
Ruthenafuran Complexes Supported by the Bipyridine-Bis(diphenylphosphino)methane Ligand Set: Synthesis and Cytotoxicity Studies


Abstract
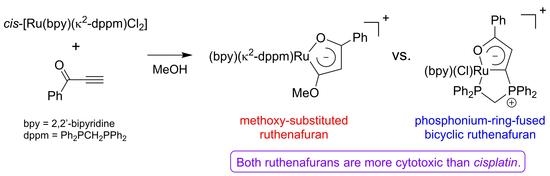
:
1. Introduction
2. Results and Discussion
2.1. Synthesis of Metal Precursors
2.2. Reactions between the Metal Precursors and Phenyl Ynone
2.3. Cytotoxicity Study
3. Materials and Methods
3.1. General Procedures
3.2. Synthesis
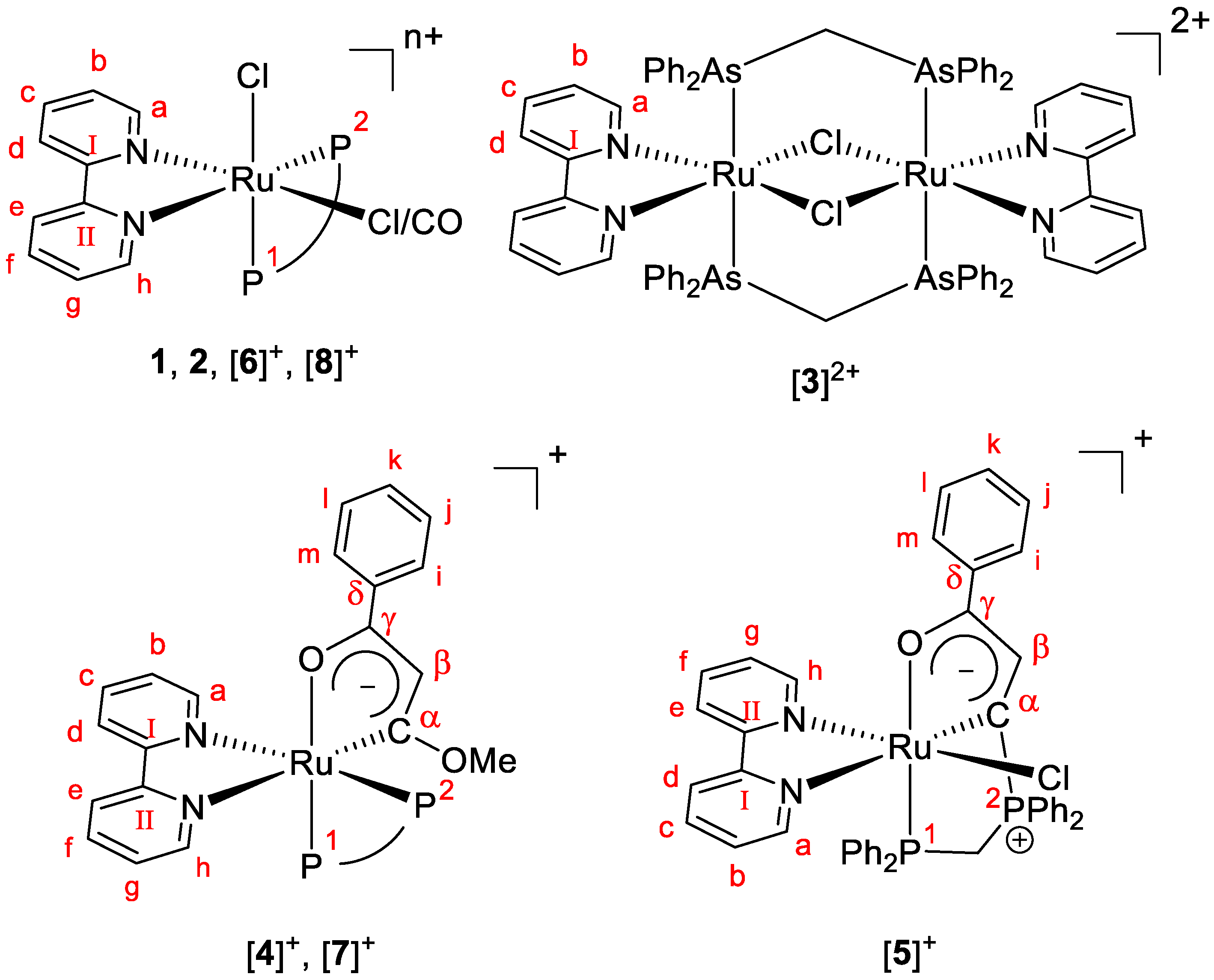
3.2.1. Synthesis of 1, 2 and [3]2+
3.2.2. Synthesis of [4](OTf) and [5](OTf)
3.2.3. Synthesis of [5](OTf) and [6](OTf)
3.2.4. Synthesis of [7](OTf)
3.2.5. Synthesis of [8](OTf)
3.3. X-ray Crystallographic Data
3.4. Cytotoxicity Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schore, N.E. Transition metal-mediated cycloaddition reactions of alkynes in organic synthesis. Chem. Rev. 1988, 88, 1081–1119. [Google Scholar] [CrossRef]
- Adams, R.D. The insertion of alkynes into metal–metal bonds and organic chemistry of the dimetallated olefin complexes. Chem. Soc. Rev. 1994, 23, 335–339. [Google Scholar] [CrossRef]
- McDonald, F.E. Alkynol endo-Cycloisomerizations and Conceptually Related Transformations. Chem. Eur. J. 1999, 5, 3103–3106. [Google Scholar] [CrossRef]
- Bubnov, Y.N.; Klimkina, E.V. Synthesis of some alkaloids using the allyl derivatives of boron (review). Chem. Heterocycl. Compd. 1999, 35, 888–899. [Google Scholar] [CrossRef]
- Nakamura, I.; Yamamoto, Y. Transition-Metal-Catalyzed Reactions in Heterocyclic Synthesis. Chem. Rev. 2004, 104, 2127–2198. [Google Scholar] [CrossRef]
- Zeni, G.; Larock, R.C. Synthesis of heterocycles via palladium pi-olefin and pi-alkyne chemistry. Chem. Rev. 2004, 104, 2285–2310. [Google Scholar] [CrossRef]
- Mori, M. Activation of nitrogen for organic synthesis. J. Organomet. Chem. 2004, 689, 4210–4227. [Google Scholar] [CrossRef]
- Nevado, C.; Echavarren, A.M. Transition Metal-Catalyzed Hydroarylation of Alkynes. Synthesis 2005, 2, 167–182. [Google Scholar] [CrossRef]
- Hashmi, A.S.K. Gold-Catalyzed Organic Reactions. Chem. Rev. 2007, 107, 3180–3211. [Google Scholar] [CrossRef]
- Lazzaroni, R.; Settambolo, R.; Rocchiccioli, S.; Guazzelli, G. From chiral and prochiral N-allylpyrroles to stereodefined pyrrole fused architectures: A particular application of the rhodium-catalyzed hydroformylation. J. Organomet. Chem. 2007, 692, 1812–1816. [Google Scholar] [CrossRef]
- Li, Z.; Brouwer, C.; He, C. Gold-Catalyzed Organic Transformations. Chem. Rev. 2008, 108, 3239–3265. [Google Scholar] [CrossRef] [PubMed]
- Patil, N.T.; Yamamoto, Y. Coinage metal-assisted synthesis of heterocycles. Chem. Rev. 2008, 108, 3395–3442. [Google Scholar] [CrossRef] [PubMed]
- Kirsh, S.F. Construction of Heterocycles by the Strategic Use of Alkyne π-Activation in Catalyzed Cascade Reactions. Synthesis 2008, 20, 3183–3204. [Google Scholar] [CrossRef]
