
Iridium(I)-Catalyzed Isoindolinone-Directed Branched-Selective Aromatic C–H Alkylation with Simple Alkenes


Shanghai Engineering Research Center of Molecular Therapeutics and New Drug Development, School of Chemistry and Molecular Engineering, East China Normal University, Shanghai 200062, China
*
Authors to whom correspondence should be addressed.
Molecules 2022, 27(6), 1923; https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27061923
Submission received: 16 February 2022
/
Revised: 5 March 2022
/
Accepted: 10 March 2022
/
Published: 16 March 2022
(This article belongs to the Section Organic Chemistry)
Abstract
:We report an iridium(I)-catalyzed branched-selective C–H alkylation of N-arylisoindolinones with simple alkenes as the alkylating agents. The amide carbonyl group of the isoindolinone motif acts as the directing group to assist the ortho C–H activation of the N-aryl ring. With this atom-economic and highly branched-selective protocol, an array of biologically relevant N-arylisoindolinones were obtained in good yields. Asymmetric control was achieved with up to 87:13 er when a BiPhePhos-like chiral ligand was employed.
1. Introduction
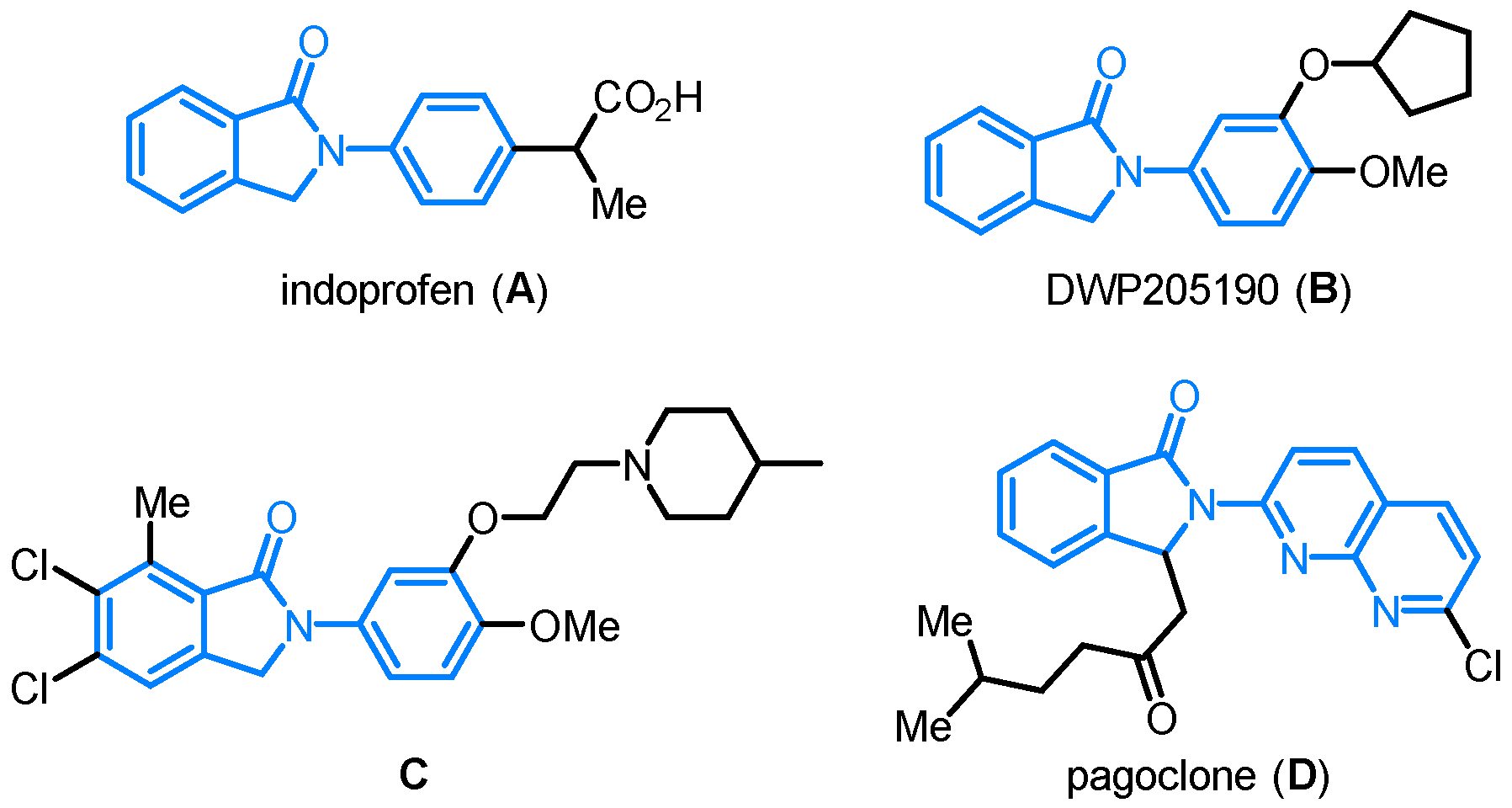
The skeleton of isoindolinone is widely present in a number of natural products, biologically active molecules and pharmaceuticals [1,2,3,4,5]. Among them, N-arylisoindolinones belong to an important class of compounds showing very broad biological activities [6,7]. For example, as shown in Figure 1, indoprofen (A) is known as a nonsteroidal anti-inflammatory drug (NSAID) and cyclo-oxygenase (COX) inhibitor [8], and DWP205190 (B) displays inhibitory activity toward tumor necrosis factor TNF-α production [9,10]. Compound C behaves as a potent and selective 5-HT2C antagonist [11]. Pagoclone (D) is a partial benzodiazepine-GABA receptor agonist that is used for the treatment of panic and other anxiety disorders [12]. As a result, the development of new methodologies toward the rapid access of N-arylisoindolinones bearing different substituting patterns is highly appealing.
