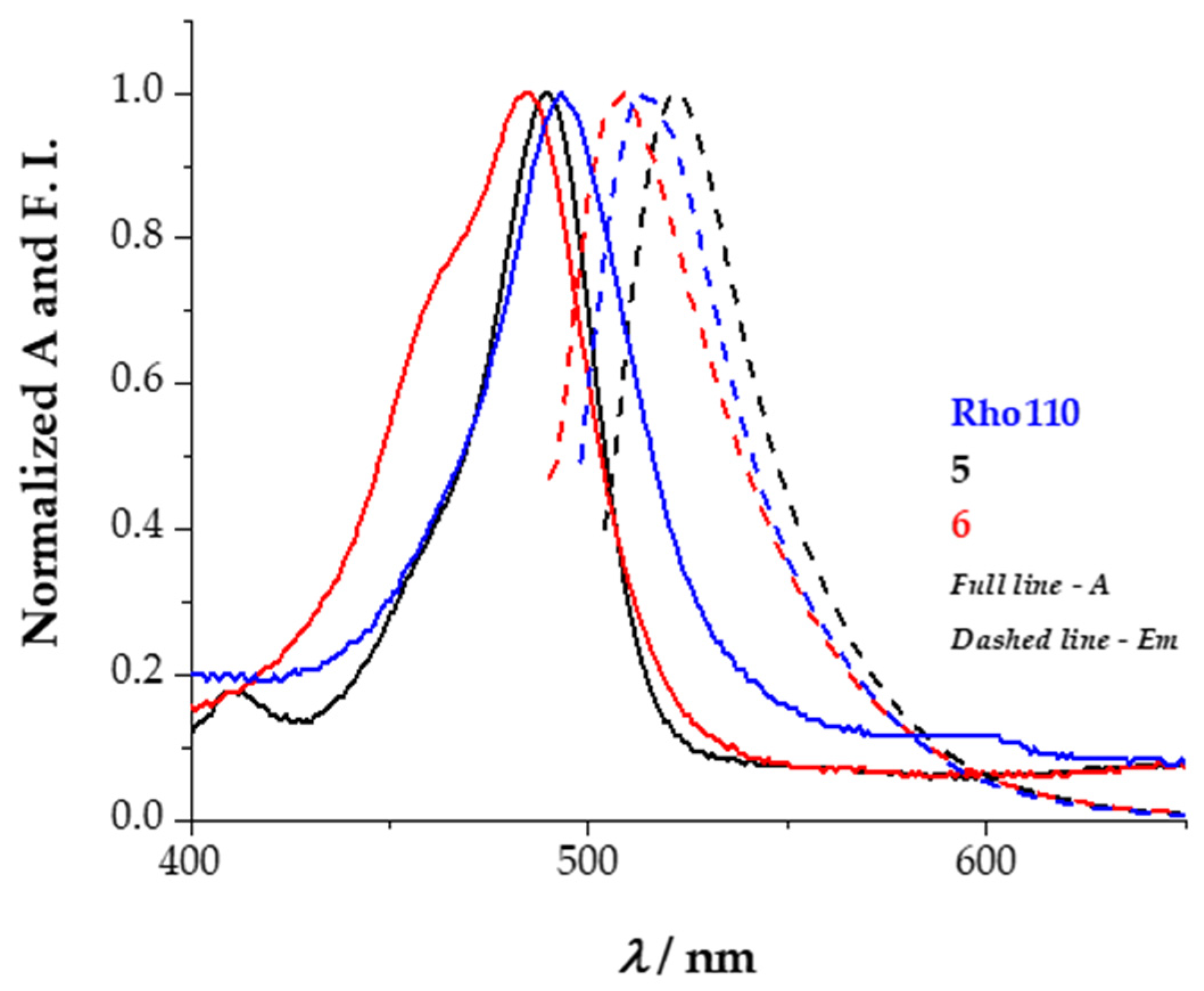
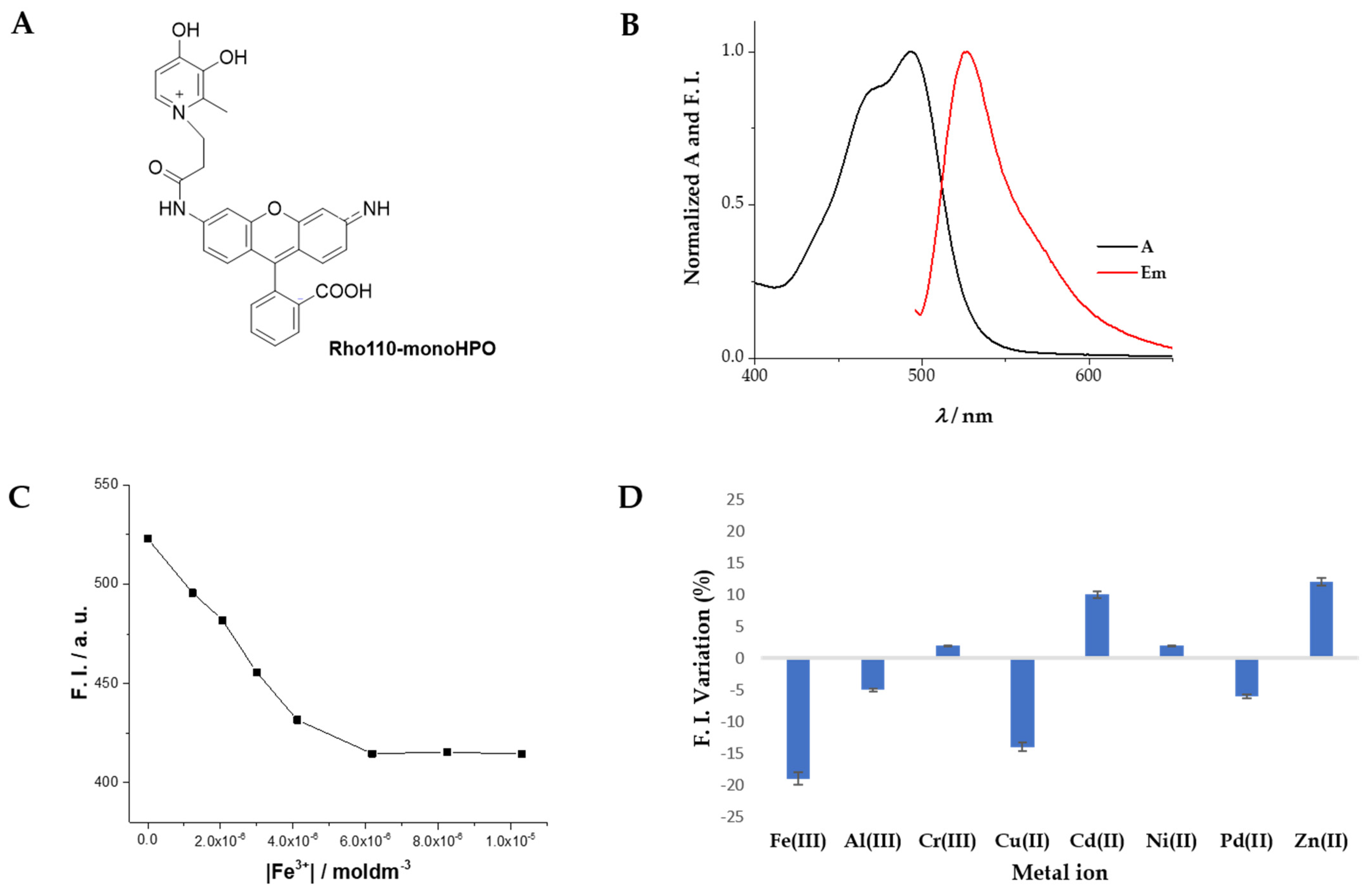
2.3. Spectroscopic Characterization
3. Materials and Methods
3.1. Synthesis of Rosamine 1 and Dihydrorosamine 2
3.2. Synthesis of Conjugates 3 and 4
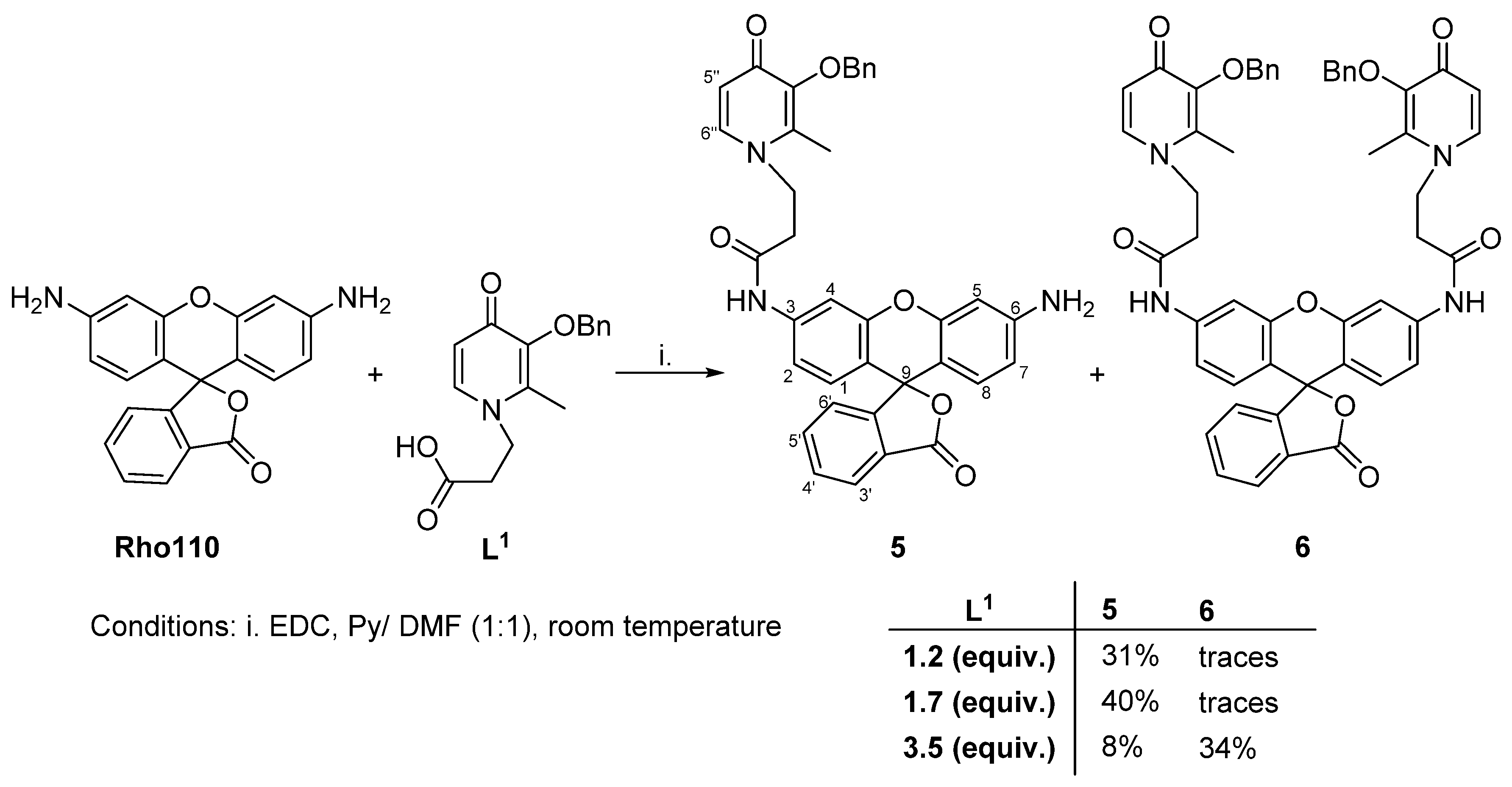
3.3. Synthesis of Conjugates 5 and 6
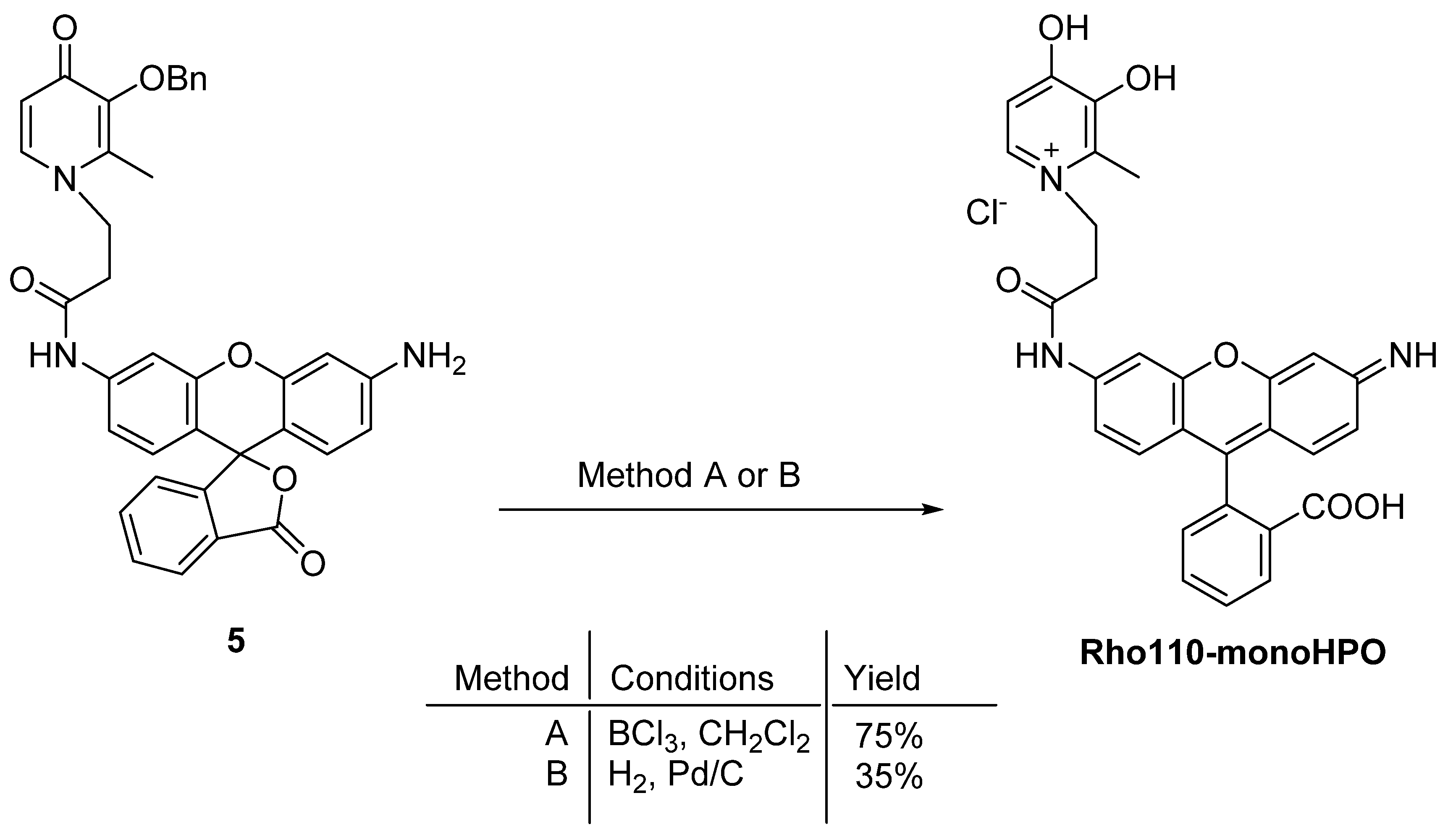
3.4. Deprotection of Benzyl Ether of 5
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Beija, M.; Afonso, C.A.M.; Martinho, J.M.G. Synthesis and applications of Rhodamine derivatives as fluorescent probes. Chem. Soc. Rev. 2009, 38, 2410. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.; Zhan, X.-Q.Q.; Bian, Q.-N.N.; Zhang, X.-J.J. Advances in modifying fluorescein and rhodamine fluorophores as fluorescent chemosensors. Chem. Commun. 2013, 49, 429–447. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, A.; Delgado-Pinar, E.; García-España, E.; Giorgi, C.; Pina, F. Highlights of metal ion-based photochemical switches. Coord. Chem. Rev. 2014, 260, 156–215. [Google Scholar] [CrossRef]
- Arai, S.; Suzuki, M.; Park, S.-J.; Yoo, J.S.; Wang, L.; Kang, N.-Y.; Ha, H.-H.; Chang, Y.-T. Mitochondria-targeted fluorescent thermometer monitors intracellular temperature gradient. Chem. Commun. 2015, 51, 8044–8047. [Google Scholar] [CrossRef] [PubMed]