- Cadierno, V.; Gimeno, J. Allenylidene and Higher Cumulenylidene Complexes. Chem. Rev. 2009, 109, 3512–3560. [Google Scholar] [CrossRef]
- Kitamura, T. Transition-Metal-Catalyzed Hydroarylation Reactions of Alkynes Through Direct Functionalization of C–H Bonds: A Convenient Tool for Organic Synthesis. Eur. J. Org. Chem. 2009, 8, 1111–1125. [Google Scholar] [CrossRef]
- Wahba, A.E.; Hamann, M.T. New One-Pot Methodologies for the Modification or Synthesis of Alkaloid Scaffolds. Mar. Drugs 2010, 8, 2395–2416. [Google Scholar] [CrossRef]
- Patil, N.T.; Kavthe, R.D.; Yamamoto, Y. Metal-Catalyzed Intramolecular Heteroatom (X) → Carbon (C) Functional Group Migration Reactions Involving Additions of X–Y Bonds Across Alkynes. Adv. Heterocycl. Chem. 2010, 101, 75–95. [Google Scholar] [CrossRef]
- Shao, Z.; Peng, F. Metal-Mediated Oxidative Cross-Coupling of Terminal Alkynes: A Promising Strategy for Alkyne Synthesis. Angew. Chem. Int. Ed. 2010, 49, 9566–9568. [Google Scholar] [CrossRef]
- Müller, T.J.J. Palladium-Copper Catalyzed Alkyne Activation as an Entry to Multicomponent Syntheses of Heterocycles. In Synthesis of Heterocycles via Multicomponent Reactions II. Topics in Heterocyclic Chemistry; Orru, R., Ruijter, E., Eds.; Springer: Berlin/Heidelberg, Germany, 2010; Volume 25, pp. 25–94. [Google Scholar]
- Vaquero, J.J.; Alvarez-Builla, J. Heterocycles Containing a Ring-Junction Nitrogen. In Modern Heterocyclic Chemistry; Alvarez-Builla, J., Vaquero, J.J., Barluenga, J., Eds.; Wiley-VCH: Weinheim, Germany, 2011; pp. 1989–2070. [Google Scholar]
- Gomez-Suarez, A.; Nolan, S.P. Dinuclear Gold Catalysis: Are Two Gold Centers Better than One? Angew. Chem. Int. Ed. 2012, 51, 8156–8159. [Google Scholar] [CrossRef]
- Marinetti, A.; Jullien, H.; Voituriez, A. Enantioselective, transition metal catalyzed cycloisomerizations. Chem. Soc. Rev. 2012, 41, 4884–4908. [Google Scholar] [CrossRef]
- Watson, I.D.G.; Toste, F.D. Catalytic enantioselective carbon-carbon bond formation using cycloisomerization reactions. Chem. Sci. 2012, 3, 2899–2919. [Google Scholar] [CrossRef]
- Zhang, D.-H.; Zhang, Z.; Shi, M. ChemInform Abstract: Transition Metal-Catalyzed Carbocyclization of Nitrogen and Oxygen-Tethered 1,n-Enynes and Diynes: Synthesis of Five or Six-Membered Heterocyclic Compounds. Chem. Commun. 2012, 48, 10271–10279. [Google Scholar] [CrossRef] [PubMed]
- Gulevich, A.V.; Dudnik, A.S.; Chernyak, N.; Gevorgyan, V. Transition metal-mediated synthesis of monocyclic aromatic heterocycles. Chem. Rev. 2013, 113, 3084–3213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiroodi, R.K.; Gevorgyan, V. Metal-catalyzed double migratory cascade reactions of propargylic esters and phosphates. Chem. Soc. Rev. 2013, 42, 4991–5001. [Google Scholar] [CrossRef]