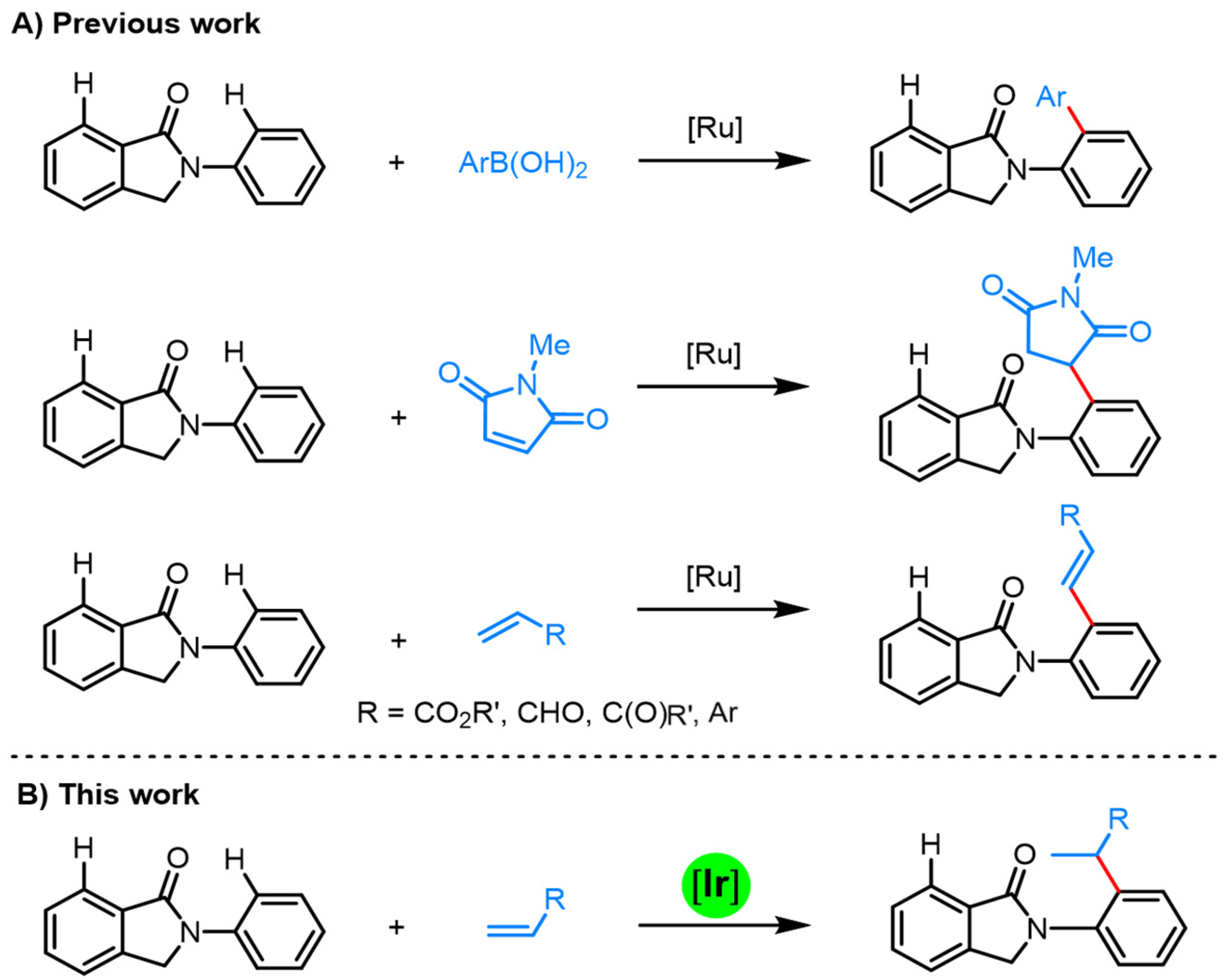
Over the past decades, transition metal-catalyzed directing group (DG)-assisted aromatic C–H activation has emerged as a very powerful synthetic protocol [13,14,15,16,17,18,19,20], thus offering numerous direct C–H functionalization strategies for biologically related molecules. In this context, the amide-carbonyl group in the N-arylisoindolinone skeleton may act as a good directing group to assist the transition metal catalyst in the ortho position C–H activation. However, there are two distinct ortho C–H sites, either from the lactam-fused benzene ring or the N-aryl ring. As a result, the site-selective control is the key for realizing such type of C–H functionalizations. Recently, Gramage-Doria and coworkers successfully realized the site-selective C–H functionalization of N-arylisoindolinones by employing ruthenium catalysis. With aryl boronic acids, maleimides or activated alkenes such as α,β-unsaturated alkenes or styrenes as the functionalizing agents (Scheme 1A), the ortho C–H functionalization of the N-aryl ring was achieved with high efficiency [21,22,23]. Inspired by these advances, we decided to develop new types of C–H functionalization strategies by focusing on the biologically relevant N-arylisoindolinone motif.
In recent years, iridium catalysts have been successfully applied to an array of DG-assisted aromatic C–H alkylation reactions [24,25,26,27,28,29,30,31,32,33,34,35,36,37,38]. By employing alkenes or vinyl ethers as the alkylating agents, significant advances have been made by Shibata, Hartwig, Nishimura, Bower and others [26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48]. This iridium-catalyzed atom-economic C–H alkylation protocol generally favors the formation of branched-selective products [49,50]. By utilizing different types of chiral ligands, a number of asymmetric versions of such transformations have been realized [28,29,31,32,33,34,37,38]. In view of these progress, we envisioned that the amide carbonyl group of the isoindolinone motif may act as an efficient directing group, to enable the development of an iridium-catalyzed branched-selective C–H alkylation reaction with simple alkenes (Scheme 1B), and asymmetric control may be achieved with suitable chiral phosphine ligands. Herein, we report the details of this study.
2. Results and Discussion
We chose N-phenylisoindolinone (1a) as the model substrate and 1-octene (2a) as the alkylating agent for our initial studies towards this iridium-catalyzed C–H functionalization reaction. To allow the rapid access of structurally diversified products without considering the stereochemistry for the initial biological evaluation purpose, racemic ligands were employed. Upon thorough condition optimizations in terms of the iridium sources, ligands, solvents and temperature, the desired branched-selective alkylation product 3a was ultimately obtained in an 87% yield (isolated yield: 81%) with a >20:1 branched selectivity (Table 1, entry 1). A BiPhenPhos-like bidentate ligand (L1), of which the chiral form has been successfully utilized to the branched-selective and enantioselective N-acetyl directed C–H alkylation of anilides with alkenes as developed by Bower and coworkers [37], which was crucial for achieving the good reactivity as well as the excellent branched-selectivity control. The use of the cationic [Ir(COD)2]BArF pre-catalyst was also crucial for this transformation. A set of control experiments was conducted to understand the role of each reactant (Table 1). Among different bidentate phosphine ligands tested, rac-BINAP showed very low efficiency while rac-BIPHEP produced the desired product 3a in a 12% yield with 10:1 branched selectivity (Table 1, entries 2 and 3). With dppf or dppb as the ligand, the desired product 3a was afforded in high branched selectivities, albeit with 20–25% yields (entries 4 and 5). On the other hand, small bite-angle ligands such as dppe showed a low branched selectivity (entry 6). These results are in accordance with literature precedents in which the branched-selective product formation is favored by larger bite-angle ligands [35,36]. It is worth mentioning that for all these entries, no other side products (i.e., the N-aryl dialkylation product, alkylation product on the ortho-position on the aryl ring of the isoindolinone) were observed. Next, the effect of solvent was investigated. While different solvents such as cyclopentyl methyl ether (CPME), toluene, 1,2-dichloroethane (DCE), chlorobenzene or m-xylene could all produce the desired product in excellent branched selectivity, a decreased reaction efficiency was observed, and the use of 1,4-dioxane as the solvent was superior (Table 1, entries 7–11).
With the optimization conditions in hand, we applied this branched-selective C–H alkylation reaction toward the synthesis of different substituted N-arylisoindolinones (Table 2). Alkyl-substituted alkenes such as 1-octene or 1-hexene produced the corresponding products in high yields (3a and 3b). Different styrene-type alkenes were then investigated and excellent branched selectivities were generally observed. Simple styrene or styrenes bearing electron-donating groups such as methyl or methoxyl at different positions were suitable substrates, yielding corresponding products with good efficiency (3d–3g). On the other hand, para- or meta-fluoro-substituted styrenes were less efficient, yielding the desired products in moderate yields (3h and 3i) (56% or 58%, respectively). N-arylisoindolinones bearing different substituents on the N-aryl ring were then tested. Both electron-donating groups such methyl or methoxyl and electron-withdrawing groups such as chloro or fluoro at different positions could all be well-tolerated, giving the corresponding products in moderate to high yields (3j–3p). Again, excellent branched selectivities were observed for these substrates, thus guaranteeing the rapid collection of a set of N-arylisoindolinones with distinct electron properties.
