
Conformer-Specific Dissociation Dynamics in Dimethyl Methylphosphonate Radical Cation

 , , , and
, , , and 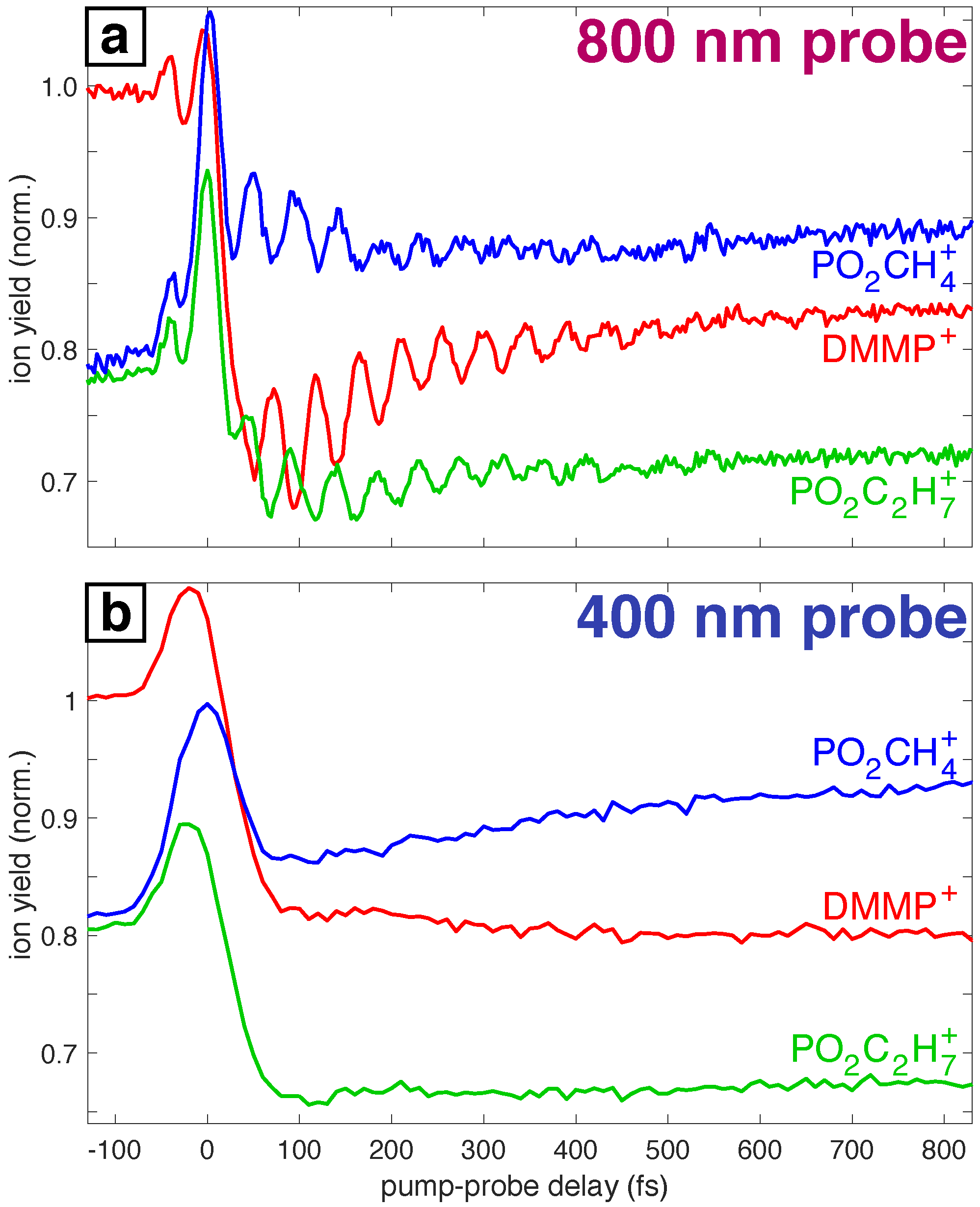{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Electronic Structure Calculations
2.2. Dynamics
2.3. Experimental Methods
3. Results
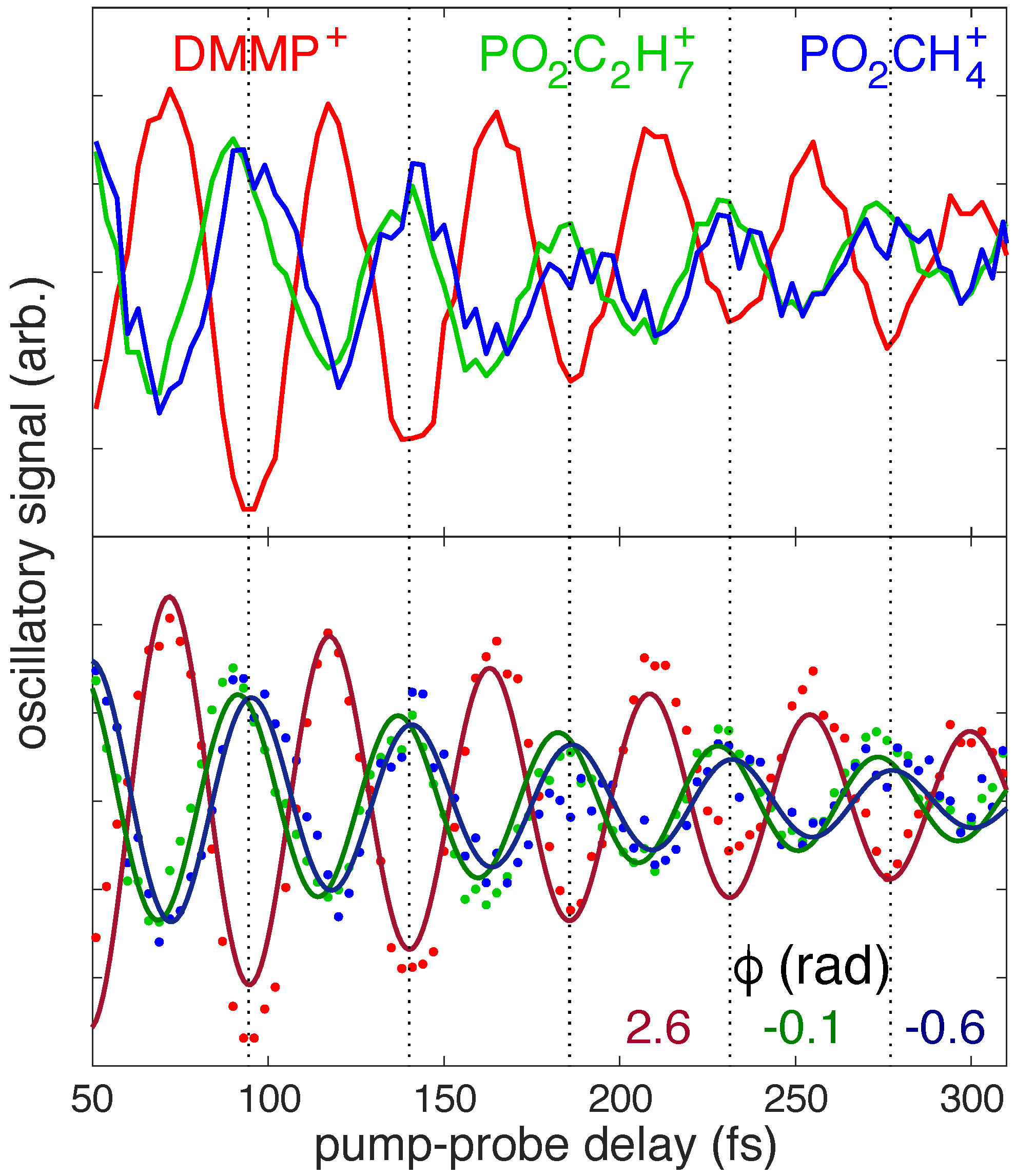
3.1. Experimental Motivation
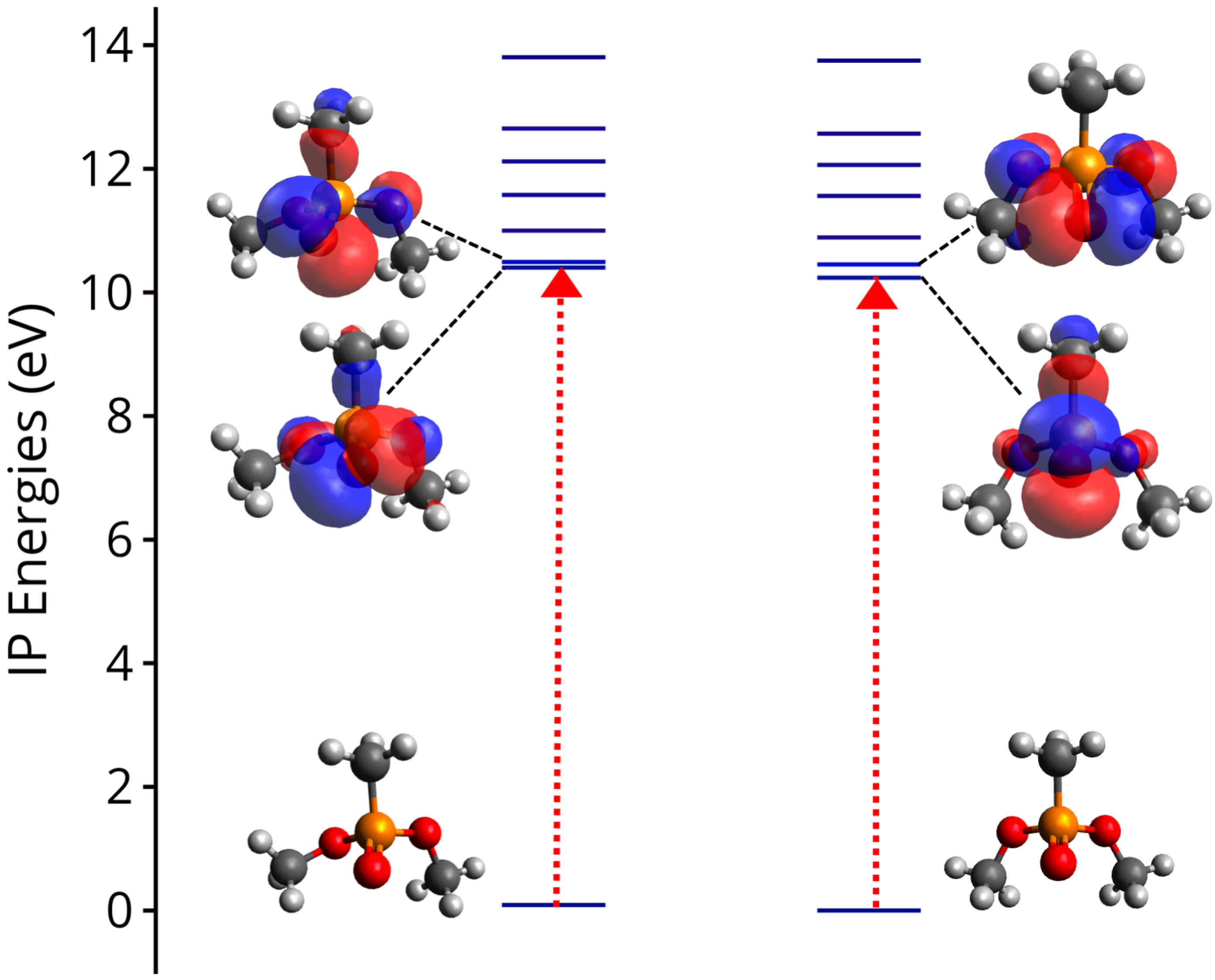
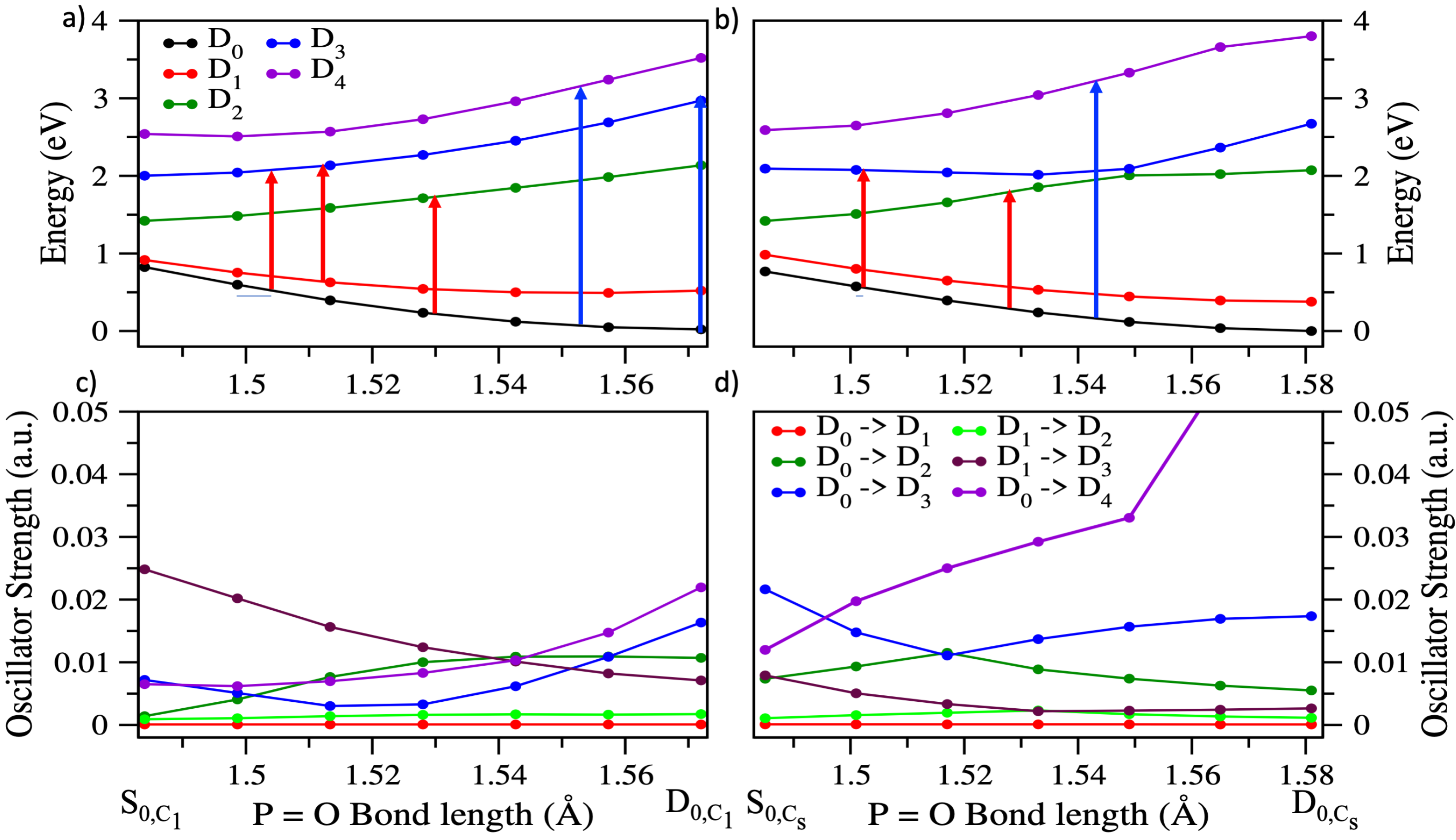
3.2. Ionization Potentials
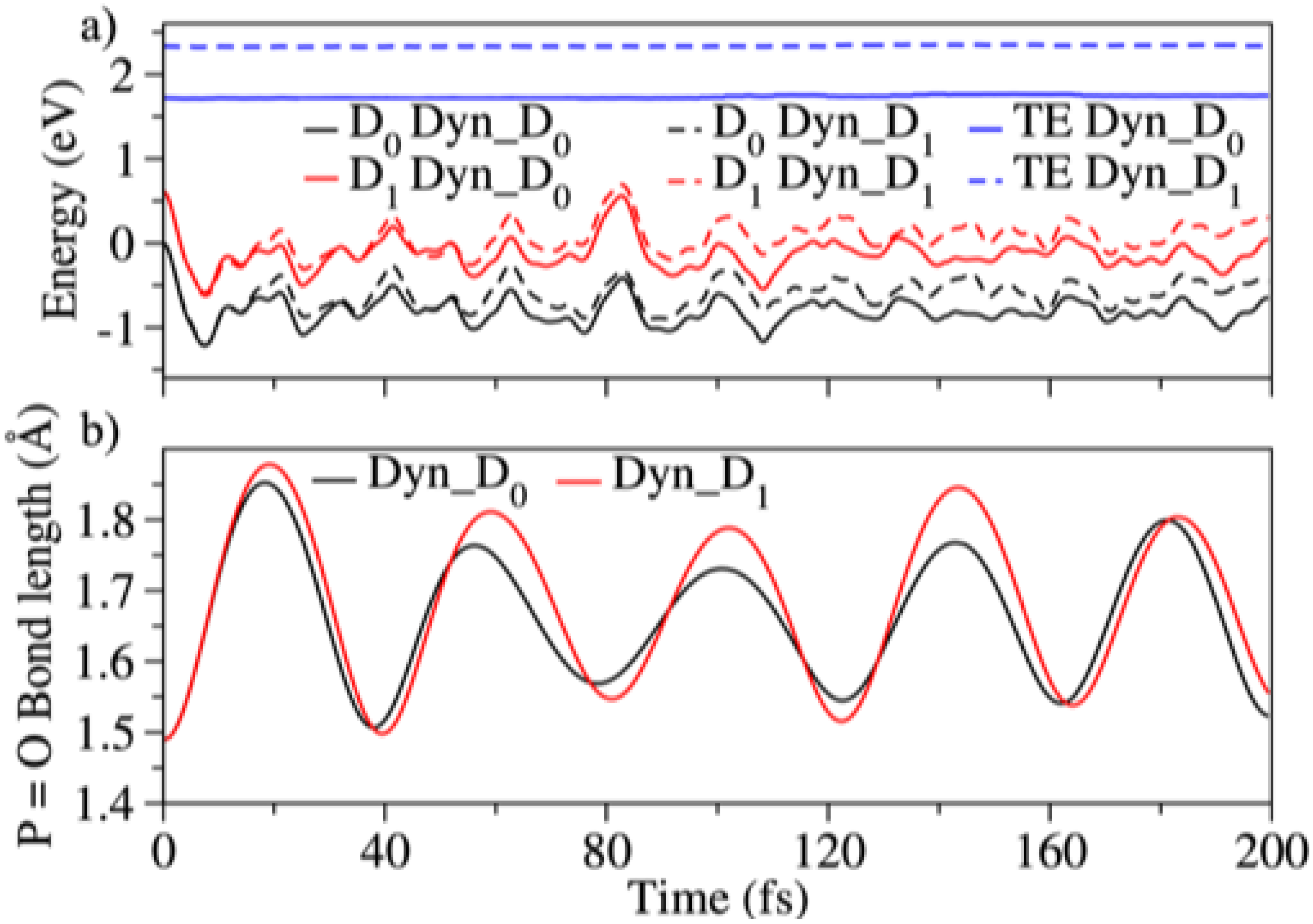