- Chinchilla, R.; Najera, C. Chemicals from Alkynes with Palladium Catalysts. Chem. Rev. 2014, 114, 1783–1826. [Google Scholar] [CrossRef] [Green Version]
- Fürstner, A. From Understanding to Prediction: Gold- and Platinum-Based π-Acid Catalysis for Target Oriented Synthesis. Acc. Chem. Res. 2014, 47, 925–938. [Google Scholar] [CrossRef]
- Fensterbank, L.; Malacria, M. Molecular Complexity from Polyunsaturated Substrates: The Gold Catalysis Approach. Acc. Chem. Res. 2014, 47, 953–965. [Google Scholar] [CrossRef]
- Dixneuf, P.H. Early Steps of Homogeneous Catalysis in Rennes: Carbon Dioxide Incorporation, Alkyne Activation and Ruthenium Catalysis. Catal. Lett. 2015, 145, 360–372. [Google Scholar] [CrossRef] [Green Version]
- Chung, L.-H.; Wong, C.-Y. Ruthenium-Induced Alkyne Cycloisomerization: Construction of Metalated Heterocycles, Revelation of Unconventional Reaction Pathways, and Exploration of Functional Applications. Chem. Eur. J. 2019, 25, 2889–2897. [Google Scholar] [CrossRef]
- Chung, L.-H.; Yeung, C.-F.; Wong, C.-Y. Ruthenium-Induced Cyclization of Heteroatom-Functionalized Alkynes: Progress, Challenges and Perspectives. Chem. Eur. J. 2020, 26, 6102–6112. [Google Scholar] [CrossRef]
- Wong, C.-Y.; Man, W.-L.; Wang, C.; Kwong, H.-L.; Wong, W.-Y.; Lau, T.-C. Proton-Bridged Dinuclear (salen)Ru Carbene Complexes: Synthesis, Structure, and Reactivity of {[(salchda)Ru═C(OR)(CH═CPh2)]2·H}+. Organometallics 2008, 27, 324–326. [Google Scholar] [CrossRef]
- Wong, C.-Y.; Lai, L.-M.; Pat, P.-K. Ruthenium Acetylide Complexes Supported by Trithiacyclononane and Aromatic Diimine: Structural, Spectroscopic, and Theoretical Studies. Organometallics 2009, 28, 5656–5660. [Google Scholar] [CrossRef]
- Wong, C.-Y.; Lai, L.-M.; Chan, S.-C.; Tai, L.-H. Photophysical and Theoretical Studies of Ruthenium(II)–Acetylide and –Cyanide Complexes with Aromatic Diimine and Trithiacyclononane. Organometallics 2010, 29, 6259–6266. [Google Scholar] [CrossRef]
- Chung, L.-H.; Wong, C.-Y. Isolation of Ruthenium–Indolizine Complexes: Insight into the Metal-Induced Cycloisomerization of Propargylic Pyridines. Organometallics 2013, 32, 3583–3586. [Google Scholar] [CrossRef]
- Chung, L.-H.; Yeung, C.-F.; Ma, D.-L.; Leung, C.-H.; Wong, C.-Y. Metal–Indolizine Zwitterion Complexes as a New Class of Organometallic Material: A Spectroscopic and Theoretical Investigation. Organometallics 2014, 33, 3443–3452. [Google Scholar] [CrossRef]
- Tsui, W.-K.; Chung, L.-H.; Tsang, W.-H.; Yeung, C.-F.; Chiu, C.-H.; Lo, H.-S.; Wong, C.-Y. Synthesis, Spectroscopic and Theoretical Studies of Ruthenafuran and Osmafuran Prepared by Activation of Ynone in Alcohol. Organometallics 2015, 34, 1005–1012. [Google Scholar] [CrossRef]