- Zhegalova, N.G.; Dergunov, S.A.; Wang, S.T.; Pinkhassik, E.; Berezin, M.Y. Design of Fluorescent Nanocapsules as Ratiometric Nanothermometers. Chem. A Eur. J. 2014, 20, 10292–10297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, M.; Guo, Y.-S.; Xu, W.-N.; Zhao, Y.-F.; Xie, H.-Y.; Li, H.-J.; Chen, X.-F.; Zhao, R.-S.; Guo, D.-S. Far-red to near-infrared fluorescent probes based on silicon-substituted xanthene dyes for sensing and imaging. TrAC Trends Anal. Chem. 2020, 122, 115704. [Google Scholar] [CrossRef]
- Wang, L.; Du, W.; Hu, Z.; Uvdal, K.; Li, L.; Huang, W. Hybrid Rhodamine Fluorophores in the Visible/NIR Region for Biological Imaging. Angew. Chem. Int. Ed. 2019, 58, 14026–14043. [Google Scholar] [CrossRef]
- Luo, X.; Qian, L.; Xiao, Y.; Tang, Y.; Zhao, Y.; Wang, X.; Gu, L.; Lei, Z.; Bao, J.; Wu, J.; et al. A diversity-oriented rhodamine library for wide-spectrum bactericidal agents with low inducible resistance against resistant pathogens. Nat. Commun. 2019, 10, 258. [Google Scholar] [CrossRef]
- Chaudhary, A.; Khurana, J.M. Advances in the Synthesis of Xanthenes: An Overview. Curr. Org. Synth. 2018, 15, 341–369. [Google Scholar] [CrossRef]