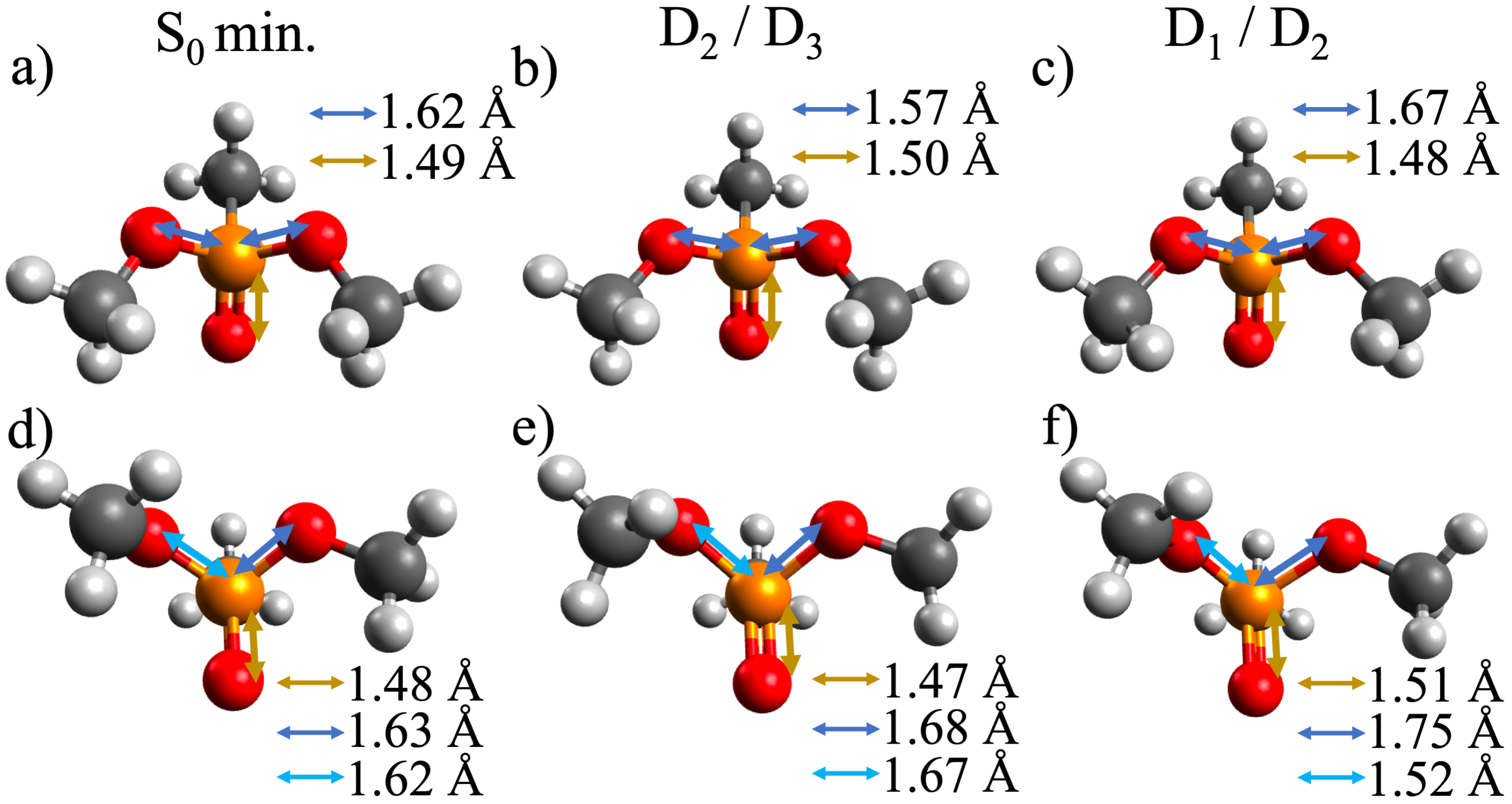
3.3. Dynamics on D and D
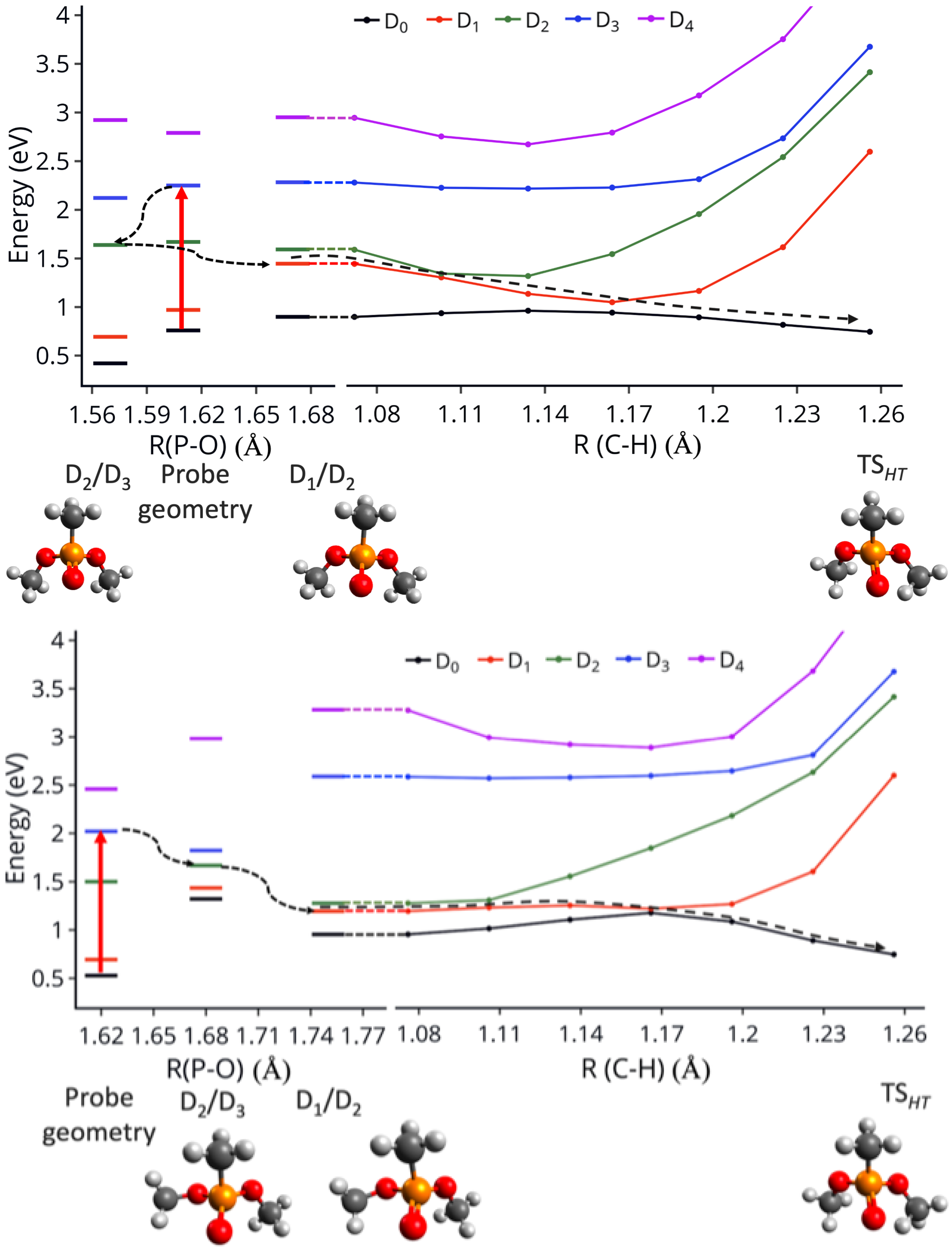
3.4. Effect of the Probe: Accessing Higher Electronic States
3.5. Differences between PO2CH4+ and PO2C2H7+: Conformational Effects
3.6. Hydrogen Transfer
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mathies, R.A. A coherent picture of vision. Nat. Chem. 2015, 7, 945–947. [Google Scholar] [CrossRef] [PubMed]
- Crespo-Hernández, C.E.; Cohen, B.; Hare, P.M.; Kohler, B. Ultrafast excited-state