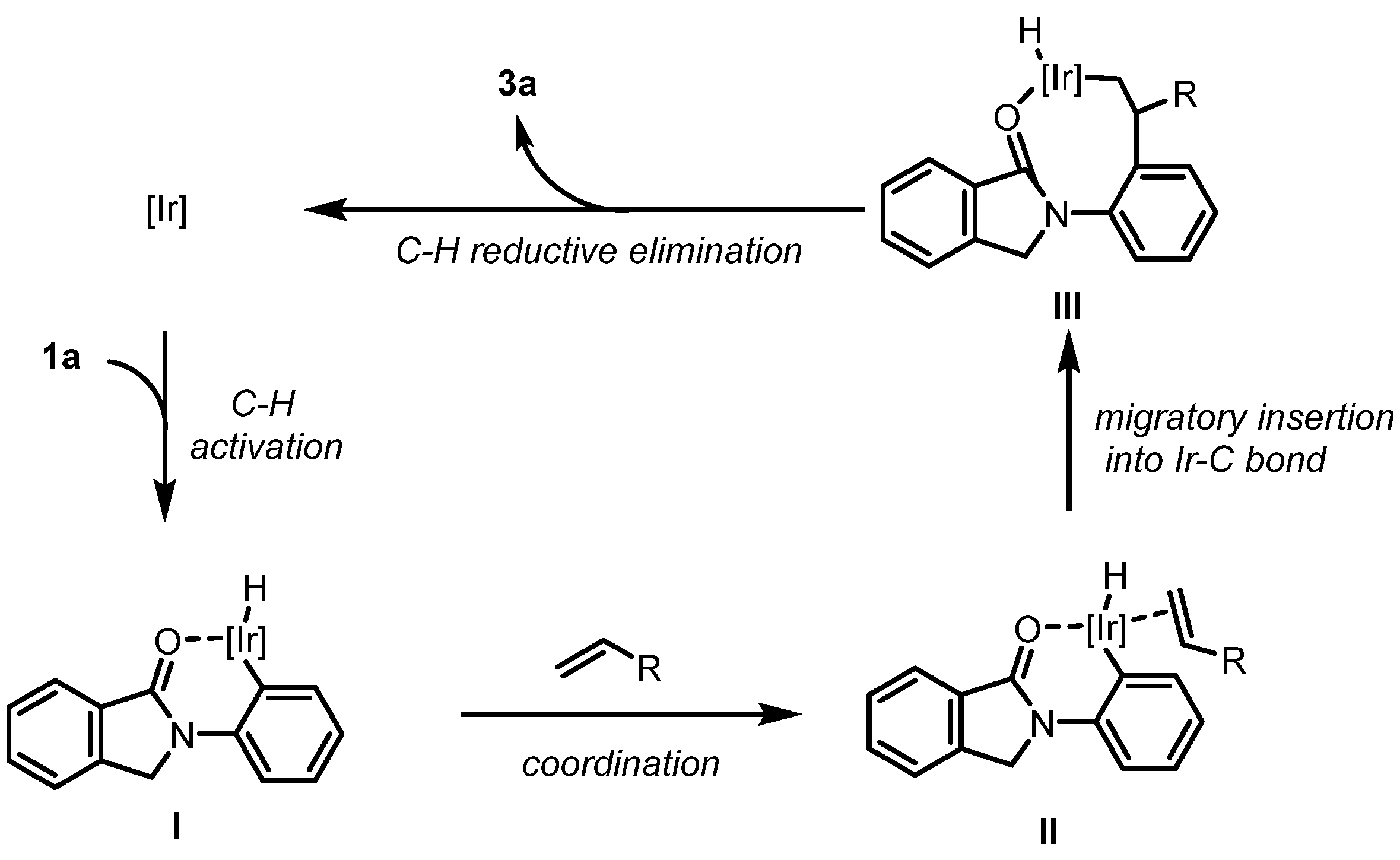
Based on our experimental results as well as literature precedents [37,49,50], a plausible mechanism by following a modified Chalk–Harrod type pathway is proposed for this iridium(I)-catalyzed branched-selective C–H alkylation reaction (Scheme 2). First, with the chelation assistance from the amide carbonyl group of the isoindolinone ring, the iridium catalyst activates the C–H bond by forming an iridium hydride species I. The alkene substrate then coordinates with intermediate I to form intermediate II. Migratory insertion of the alkene moiety into the Ir–C bond leads to intermediate III, by which the branched-selective product formation is favored. Intermediate III then undergoes C–H reductive elimination to deliver both the iridium(I) catalyst as well as the desired product 3a.
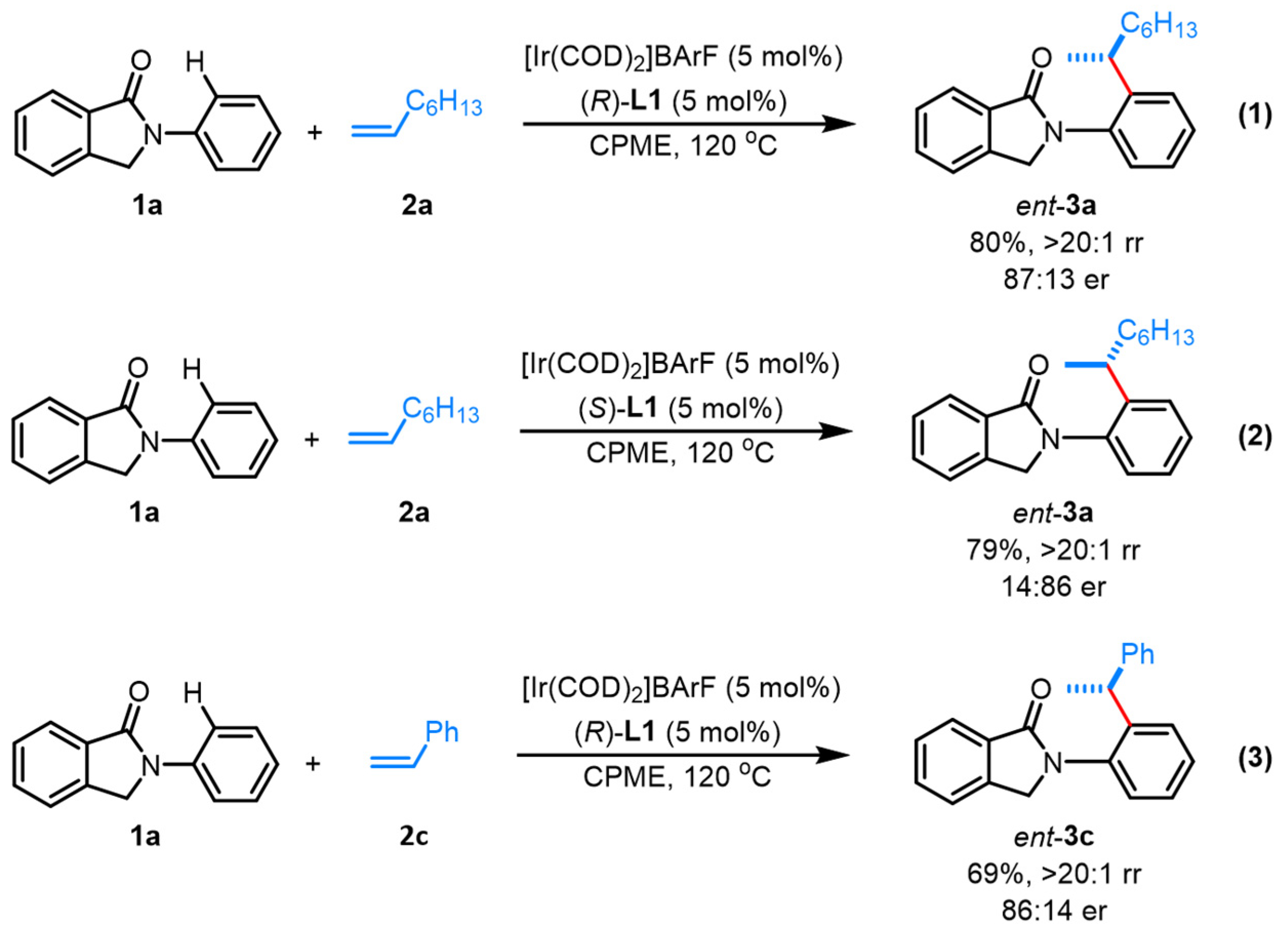
With excellent branched-selective control being achieved, attempts for asymmetric control for this iridium-catalyzed alkylation were conducted with the screening of a handful of readily available chiral bidentate phosphine ligands. With 1-octene (2a) as the alkylating agents, a thorough condition optimization was conducted (see Supplementary Materials). Finally, with chiral BiPhePhos-like (R)-L1 as the ligand and CPME as the solvent, the corresponding optically active product 3a was obtained with excellent branched selectivity and high enantioselectivity (87:13 er) (Scheme 3, Equation (1)). Styrene (2c) was also applicable toward this asymmetric protocol, yielding corresponding product 3c in 86:14 er (Scheme 3, Equation (3)). Furthermore, when (S)-L1 was utilized as the ligand, the corresponding enantiomer of 3a was obtained with 14:86 er (Scheme 3, Equation (2)), thus allowing the rapid access of both enantiomers of 3a by switching the absolute configuration of the ligand. These results further illustrate the novel structural nature of the BiPhePhos-type ligand for the efficient asymmetric control on such types of alkylation reactions. This strategy also offers a powerful strategy for potential biological evaluation of structure-diversified chiral isoindolinone skeletons.