- Lopez Arbeloa, F.; Lopez Arbeloa, T.; Tapia Estevez, M.J.; Lopez Arbeloa, I. Photophysics of rhodamines: Molecular structure and solvent effects. J. Phys. Chem. 1991, 95, 2203–2208. [Google Scholar] [CrossRef]
- Jiao, G.-S.; Castro, J.C.; Thoresen, L.H.; Burgess, K. Microwave-Assisted Syntheses of Regioisomerically Pure Bromorhodamine Derivatives. Org. Lett. 2003, 5, 3675–3677. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, I.C.S.; Amorim, A.L.; Queirós, C.; Lopes, S.C.; Gameiro, P.; de Castro, B.; Rangel, M.; Silva, A.M.G. Microwave-Assisted Synthesis and Spectroscopic Properties of 4′-Substituted Rosamine Fluorophores and Naphthyl Analogues. Eur. J. Org. Chem. 2012, 2012, 5810–5817. [Google Scholar] [CrossRef]
- Kraus, G.A.; Guney, T.; Kempema, A.; Hyman, J.M.; Parvin, B. Efficient synthesis of fluorescent rosamines: Multifunctional platforms for cellular imaging. Tetrahedron Lett. 2014, 55, 1549–1551. [Google Scholar] [CrossRef]
- Arambula, C.; Rodrigues, J.; Koh, J.J.; Woydziak, Z. Synthesis of Rhodamines and Rosamines Using 3,6-Difluoroxanthone as a Common Intermediate. J. Org. Chem. 2021, 86, 17856–17865. [Google Scholar] [CrossRef] [PubMed]