dynamics in nucleic acids. Chem. Rev. 2004, 104, 1977–2020. [Google Scholar] [CrossRef]
- Calegari, F.; Ayuso, D.; Trabattoni, A.; Belshaw, L.; De Camillis, S.; Anumula, S.; Frassetto, F.; Poletto, L.; Palacios, A.; Decleva, P.; et al. Ultrafast electron dynamics in phenylalanine initiated by attosecond pulses. Science 2014, 346, 336–339. [Google Scholar] [CrossRef] [Green Version]
- Attar, A.R.; Bhattacherjee, A.; Pemmaraju, C.; Schnorr, K.; Closser, K.D.; Prendergast, D.; Leone, S.R. Femtosecond X-ray spectroscopy of an electrocyclic ring-opening reaction. Science 2017, 356, 54–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, Y. Photochemical α-cleavage of ketones: Revisiting acetone. Photochem. Photobiol. Sci. 2004, 3, 6–16. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, M.; Garavelli, M.; Cerullo, G. Ultrafast spectroscopy: State of the art and open challenges. J. Am. Chem. Soc. 2019, 142, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Pearson, B.J.; Nichols, S.R.; Weinacht, T. Molecular fragmentation driven by ultrafast dynamic ionic resonances. J. Chem. Phys. 2007, 127, 131101. [Google Scholar] [CrossRef] [PubMed]
- Bohinski, T.; Moore Tibbetts, K.; Tarazkar, M.; Romanov, D.; Matsika, S.; Levis, R. Measurement of ionic resonances in alkyl phenyl ketone cations via infrared strong field mass spectrometry. J. Phys. Chem. A 2013, 117, 12374–12381. [Google Scholar] [CrossRef]
- Bohinski, T.; Moore Tibbetts, K.; Tarazkar, M.; Romanov, D.A.; Matsika, S.; Levis, R.J. Strong field adiabatic