3. Experimental Section
Unless otherwise stated, 1H-NMRand 13C-NMRspectra were recorded on a Bruker (400 MHz) spectrometer. Chemical shifts were reported in parts per million (ppm), and the residual solvent peak was used as an internal reference: proton (chloroform δ 7.26), carbon (chloroform δ 77.0) or tetramethylsilane (TMS δ 0.00) was used as a reference. Data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, dd = doublet of doublets, td = triplet of doublets, dt = doublet of triplets, ddd = doublet of doublet of doublets, m = multiplet, bs = broad singlet, etc.), coupling constants (Hz) and integration. High-resolution mass spectra (HRMS) were obtained on IonSpec FT-ICR or Waters Micromass Q-TOF micro Synapt high-definition mass spectrometer. Optical rotation was determined on RUDOLPH AUTOPOL-VI apparatus. Melting points were measured on INESA WRR-Y melting point apparatus. Flash chromatography was carried out with 300–400 mesh silica gel. All the key reactions were carried out under nitrogen atmosphere with a stir bar in a sealed vial. 1,4-dioxane (99.5%, extra dry, stabilized) used for the key reactions was purchased from Acros and degassed with nitrogen before use. [Ir(COD)2] BArF was prepared according to literature methods. All the ligands were purchased from Strem Chemicals and were used as received. All starting materials 1a–p were prepared according to literature methods [22,23,24].
General Procedure for the Synthesis of Product (3a–3p)
In a N2-filled glovebox, a 4 mL baked vial charged with a stir bar and 6.4 mg of [Ir(COD)2]BArF (0.005 mmol), 4.0 mg of rac-L1 (0.005 mmol) was added. Then, 0.2 mL of 1, 4-dioxane was added into the vial and the resulting solution was stirred for 5 min. Then, 1a (21 mg, 0.1 mmol) and 2a (56 mg, 0.5 mmol) were added. The vial was tightly capped with a screw cap and then removed from the glovebox and placed in a pre-heated aluminum block at 120 °C for 48 h. The reaction mixture was directly purified by column chromatography on silica gel with EtOAc/PE mixture as an eluent.
2-(2-(octan-2-yl)phenyl)isoindolin-1-one (3a)
White foam, 26.0 mg, 81% yield, 1H-NMR(400 MHz, CDCl3) δ 7.96 (d, J = 7.5 Hz, 1H), 7.64–7.58 (m, 1H), 7.56–7.50 (m, 2H), 7.41–7.38 (m, 2H), 7.30–7.25 (m, 1H), 7.21 (d, J = 7.6 Hz, 1H), 4.81–4.59 (m, 2H), 2.82–2.71 (m, 1H), 1.64–1.46 (m, 2H), 1.31–1.06 (m, 11H), 0.80 (t, J = 6.4 Hz, 3H). 13C-NMR(101 MHz, CDCl3) δ 167.28, 145.51, 140.45, 135.23, 131.41, 130.60, 127.75, 127.24, 127.02, 125.95, 125.62, 123.23, 121.73, 53.18, 37.05, 32.72, 30.65, 28.37, 26.80, 21.59, 21.28, 13.00. HRMS (ESI) calcd. for C22H28NO [M + H]+: 322.2081, Found: 322.2076. Chiralcel IC column, n-hexane/i-PrOH = 100:10, flow rate = 1.0 mL/min, λ = 254 nm, tR (major isomer) = 25.168 min, tR (minor isomer) = 21.322 min. er: 87:13.
2-(2-(hexan-2-yl)phenyl)isoindolin-1-one (3b)
White foam, 24.9 mg, 85% yield, 1H-NMR(400 MHz, CDCl3) δ 7.89 (d, J = 7.4 Hz, 1H), 7.58–7.51 (m, 1H), 7.49–7.42 (m, 2H), 7.34–7.30 (m, 2H), 7.23–7.20 (m, 1H), 7.14 (d, J = 7.6 Hz, 1H), 4.77–4.51 (m, 2H), 2.79–2.62 (m, 1H), 1.61–1.40 (m, 2H), 1.25–0.95 (m, 7H), 0.73 (t, J = 7.0 Hz, 3H). 13C-NMR(101 MHz, CDCl3) δ 167.28, 145.50, 140.46, 135.25, 131.44, 130.59, 127.78, 127.26, 127.04, 125.97, 125.65, 123.29, 121.73, 53.19, 36.73, 32.79, 29.05, 21.77, 21.43, 12.92. HRMS (ESI) calcd. for C20H24NO [M + H]+: 294.1706, Found: 294.1700.
2-(2-(1-phenylethyl)phenyl)isoindolin-1-one (3c)
White solid, 22.5 mg, 72% yield, m.p. = 130–132 °C. 1H-NMR(400 MHz, CDCl3) δ 7.96 (d, J = 7.1 Hz, 1H), 7.59–7.48 (m, 3H), 7.45–7.38 (m, 1H), 7.36–7.27 (m, 2H), 7.17 (d, J = 7.7 Hz, 1H), 7.05 (s, 3H), 6.95 (s, 2H), 4.54–4.31 (m, 2H), 3.53 (s, 1H), 1.61 (d, J = 7.0 Hz, 3H). 13C-NMR(101 MHz, CDCl3) δ 167.99, 146.41, 144.76, 141.97, 136.86, 132.21, 131.55, 128.59, 128.24, 128.22, 128.13, 127.93, 127.38, 127.35, 125.87, 124.13, 122.53, 53.40, 39.77, 21.85. HRMS (ESI) calcd. for C22H20NO [M+H]+: 314.1514, Found: 314.1562. Chiralcel IC column, n-hexane/i-PrOH = 100:10, flow rate = 1.0 mL/min, λ = 254 nm, tR (major isomer) = 19.801 min, tR (minor isomer) = 20.807 min. er: 86:14.
2-(2-(1-(p-tolyl)ethyl)phenyl)isoindolin-1-one (3d)
White solid, 28.4 mg, 87% yield, m.p. = 140–142 °C. 1H-NMR(400 MHz, CDCl3) δ 7.96 (d, J = 7.2 Hz, 1H), 7.60–7.48 (m, 3H), 7.43–7.37 (m, 1H), 7.35–7.28 (m, 2H), 7.17 (d, J = 7.6 Hz, 1H), 6.87 (s, 4H), 4.44 (d, J = 16.9 Hz, 1H), 4.34 (q, J = 6.9 Hz, 1H), 3.67 (s, 1H), 2.27 (s, 3H), 1.59 (d, J = 7.0 Hz, 3H). 13C-NMR(101 MHz, CDCl3) δ 167.00, 143.95, 142.27, 140.94, 135.70, 134.31, 131.21, 130.50, 127.83, 127.57, 127.18, 127.08, 126.93, 126.23, 126.18, 123.11, 121.45, 52.48, 38.22, 20.89, 19.88. HRMS (ESI) calcd. for C23H22NO [M + H]+: 328.1701, Found: 328.1708.
2-(2-(1-(4-methoxyphenyl)ethyl)phenyl)isoindolin-1-one (3e)
White solid, 29.8 mg, 87% yield, m.p. = 96–98 °C. 1H-NMR(400 MHz, CDCl3) δ 7.97 (d, J = 7.3 Hz, 1H), 7.60–7.46 (m, 3H), 7.43–7.37 (m, 1H), 7.36–7.28 (m, 2H), 7.17 (d, J = 7.7 Hz, 1H), 6.88 (s, 2H), 6.62 (d, J = 7.9 Hz, 2H), 4.45 (d, J = 16.9 Hz, 1H), 4.34 (q, J = 7.0 Hz, 1H), 3.70 (s, 4H), 1.58 (d, J = 7.1 Hz, 3H). 13C-NMR(101 MHz, CDCl3) δ 166.93, 156.63, 143.99, 140.89, 137.42, 135.67, 131.22, 130.50, 127.55, 127.21, 127.19, 127.09, 126.81, 126.22, 123.08, 121.51, 112.53, 54.17, 52.47, 37.79, 20.96. HRMS (ESI) calcd. for C23H22NO2 [M + H]+: 344.1651, Found: 344.1679.