- Queirós, C.; Leite, A.; Couto, M.G.M.; Moniz, T.; Cunha-Silva, L.; Gameiro, P.; Silva, A.M.G.; Rangel, M. Tuning the limits of pH interference of a rhodamine ion sensor by introducing catechol and 3-hydroxy-4-pyridinone chelating units. Dye. Pigment. 2014, 110, 193–202. [Google Scholar] [CrossRef]
- Irto, A.; Cardiano, P.; Chand, K.; Cigala, R.M.; Crea, F.; De Stefano, C.; Gattuso, G.; Sammartano, S.; Santos, M.A. Complexation of environmentally and biologically relevant metals with bifunctional 3-hydroxy-4-pyridinones. J. Mol. Liq. 2020, 319, 114349. [Google Scholar] [CrossRef]
- Cilibrizzi, A.; Abbate, V.; Chen, Y.-L.; Ma, Y.; Zhou, T.; Hider, R.C. Hydroxypyridinone Journey into Metal Chelation. Chem. Rev. 2018, 118, 7657–7701. [Google Scholar] [CrossRef]
- Sharma, S.; Baral, M.; Kanungo, B.K. Recent advances in therapeutical applications of the versatile hydroxypyridinone chelators. J. Incl. Phenom. Macrocycl. Chem. 2022. [Google Scholar] [CrossRef]
- Jiang, X.; Zhou, T.; Bai, R.; Xie, Y. Hydroxypyridinone-Based Iron Chelators with Broad-Ranging Biological Activities. J. Med. Chem. 2020, 63, 14470–14501. [Google Scholar] [CrossRef]
- Chandran, S.S.; Dickson, K.A.; Raines, R.T. Latent Fluorophore Based on the Trimethyl Lock. J. Am. Chem. Soc. 2005, 127, 1652–1653. [Google Scholar] [CrossRef]