- Yeung, C.-F.; Chung, L.-H.; Lo, H.-S.; Chiu, C.-H.; Cai, J.; Wong, C.-Y. Isolation of Ruthenium–Indoline and –Indole Zwitterion Complexes: Insight into the Metal-Induced Cyclization of Aniline-Tethered Alkynes and Strategy to Lower the Activation Barrier of Metal–Vinylidene Formation. Organometallics 2015, 34, 1963–1968. [Google Scholar] [CrossRef]
- Chung, L.-H.; Ng, S.-W.; Yeung, C.-F.; Shek, H.-L.; Tse, S.-Y.; Lo, H.-S.; Chan, S.-C.; Tse, M.-K.; Yiu, S.-M.; Wong, C.-Y. Ruthenium–indolizinone complexes as a new class of metalated heterocyclic compounds: Insight into unconventional alkyne activation pathways, revelation of unexpected electronic properties and exploration of medicinal application. Dalton Trans. 2018, 47, 12838–12842. [Google Scholar] [CrossRef]
- Chung, L.-H.; Yeung, C.-F.; Shek, H.-L.; Wong, C.-Y. Isolation of a C3-metalated indolizine complex and a phosphonium ring-fused bicyclic metallafuran from the osmium-induced transformation of pyridine-tethered alkynes. Faraday Discuss. 2019, 220, 196–207. [Google Scholar] [CrossRef]
- Yeung, C.-F.; Chung, L.-H.; Ng, S.-W.; Shek, H.-L.; Tse, S.-Y.; Chan, S.-C.; Tse, M.-K.; Yiu, S.-M.; Wong, C.-Y. Phosphonium-Ring-Fused Bicyclic Metallafuran Complexes of Ruthenium and Osmium. Chem. Eur. J. 2019, 25, 9159–9163. [Google Scholar] [CrossRef]
- Chan, S.-C.; Yeung, C.-F.; Shek, H.-L.; Ng, S.-W.; Tse, S.-Y.; Tse, M.-K.; Yiu, S.-M.; Wong, C.-Y. Iron(II)-induced cycloisomerization of alkynes via “non-vinylidene” pathways for iron(II)-indolizine and -indolizinone complexes. Chem. Commun. 2020, 56, 12644–12647. [Google Scholar] [CrossRef] [PubMed]
- Yeung, C.-F.; Chung, L.-H.; Tse, S.-Y.; Shek, H.-L.; Tse, M.-K.; Yiu, S.-M.; Wong, C.-Y. Conventional and unconventional alkyne activations by Ru and Os for unprecedented dimetalated quinolizinium complexes. Chem. Commun. 2020, 56, 8908–8911. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.-W.; Tse, S.-Y.; Yeung, C.-F.; Chung, L.-H.; Tse, M.-K.; Yiu, S.-M.; Wong, C.-Y. Ru(II)- and Os(II)-Induced Cycloisomerization of Phenol-Tethered Alkyne for Functional Chromene and Chromone Complexes. Organometallics 2020, 39, 1299–1309. [Google Scholar] [CrossRef]
- Shek, H.-L.; Yeung, C.-F.; Chung, L.-H.; Wong, C.-Y. A focused review on the unconventional alkyne activations by ruthenium(II) and osmium(II) complexes supported by 1,2-bis(diphenylphosphino)methane (dppm). Polyhedron 2021, 197, 115023. [Google Scholar] [CrossRef]
- Yeung, C.-F.; Shek, H.-L.; Yiu, S.-M.; Tse, M.-K.; Wong, C.-Y. Controlled Activation of Dipicolinyl-Substituted Propargylic Alcohol by Ru(II) and Os(II) for Unprecedented Indolizine-Fused Metallafuran Complexes. Organometallics 2021, 40, 2458–2466. [Google Scholar] [CrossRef]