2-(2-(1-(2-methoxyphenyl)ethyl)phenyl)isoindolin-1-one (3f)
White solid, 25.7 mg, 75% yield, m.p. = 157–159 °C. 1H-NMR(400 MHz, CDCl3) δ 7.96 (d, J = 7.1 Hz, 1H), 7.61–7.47 (m, 3H), 7.44–7.37 (m, 1H), 7.33–7.27 (m, 2H), 7.14 (d, J = 7.6 Hz, 1H), 7.08–6.98 (m, 1H), 6.88 (d, J = 7.4 Hz, 1H), 6.82–6.74 (m, 1H), 6.47 (d, J = 8.1 Hz, 1H), 4.84 (q, J = 6.9 Hz, 1H), 4.35 (d, J = 16.9 Hz, 1H), 3.42 (d, J = 16.8 Hz, 1H), 3.10 (s, 3H), 1.54 (d, J = 7.0 Hz, 3H). 13C-NMR(101 MHz, CDCl3) δ 167.94, 156.09, 145.51, 142.09, 136.87, 135.06, 132.60, 131.21, 128.42, 128.30, 128.12, 127.86, 127.60, 127.08, 126.82, 124.03, 122.42, 120.29, 109.77, 54.38, 52.95, 31.96, 20.53. HRMS (ESI) calcd. for C23H22NO2 [M + H]+: 344.1651, Found: 344.1679.
2-(2-(1-(m-tolyl)ethyl)phenyl)isoindolin-1-one (3g)
White solid, 23.5 mg, 72% yield, m.p. = 119–121 °C. 1H-NMR(400 MHz, CDCl3) δ 7.97 (d, J = 6.7 Hz, 1H), 7.60–7.48 (m, 3H), 7.46–7.38 (m, 1H), 7.35–7.24 (m, 2H), 7.16 (d, J = 7.3 Hz, 1H), 6.98–6.92 (m, 1H), 6.83 (d, J = 6.6 Hz, 1H), 6.70 (s, 2H), 4.45–4.08 (m, 2H), 3.34 (s, 1H), 1.94 (s, 3H), 1.59 (d, J = 6.9 Hz, 3H). 13C-NMR(101 MHz, CDCl3) δ 167.86, 146.42, 144.73, 142.07, 137.89, 136.92, 132.32, 131.48, 128.52, 128.39, 128.25, 128.07, 128.05, 127.72, 127.31, 126.49, 124.17, 124.11, 122.43, 53.31, 39.92, 21.72, 21.00. HRMS (ESI) calcd. for C23H22NO [M + H]+: 328.1701, Found: 328.1708.
2-(2-(1-(4-fluorophenyl)ethyl)phenyl)isoindolin-1-one (3h)
White solid, 19.2 mg, 58% yield, m.p. = 134–136 °C. 1H-NMR(400 MHz, CDCl3) δ 7.96 (d, J = 7.2 Hz, 1H), 7.66–7.28 (m, 6H), 7.18 (d, J = 7.5 Hz, 1H), 6.93 (s, 2H), 6.84–6.70 (m, 2H), 4.56–4.28 (m, 2H), 3.65 (s, 1H), 1.58 (d, J = 7.0 Hz, 3H). 13C-NMR(101 MHz, CDCl3) δ 167.94, 161.09 (d, J = 244.1 Hz), 144.57, 142.04, 141.78, 136.74, 132.15, 131.69, 128.77, 128.69, 128.66, 128.24, 127.88, 127.50, 124.16, 122.60, 114.93 (d, J = 21.1 Hz), 53.47, 38.94, 21.99. HRMS (ESI) calcd. for C22H19FNO [M + H]+: 332.1451, Found: 332.1478.
2-(2-(1-(3-fluorophenyl)ethyl)phenyl)isoindolin-1-one (3i)
White solid, 18.5 mg, 56% yield, m.p. = 121–123 °C. 1H-NMR(400 MHz, CDCl3) δ 7.96 (d, J = 7.4 Hz, 1H), 7.59–7.38 (m, 4H), 7.37–7.31 (m, 2H), 7.19 (d, J = 7.7 Hz, 1H), 7.04–6.97 (m, 1H), 6.79–6.65 (m, 3H), 4.49 (d, J = 16.8 Hz, 1H), 4.41 (q, J = 7.1 Hz, 1H), 3.64 (s, 1H), 1.59 (d, J = 7.1 Hz, 3H). 13C-NMR(101 MHz, CDCl3) δ 167.96, 162.77 (d, J = 245.8 Hz), 149.14, 144.08, 141.76, 136.80, 132.10, 131.70, 129.61 (d, J = 8.2 Hz), 128.70, 128.26, 128.22, 127.94, 127.63, 124.18, 123.20, 122.56, 114.16 (d, J = 21.4 Hz), 112.77 (d, J = 21.2 Hz), 53.45, 39.46, 21.70. HRMS (ESI) calcd. for C22H19FNO [M + H]+: 332.1451, Found: 332.1478.
2-(4-methyl-2-(1-phenylethyl)phenyl)isoindolin-1-one (3j)
White solid, 20.3 mg, 62% yield, m.p. = 146–148 °C. 1H-NMR(400 MHz, CDCl3) δ 7.96 (d, J = 6.9 Hz, 1H), 7.59–7.47 (m, 2H), 7.36–7.28 (m, 2H), 7.17–6.93 (m, 7H), 4.48–4.24 (m, 2H), 3.53 (s, 1H), 2.42 (s, 3H), 1.61 (d, J = 7.1 Hz, 3H). 13C-NMR(101 MHz, CDCl3) δ 168.09, 146.53, 144.30, 141.99, 138.39, 134.20, 132.32, 131.47, 128.62, 128.21, 128.07, 128.04, 127.99, 127.37, 125.83, 124.09, 122.52, 53.43, 39.68, 21.87, 21.52. HRMS (ESI) calcd. for C23H22NO [M + H]+: 328.1701, Found: 328.1708.
2-(4-methoxy-2-(1-phenylethyl)phenyl)isoindolin-1-one (3k)
White solid, 27.4 mg, 80% yield, m.p. = 140–142 °C. 1H-NMR(400 MHz, CDCl3) δ 7.94–7.83 (m, 1H), 7.50–7.39 (m, 2H), 7.21 (s, 1H), 7.05–6.74 (m, 8H), 4.34–4.21 (m, 2H), 3.77 (s, 3H), 3.29 (s, 1H), 1.51 (d, J = 7.1 Hz, 3H). 13C-NMR(101 MHz, CDCl3) δ 168.22, 159.53, 146.09, 141.97, 134.83, 132.26, 131.48, 129.63, 129.23, 128.25, 128.06, 127.32, 125.92, 124.07, 122.50, 114.30, 111.65, 55.50, 53.52, 39.94, 21.84. HRMS (ESI) calcd. for C23H22NO2 [M + H]+: 344.1651, Found: 344.1679.