- Fan, J.; Ye, Y.; Chu, G.; Zhang, Z.; Fu, Y.; Li, Y.-M.; Shi, J. Semisynthesis of Ubiquitin and SUMO-Rhodamine 110-Glycine through Aminolysis of Boc-Protected Thioester Counterparts. J. Org. Chem. 2019, 84, 14861–14867. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.M.G.; Leite, A.; Gonzalez, P.; Domingues, M.R.M.; Gameiro, P.; de Castro, B.; Rangel, M. Use of a porphyrin platform and 3,4-HPO chelating units to synthesize ligands with N4 and O4 coordination sites. Tetrahedron 2011, 67, 7821–7828. [Google Scholar] [CrossRef]
- Guillén, M.; Gámez, F.; Suárez, B.; Queirós, C.; Silva, A.; Barranco, Á.; Sánchez-Valencia, J.; Pedrosa, J.; Lopes-Costa, T. Preparation and Optimization of Fluorescent Thin Films of Rosamine-SiO2/TiO2 Composites for NO2 Sensing. Materials 2017, 10, 124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karpiuk, J.; Grabowski, Z.R.; De Schryver, F.C. Photophysics of the Lactone Form of Rhodamine 101. J. Phys. Chem. 1994, 98, 3247–3256. [Google Scholar] [CrossRef]
- Sinel’nikov, A.N.; Artyukhov, V.Y. Fluorescence of the lactone form of rhodamine B. Russ. J. Phys. Chem. A 2013, 87, 1409–1416. [Google Scholar] [CrossRef]
- Liu, Z.D.; Hider, R.C. Design of clinically useful iron(III)-selective chelators. Med. Res. Rev. 2002, 22, 26–64. [Google Scholar] [CrossRef]