- Dierkes, P.; Van Leeuwen, P.W.N.M. The bite angle makes the difference: A practical ligand parameter for diphosphine ligands. J. Chem. Soc. Dalton Trans. 1999, 10, 1519–1529. [Google Scholar] [CrossRef]
- Bianchini, C.; Innocenti, P.; Peruzzini, M.; Romerosa, A.; Zanobini, F. C–C Bond Formation between Vinylidene and Alkynyl Ligands at Ruthenium(II) Leading to either Enynyl or Dienynyl Complexes. Organometallics 1996, 15, 272–285. [Google Scholar] [CrossRef]
- Bianchini, C.; Casares, J.A.; Peruzzini, M.; Romerosa, A.; Zanobini, F. The Mechanism of the Ru-Assisted C-C Bond Cleavage of Terminal Alkynes by Water. J. Am. Chem. Soc. 1996, 118, 4585–4595. [Google Scholar] [CrossRef]
- Clarke, M.J. Ruthenium Metallopharmaceuticals. Coord. Chem. Rev. 2003, 236, 209–233. [Google Scholar] [CrossRef]
- Brabec, V.; Novakova, O. DNA binding mode of ruthenium complexes and relationship to tumor cell toxicity. Drug Resist. Updates 2006, 9, 111–122. [Google Scholar] [CrossRef]
- Kostova, I. Ruthenium complexes as anticancer agents. Curr. Med. Chem. 2006, 13, 1085–1107. [Google Scholar] [CrossRef] [PubMed]
- Gasser, G.; Metzler-Nolte, N. The potential of organometallic complexes in medicinal chemistry. Curr. Opin. Chem. Biol. 2012, 16, 84–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gill, M.R.; Thomas, J.A. Ruthenium(II) polypyridyl complexes and DNA—from structural probes to cellular imaging and therapeutics. Chem. Soc. Rev. 2012, 41, 3179–3192. [Google Scholar] [CrossRef] [PubMed]
- Kandioller, W.; Balsano, E.; Meier, S.M.; Jungwirth, U.; Goschl, S.; Roller, A.; Jakupec, M.A.; Berger, W.; Keppler, B.K.; Hartinger, C.G. Organometallic anticancer complexes of lapachol: Metal centre-dependent formation of reactive oxygen species and correlation with cytotoxicity. Chem. Commun. 2013, 49, 3348–3350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furrer, J.; Süss-Fink, G. Thiolato-bridged dinuclear arene ruthenium complexes and their potential as anticancer drugs. Coord. Chem. Rev. 2016, 309, 36–50. [Google Scholar] [CrossRef]
- Kwong, W.-L.; Lam, K.-Y.; Lok, C.-N.; Lai, Y.-T.; Lee, P.-Y.; Che, C.-M. A Macrocyclic Ruthenium(III) Complex Inhibits Angiogenesis with Down-Regulation of Vascular Endothelial Growth Factor Receptor-2 and Suppresses Tumor Growth In Vivo. Angew. Chem. Int. Ed. 2016, 55, 13524–13528. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Ma, R.; Wang, Z.; Yiu, S.-M.; Zhu, G. Heterodinuclear Pt(IV)–Ru(II) anticancer prodrugs to combat both drug resistance and tumor metastasis. Chem. Commun. 2016, 52, 10735–10738. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, F.P.; Carneiro, Z.A.; Mascharak, P.; Curti, C.; da Silva, R.S. Incorporation of a ruthenium nitrosyl complex into liposomes, the nitric oxide released from these liposomes and HepG2 cell death mechanism. Coord. Chem. Rev. 2016, 306, 701–707. [Google Scholar] [CrossRef]