2-(4-fluoro-2-(1-phenylethyl)phenyl)isoindolin-1-one (3l)
White solid, 27.1 mg, 82% yield, m.p. = 129–131 °C. 1H-NMR(400 MHz, CDCl3) δ 7.87 (d, J = 7.1 Hz, 1H), 7.52–7.41 (m, 2H), 7.25–6.81 (m, 9H), 4.46–4.21 (m, 2H), 3.37 (s, 1H), 1.50 (d, J = 7.1 Hz, 3H). 13C-NMR(101 MHz, CDCl3) δ 167.03, 161.45 (d, J = 247.4 Hz), 146.34, 144.57, 140.83, 131.69, 130.91, 130.64, 128.80 (d, J = 9.0 Hz), 127.30, 127.15, 126.21, 125.10, 123.10, 121.50, 114.00 (d, J = 22.7 Hz), 113.09 (d, J = 22.6 Hz), 52.32, 38.87, 20.69. HRMS (ESI) calcd. for C22H19FNO [M + H]+: 332.1451, Found: 332.1478.
2-(5-methyl-2-(1-phenylethyl)phenyl)isoindolin-1-one (3m)
White solid, 19.6 mg, 60% yield, m.p. = 157–159 °C. 1H-NMR(400 MHz, CDCl3) δ 7.96 (d, J = 7.1 Hz, 1H), 7.60–7.46 (m, 2H), 7.39 (d, J = 4.5 Hz, 1H), 7.33–7.18 (m, 2H), 7.13–6.89 (m, 6H), 4.51–4.21 (m, 2H), 3.53 (s, 1H), 2.34 (s, 3H), 1.59 (d, J = 7.1 Hz, 3H). 13C-NMR(101 MHz, CDCl3) δ 167.02, 145.58, 140.91, 140.59, 136.14, 135.52, 131.21, 130.46, 128.34, 127.79, 127.15, 127.05, 126.74, 126.27, 124.75, 123.07, 121.46, 52.35, 38.41, 20.86, 19.77. HRMS (ESI) calcd. for C23H22NO [M + H]+: 328.1701, Found: 328.1708.
2-(5-methoxy-2-(1-phenylethyl)phenyl)isoindolin-1-one (3n)
White solid, 28.1 mg, 82% yield, m.p. = 170–172 °C. 1H-NMR(400 MHz, CDCl3) δ 7.95 (d, J = 6.3 Hz, 1H), 7.59–7.47 (m, 2H), 7.41 (d, J = 7.3 Hz, 1H), 7.28 (d, J = 6.1 Hz, 1H), 7.08–6.88 (m, 6H), 6.72 (s, 1H), 4.49–4.22 (m, 2H), 3.79 (s, 3H), 3.55 (s, 1H), 1.58 (d, J = 6.4 Hz, 3H). 13C-NMR(101 MHz, CDCl3) δ 167.93, 158.65, 146.79, 141.92, 137.57, 136.83, 132.16, 131.57, 128.64, 128.18, 128.13, 127.25, 125.79, 124.13, 122.54, 114.12, 113.79, 55.45, 53.32, 39.18, 22.02. HRMS (ESI) calcd. for C23H22NO2 [M + H]+: 344.1651, Found: 344.1679.
2-(5-chloro-2-(1-phenylethyl)phenyl)isoindolin-1-one (3o)
White solid, 16.7 mg, 48% yield, m.p. = 169–172 °C. 1H-NMR(400 MHz, CDCl3) δ 7.88 (d, J = 7.2 Hz, 1H), 7.55–7.41 (m, 2H), 7.41–7.30 (m, 2H), 7.22 (d, J = 7.1 Hz, 1H), 7.11 (s, 1H), 6.98 (s, 3H), 6.84 (s, 2H), 4.39–4.22 (m, 2H), 3.43 (s, 1H), 1.51 (d, J = 7.1 Hz, 3H). 13C-NMR(101 MHz, CDCl3) δ 166.83, 144.86, 142.50, 140.79, 136.96, 131.42, 130.76, 130.74, 128.03, 127.62, 127.43, 127.27, 127.22, 126.20, 125.03, 123.16, 121.54, 52.14, 38.45, 20.71. HRMS (ESI) calcd. for C22H19ClNO [M + H]+: 348.1155, Found: 348.1168.
2-(5-fluoro-4-methyl-2-(1-phenylethyl)phenyl)isoindolin-1-one (3p)
White solid, 20.7 mg, 61% yield, m.p. = 130–132 °C. 1H-NMR(400 MHz, CDCl3) δ 7.87 (d, J = 7.2 Hz, 1H), 7.52–7.39 (m, 2H), 7.24–7.19 (m, 2H), 6.98 (s, 3H), 6.85 (s, 2H), 6.77 (d, J = 9.6 Hz, 1H), 4.41–4.15 (m, 2H), 3.46 (s, 1H), 2.25 (s, 3H), 1.50 (d, J = 7.0 Hz, 3H). 13C-NMR(101 MHz, CDCl3) δ 166.96, 158.63 (d, J = 245.6 Hz), 145.26, 140.79, 139.20, 134.23 (d, J = 9.2 Hz), 130.89, 130.66, 129.45 (d, J = 5.7 Hz), 127.21, 127.15, 126.20, 124.89, 124.08 (d, J = 16.7 Hz), 123.13, 121.52, 113.85 (d, J = 22.7 Hz), 52.18, 38.20, 20.92, 13.64 (d, J = 2.9 Hz). HRMS (ESI) calcd. for C23H21FNO [M + H]+: 346.1607, Found: 346.1635.
4. Conclusions
In summary, we have developed an iridium-catalyzed branched-selective C–H alkylation of N-arylisoindolinones with simple alkenes. With the assistance of the isoindolinone skeleton as a directing group, site-selective C–H activation was achieved at the ortho-position of the N-aryl ring. An array of biological N-arylisoindolinones were obtained in good yields and excellent branched selectivities. With a BiPhePhos-type chiral ligand, efficient asymmetric control was achieved with up to 87:13 er. The application of this methodology toward other types of biologically-relevant structures as well as medicinal studies of the resulted N-arylisoindolinones are currently ongoing.
Supplementary Materials
The following supporting information can be downloaded at: https://0-www-mdpi-com.brum.beds.ac.uk/article/10.3390/molecules27061923/s1, The chiral ligand screening, 1H-NMR and 13C-NMRspectra for all new compounds. HPLC spectra for 3a and 3c.
Author Contributions
Conceptualization, M.X. and D.X.; methodology, M.X.; validation, M.X., Y.S., J.T., F.Y. and D.X.; formal analysis, M.X. and D.X.; investigation, D.X.; data curation, M.X., Y.S., J.T., F.Y. and D.X.; writing—original draft preparation, D.X.; writing—review and editing, D.X.; visualization, M.X.; supervision, J.T., F.Y. and D.X.; All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China (21772043), the Shanghai Pujiang Program (No. 19PJ1403000).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The date presents in study are available in Supplementary Materials.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds are not available from the authors.