- Irving, H.; Williams, R.J.P. Order of Stability of Metal Complexes. Nature 1948, 162, 746–747. [Google Scholar] [CrossRef]
- Dehkordi, L.S.; Liu, Z.D.; Hider, R.C. Basic 3-hydroxypyridin-4-ones: Potential antimalarial agents. Eur. J. Med. Chem. 2008, 43, 1035–1047. [Google Scholar] [CrossRef]
- Merkofer, M.; Kissner, R.; Hider, R.; Koppenol, W. Redox Properties of the Iron Complexes of Orally Active Iron ChelatorsCP20, CP502, CP509, andICL670. Helv. Chim. Acta 2004, 87, 3021–3034. [Google Scholar] [CrossRef]
- Rangel, M.; Leite, A.; Silva, A.M.N.; Moniz, T.; Nunes, A.; Amorim, M.J.; Queirós, C.; Cunha-Silva, L.; Gameiro, P.; Burgess, J. Distinctive EPR signals provide an understanding of the affinity of bis-(3-hydroxy-4-pyridinonato) copper(ii) complexes for hydrophobic environments. Dalton Trans. 2014, 43, 9722–9731. [Google Scholar] [CrossRef]
- Santos, M.A.; Gil, M.; Marques, S.; Gano, L.; Cantinho, G.; Chaves, S. N-Carboxyalkyl derivatives of 3-hydroxy-4-pyridinones: Synthesis, complexation with Fe(III), Al(III) and Ga(III) and in vivo evaluation. J. Inorg. Biochem. 2002, 92, 43–54. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | λabs/nm | ε × 103/M−1⋅cm−1 | λem/nm | Stokes Shift/nm |
|---|---|---|---|---|
| 1 | 511 | 3.3 | 526 | 15 |