- Zeng, L.; Gupta, P.; Chen, Y.; Wang, E.; Ji, L.; Chao, H.; Chen, Z.-S. The development of anticancer ruthenium(II) complexes: From single molecule compounds to nanomaterials. Chem. Soc. Rev. 2017, 46, 5771–5804. [Google Scholar] [CrossRef]
- Coverdale, J.P.C.; LaroiyaMcCarron, T.; Romero-Canelón, I. Designing Ruthenium Anticancer Drugs: What Have We Learnt from the Key Drug Candidates? Inorganics 2019, 7, 31. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.-Y.; Kim, C.-Y.; Nam, T.-G. Ruthenium Complexes as Anticancer Agents: A Brief History and Perspectives. Drug Des. Dev. Ther. 2020, 14, 5375–5392. [Google Scholar] [CrossRef] [PubMed]
- Toyama, M.; Inoue, K.-I.; Iwamatsu, S.; Nagao, N. Syntheses and Crystal Structures of Mono(2,2′-bipyridine)dichlorobis-(dimethyl sulfoxide-S)ruthenium(II) Complexes, [RuCl2(bpy)(dmso-S)2]. Bull. Chem. Soc. Jpn. 2006, 79, 1525–1534. [Google Scholar] [CrossRef]
- Schraml, J.; Čapka, M.; Blechta, V. 31P and 13C NMR spectra of cyclohexylphenylphosphines, tricyclohexylphosphine and triphenylphosphine. Magn. Reson. Chem. 1992, 30, 544–547. [Google Scholar] [CrossRef]
- Chamchoy, K.; Pakotiprapha, D.; Pumirat, P.; Leartsakulpanich, U.; Boonyuen, U. Application of WST-8 based colorimetric NAD(P)H detection for quantitative dehydrogenase assays. BMC Biochem. 2019, 20, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | IC50 (μM) |
|---|---|
| [4](OTf) | 2.53 ± 0.40 |
| [5](OTf) | 1.60 ± 0.20 |
| [7](OTf) | 2.83 ± 0.45 |
| [9](OTf) | 0.84 ± 0.08 |
| cis-[Pt(NH3)2Cl2] | 21.8 ± 1.34 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yeung, C.-F.; Tang, S.-H.; Yang, Z.; Li, T.-Y.; Li, K.-K.; Chan, Y.-M.; Shek, H.-L.; Io, K.-W.; Tam, K.-T.; Yiu, S.-M.; et al. Ruthenafuran Complexes Supported by the Bipyridine-Bis(diphenylphosphino)methane Ligand Set: Synthesis and Cytotoxicity Studies. Molecules 2022, 27, 1709. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27051709
Yeung C-F, Tang S-H, Yang Z, Li T-Y, Li K-K, Chan Y-M, Shek H-L, Io K-W, Tam K-T, Yiu S-M, et al. Ruthenafuran Complexes Supported by the Bipyridine-Bis(diphenylphosphino)methane Ligand Set: Synthesis and Cytotoxicity Studies. Molecules. 2022; 27(5):1709. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27051709
Chicago/Turabian StyleYeung, Chi-Fung, Sik-Him Tang, Zhe Yang, Tsun-Yin Li, Ka-Kit Li, Yuen-Man Chan, Hau-Lam Shek, Kai-Wa Io, King-Ting Tam, Shek-Man Yiu, and et al. 2022. "Ruthenafuran Complexes Supported by the Bipyridine-Bis(diphenylphosphino)methane Ligand Set: Synthesis and Cytotoxicity Studies" Molecules 27, no. 5: 1709. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27051709