References
- Anzini, M.; Cappelli, A.; Vomero, S.; Giorgi, G.; Langer, T.; Bruni, G.; Romeo, M.R.; Basile, A.S. Molecular Basis of Peripheral vs Central Benzodiazepine Receptor Selectivity in a New Class of Peripheral Benzodiazepine Receptor Ligands Related to Alpidem. J. Med. Chem. 1996, 39, 4275–4284. [Google Scholar] [CrossRef] [PubMed]
- Donohoe, T.J. Product class 14:1H- and 2H-isoindoles. Sci. Synth. 2001, 10, 653–692. [Google Scholar]
- Comins, D.L.; Schilling, S.; Zhang, Y. Asymmetric Synthesis of 3-Substituted Isoindolinones: Application to the Total Synthesis of (+)-Lennoxamine. Org. Lett. 2005, 7, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Nomura, S.; Endo-Umeda, K.; Aoyama, A.; Makishima, M.; Hashimoto, Y.; Ishikawa, M. Styrylphenylphthalimides as Novel Transrepression-Selective Liver X Receptor (LXR) Modulators. ACS Med. Chem. Lett. 2015, 6, 902–907. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Shinji, C.; Ogura, K.; Shimizu, M.; Maeda, S.; Sato, M.; Yoshida, M.; Hashimoto, Y.; Miyachi, H. Design, synthesis, and evaluation of isoindolinone-hydroxamic acid derivatives as histone deacetylase (HDAC) inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 4895–4900. [Google Scholar] [CrossRef]
- Speck, K.; Magauer, T. The chemistry of isoindole natural products. Beilstein. J. Org. Chem. 2013, 9, 2048–2078. [Google Scholar] [CrossRef]
- Bhatia, R.K. Isoindole Derivatives: Propitious Anticancer Structural Motifs. Curr. Top. Med. Chem. 2016, 17, 189–207. [Google Scholar] [CrossRef]
- Lunn, M.R.; Root, D.E.; Martino, A.M.; Flaherty, S.P.; Kelley, B.P.; Coovert, D.D.; Burghes, A.H.; Man, T.M.; Morris, G.E.; Zhou, J.; et al. Indoprofen Upregulates the Survival Motor Neuron Protein through a CyclooxygenaseIndependent Mechanism. Chem. Biol. 2004, 11, 1489–1493. [Google Scholar] [CrossRef] [Green Version]
- Miyachi, H.; Azuma, A.; Hioki, E.; Kobayashi, Y.; Iwasaki, S.; Hashimoto, Y. Inducer-specific regulators of tumor necrosis factor alpha production. Chem. Pharm. Bull. 1996, 44, 1980–1982. [Google Scholar] [CrossRef] [Green Version]
- Park, J.S.; Moon, S.C.; Baik, K.U.; Cho, J.Y.; Yoo, E.S.; Byun, Y.S.; Park, M.H. Synthesis and SAR studies for the inhibition of TNF-α production. Part 2. 2-[3-(Cyclopentyloxy)-4-methoxyphenyl]-substituted-1-isoindolinone derivatives. Arch. Pharmacal Res. 2002, 25, 137–142. [Google Scholar] [CrossRef]
- Hamprecht, D.; Micheli, F.; Tedesco, G.; Checchia, A.; Donati, D.; Petrone, M.; Terreni, S.; Wood, M. Isoindolone derivatives, a new class of 5-HT2C antagonists: Synthesis and biological evaluation. Bioorg. Med. Chem. Lett. 2007, 17, 428–433. [Google Scholar] [CrossRef] [PubMed]
- Maguire, G.; Franklin, D. New Molecular Targets for Antianxiety Interventions. J. Clin. Psychopharmacol. 2010, 30, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.M.; Morton, D. Recent Advances in C−H Functionalization. J. Org. Chem. 2016, 81, 343–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandeepan, P.; Ackermann, L. Transient Directing Groups for Transformative C–H Activation by Synergistic Metal Catalysis. Chem. 2018, 4, 199–222. [Google Scholar] [CrossRef] [Green Version]
- Hummel, J.R.; Boerth, J.A.; Ellman, J.A. Transition-Metal-Catalyzed C–H Bond Addition to Carbonyls, Imines, and Related Polarized π Bonds. Chem. Rev. 2017, 117, 9163–9227. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.S.; Park, W.J.; Jun, C.H. Metal-Organic Cooperative Catalysis in C–H and C–C Bond Activation. Chem. Rev. 2017, 117, 8977–9015. [Google Scholar] [CrossRef] [PubMed]
- Luo, F. Progress in Transition Metal Catalyzed C–H Functionalization Directed by Carboxyl Group. Chin. J. Org. Chem. 2019, 39, 3084–3104. [Google Scholar] [CrossRef]
- Rej, S.; Ano, Y.; Chatani, N. Bidentate Directing Groups: An Efficient Tool in C–H Bond Functionalization Chemistry for the Expedient Construction of C–C Bonds. Chem. Rev. 2020, 120, 1788–1887. [Google Scholar] [CrossRef]
- Wang, S.; Zhu, L. Recent Advances in Directing Group—Induced C–H Activation Reactions. Chin. J. Org. Chem. 2018, 38, 291–303. [Google Scholar]
- Wencel-Delord, J.; Liu, F.; Glorius, F. Towards mild metal-catalyzed C–H bond activation. Chem. Soc. Rev. 2011, 40, 4740–4761. [Google Scholar] [CrossRef]
- Yuan, Y.C.; Bruneau, C.; Roisnel, T.; Gramage-Doria, R. Site-Selective Ruthenium-Catalyzed C−H Bond Arylations with Boronic Acids: Exploiting Isoindolinones as a Weak Directing Group. J. Org. Chem. 2019, 84, 12893–12903. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Y.C.; Bruneau, C.; Roisnel, T.; Gramage-Doria, R. C−H Bond Alkylation of Cyclic Amides with Maleimides via a SiteSelective-Determining Six-Membered Ruthenacycle. J. Org. Chem. 2019, 84, 16183–16191. [Google Scholar] [CrossRef]
- Yuan, Y.C.; Bruneau, C.; Roisnel, T.; Gramage-Doria, R. Site-selective Ru-catalyzed C–H bond alkenylation with biologically relevant isoindolinones: A case of catalyst performance controlled by subtle stereo-electronic effects of the weak directing group. Catal. Sci. Technol. 2019, 9, 4711–4717. [Google Scholar] [CrossRef]
- Crisenza, G.E.M.; Bower, J.F. Branch Selective Murai-type Alkene Hydroarylation Reactions. Chem. Lett. 2016, 45, 2–9. [Google Scholar] [CrossRef] [Green Version]
- Fernández, D.F.; López, F. Catalytic addition of C–H bonds across C–C unsaturated systems promoted by iridium(I) and its group IX congeners. Chem. Soc. Rev. 2020, 49, 7378–7405. [Google Scholar] [CrossRef]
- Tsuchikama, K.; Kasagawa, M.; Hashimoto, Y.K.; Endo, K.; Shibata, T. Cationic iridium-BINAP complex-catalyzed addition of aryl ketones to alkynes and alkenes via directed C–H bond cleavage. J. Organomet. Chem. 2008, 693, 3939–3942. [Google Scholar] [CrossRef]
- Pan, S.; Ryu, N.; Shibata, T. Ir(I)-Catalyzed C–H Bond Alkylation of C2-Position of Indole with Alkenes: Selective Synthesis of Linear or Branched 2-Alkylindoles. J. Am. Chem. Soc. 2012, 134, 17474–17477. [Google Scholar] [CrossRef]
- Sevov, C.S.; Hartwig, J.F. Iridium-Catalyzed Intermolecular Asymmetric Hydroheteroarylation of Bicycloalkenes. J. Am. Chem. Soc. 2013, 135, 2116–2119. [Google Scholar] [CrossRef] [Green Version]
- Sevov, C.S.; Hartwig, J.F. Iridium-Catalyzed Oxidative Olefination of Furans with Unactivated Alkenes. J. Am. Chem. Soc. 2014, 136, 10625–10631. [Google Scholar] [CrossRef] [Green Version]
- Ebe, Y.; Nishimura, T. Iridium-Catalyzed Branch-Selective Hydroarylation of Vinyl Ethers via C–H Bond Activation. J. Am. Chem. Soc. 2015, 137, 5899–5902. [Google Scholar] [CrossRef]