| 2 | 507 | 1.4 | 530 | 23 |
| 3 | 277 | 46.3 | - | - |
| 4 | 270 | 40.2 | - | - |
| Rho110 | 498 | 89.0 | 515 | 17 |
| 5 (spirolactone) | 270 | 2.1 | - | - |
| 6 (spirolactone) | 262 | 79.8 | - | - |
| 5 (quinoid) | 489 | 0.8 | 521 | 32 |
| 6 (quinoid) | 485 | 18.7 | 508 | 23 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Queirós, C.; Vinhas, S.; Oliveira, J.; Leite, A.; Silva, A.M.G.; Rangel, M. Functionalization of Rhodamine Platforms with 3-Hydroxy-4-pyridinone Chelating Units and Its Fluorescence Behavior towards Fe(III). Molecules 2022, 27, 1567. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27051567
Queirós C, Vinhas S, Oliveira J, Leite A, Silva AMG, Rangel M. Functionalization of Rhodamine Platforms with 3-Hydroxy-4-pyridinone Chelating Units and Its Fluorescence Behavior towards Fe(III). Molecules. 2022; 27(5):1567. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27051567
Chicago/Turabian StyleQueirós, Carla, Sílvia Vinhas, Jéssica Oliveira, Andreia Leite, Ana M. G. Silva, and Maria Rangel. 2022. "Functionalization of Rhodamine Platforms with 3-Hydroxy-4-pyridinone Chelating Units and Its Fluorescence Behavior towards Fe(III)" Molecules 27, no. 5: 1567. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27051567