- Shirai, T.; Yamamoto, Y. Cationic Iridium/S-Me-BIPAM-Catalyzed Direct Asymmetric Intermolecular Hydroarylation of Bicycloalkenes. Angew. Chem. Int. Ed. 2015, 54, 9894–9897. [Google Scholar] [CrossRef]
- Hatano, M.; Ebe, Y.; Nishimura, T.; Yorimitsu, H. Asymmetric Alkylation of N-Sulfonylbenzamides with Vinyl Ethers via C–H Bond Activation Catalyzed by Hydroxoiridium/Chiral Diene Complexes. J. Am. Chem. Soc. 2016, 138, 4010–4013. [Google Scholar] [CrossRef]
- Yamauchi, D.; Nishimura, T.; Yorimitsu, H. Asymmetric hydroarylation of vinyl ethers catalyzed by a hydroxoiridium complex: Azoles as effective directing groups. Chem. Commun. 2017, 53, 2760–2763. [Google Scholar] [CrossRef]
- Ebe, Y.; Onoda, M.; Nishimura, T.; Yorimitsu, H. Iridium-Catalyzed Regio- and Enantioselective Hydroarylation of Alkenyl Ethers by Olefin Isomerization. Angew. Chem. Int. Ed. 2017, 56, 1–6. [Google Scholar]
- Crisenza, G.E.; McCreanor, N.G.; Bower, J.F. Branch-Selective, Iridium-Catalyzed Hydroarylation of Monosubstituted Alkenes via a Cooperative Destabilization Strategy. J. Am. Chem. Soc. 2014, 136, 10258–10261. [Google Scholar] [CrossRef]
- Crisenza, G.E.; Sokolova, O.O.; Bower, J.F. Branch-Selective Alkene Hydroarylation by Cooperative Destabilization: Iridium-Catalyzed ortho-Alkylation of Acetanilides. Angew. Chem. Int. Ed. 2015, 54, 14866–14870. [Google Scholar] [CrossRef] [Green Version]
- Greĺaud, S.; Cooper, P.; Feron, L.J.; Bower, J.F. Branch-Selective and Enantioselective Iridium-Catalyzed Alkene Hydroarylation via Anilide-Directed C–H Oxidative Addition. J. Am. Chem. Soc. 2018, 140, 9351–9356. [Google Scholar] [CrossRef] [Green Version]
- Romero-Arenas, A.; Hornillos, V.; Iglesias-Sigüenza, J.; Fernańdez, R.; López-Serrano, J.; Ros, A.; Lassaletta, J.M. Ir-Catalyzed Atroposelective Desymmetrization of Heterobiaryls: Hydroarylation of Vinyl Ethers and Bicycloalkenes. J. Am. Chem. Soc. 2020, 142, 2628–2639. [Google Scholar] [CrossRef]
- Jordan, R.F.; Taylor, D.F. Zirconium-catalyzed coupling of propene and alpha picoline. J. Am. Chem. Soc. 1989, 111, 778–779. [Google Scholar] [CrossRef]
- Kuninobu, Y.; Matsuki, T.; Takai, K. Rhenium-Catalyzed Regioselective Alkylation of Phenols. J. Am. Chem. Soc. 2009, 131, 9914–9915. [Google Scholar] [CrossRef]
- Gao, K.; Yoshikai, N. Regioselectivity-Switchable Hydroarylation of Styrenes. J. Am. Chem. Soc. 2011, 133, 400–402. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.S.; Yoshikai, N. Aldimine-Directed Branched-Selective Hydroarylation of Styrenes. Angew. Chem., Int. Ed. 2013, 52, 1240–1244. [Google Scholar] [CrossRef] [PubMed]
- Song, G.Y.; Wylie, W.N.O.; Hou, Z.M. Enantioselective C–H Bond Addition of Pyridines to Alkenes Catalyzed by Chiral Half-Sandwich Rare-Earth Complexes. J. Am. Chem. Soc. 2014, 136, 12209–12212. [Google Scholar] [CrossRef]
- Banerjee, A.; Santra, S.K.; Mohanta, P.R.; Patel, B.K. Cobalt-Catalyzed Enantioselective Directed C–H Alkylation of Indole with Styrenes. Org. Lett. 2015, 17, 22–25. [Google Scholar]
- Zell, D.; Bursch, M.; Müller, V.; Grimme, S.; Ackermann, L. Full Selectivity Control in Cobalt(III)-Catalyzed C−H Alkylations by Switching of the C−H Activation Mechanism. Angew. Chem. Int. Ed. 2017, 56, 10378–10382. [Google Scholar] [CrossRef] [PubMed]
- Loup, J.; Zell, D.; Ackermann, L. Asymmetric Iron-Catalyzed C−H Alkylation Enabled by Remote Ligand meta-Substitution. Angew. Chem. Int. Ed. 2017, 56, 14197–14201. [Google Scholar] [CrossRef]
- Pesciaioli, F.; Dhawa, U.; Ackermann, L. Enantioselective Cobalt(III)-Catalyzed C−H Activation Enabled by Chiral Carboxylic Acid Cooperation. Angew. Chem. Int. Ed. 2018, 57, 15425–15429. [Google Scholar] [CrossRef]
- Liu, Y.H.; Xie, P.P.; Shi, B.F. Cp*Co(III)-Catalyzed Enantioselective Hydroarylation of Unactivated Terminal Alkenes via C–H Activation. J. Am. Chem. Soc. 2021, 143, 19112–19120. [Google Scholar] [CrossRef]
- Huang, G.P.; Liu, P. Mechanism and Origins of Ligand-Controlled Linear Versus Branched Selectivity of Iridium-Catalyzed Hydroarylation of Alkenes. ACS Catal. 2016, 6, 809–820. [Google Scholar] [CrossRef]
- Zhang, M.; Hu, L.; Cao, Y.; Huang, G.P. Mechanism and Origins of Regio- and Enantioselectivities of Iridium-Catalyzed Hydroarylation of Alkenyl Ethers. J. Org. Chem. 2018, 83, 2937–2947. [Google Scholar] [CrossRef]
Figure 1.
Representative examples of biologically active N-arylisoindolinones.

Scheme 1.
Isoindolinone-directed site-selective C–H activation. (A) Previous work; (B) This work.

Scheme 2.
Plausible mechanism.

Scheme 3.
Asymmetric alkylation with chiral ligand.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Condition optimizations a.

| Entry | Ligand (5 mol%) | Solvent | Yield (%) b | rr (b/l) c |
|---|---|---|---|---|
| 1 | rac-L1 | 1,4-dioxane | 87 (81) | >20:1 |
| 2 | rac-BINAP | 1,4-dioxane | <5 | -- |
| 3 | rac-BIPHEP | 1,4-dioxane | 12 | 10:1 |
| 4 | dppf | 1,4-dioxane | 25 | 11:1 |
| 5 | dppb | 1,4-dioxane | 20 | >20:1 |
| 6 | dppe | 1,4-dioxane | 10 | 6:1 |
| 7 | rac-L1 | CPME | 78 | >20:1 |
| 8 | rac-L1 | toluene | 51 | >20:1 |
| 9 | rac-L1 | DCE | 48 | >20:1 |
| 10 | rac-L1 | PhCl | 46 | >20:1 |
| 11 | rac-L1 | m-xylene | 63 | >20:1 |
a Reaction conditions: 1a (0.1 mmol), 2a (0.5 mmol,), [Ir(COD)2]BArF (5 mol%), ligand (5 mol%), solvent (0.2 mL), 120 °C, 48 h, b Yield was determined by GC analysis of the crude reaction mixture with dodecane as internal standard; isolated yield shown in parentheses. c The branched/linear (b/l) ratio was determined by GC analysis of the crude reaction mixture.
Table 2.
Substrate scope (3a–p) a.

 |  |  |  |
 |  |  |  |
 |  |  |  |
 |  |  |  |
a Reaction conditions: 1 (0.1 mmol), 2 (0.5 mmol,), [Ir(COD)2]BArF (5 mol%), rac-L1 (5 mol%), 1,4-dioxane (0.2 mL), 120 °C, 48 h. Isolated yield for branched-selective product. The (b/l) ratio was >20:1 for all.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Xiong, M.; Shu, Y.; Tang, J.; Yang, F.; Xing, D. Iridium(I)-Catalyzed Isoindolinone-Directed Branched-Selective Aromatic C–H Alkylation with Simple Alkenes. Molecules 2022, 27, 1923. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27061923
AMA Style
Xiong M, Shu Y, Tang J, Yang F, Xing D. Iridium(I)-Catalyzed Isoindolinone-Directed Branched-Selective Aromatic C–H Alkylation with Simple Alkenes. Molecules. 2022; 27(6):1923. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27061923
Chicago/Turabian StyleXiong, Maoqian, Yuhang Shu, Jie Tang, Fan Yang, and Dong Xing. 2022. "Iridium(I)-Catalyzed Isoindolinone-Directed Branched-Selective Aromatic C–H Alkylation with Simple Alkenes" Molecules 27, no. 6: 1923. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27061923