ionization prepares a launch state for coherent control. J. Phys. Chem. Lett. 2014, 5, 4305–4309. [Google Scholar] [CrossRef] [PubMed]
- Assmann, M.; Weinacht, T.; Matsika, S. Surface Hopping Investigation of the Relaxation Dynamics in Radical Cations. J. Chem. Phys. 2016, 144, 034301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ampadu Boateng, D.; Gutsev, G.L.; Jena, P.; Tibbetts, K.M. Dissociation dynamics of 3-and 4-nitrotoluene radical cations: Coherently driven C–NO2 bond homolysis. J. Chem. Phys. 2018, 148, 134305. [Google Scholar] [CrossRef] [Green Version]
- Moore Tibbetts, K. Coherent vibrational and dissociation dynamics of polyatomic radical cations. Chem. Eur. J. 2019, 25, 8431–8439. [Google Scholar] [CrossRef]
- Boateng, D.A.; Gutsev, G.L.; Jena, P.; Tibbetts, K.M. Ultrafast coherent vibrational dynamics in dimethyl methylphosphonate radical cation. Phys. Chem. Chem. Phys. 2018, 20, 4636–4640. [Google Scholar] [CrossRef] [PubMed]
- Lezius, M.; Blanchet, V.; Rayner, D.; Villeneuve, D.; Stolow, A.; Ivanov, M.Y. Nonadiabatic multielectron dynamics in strong field molecular ionization. Phys. Rev. Lett. 2001, 86, 51. [Google Scholar] [CrossRef] [PubMed]
- Markevitch, A.N.; Romanov, D.A.; Smith, S.M.; Schlegel, H.B.; Ivanov, M.Y.; Levis, R.J. Sequential nonadiabatic excitation of large molecules and ions driven by strong laser fields. Phys. Rev. A 2004, 69, 013401. [Google Scholar] [CrossRef] [Green Version]
- Tolstikhin, O.I.; Morishita, T. Adiabatic theory of ionization by intense laser pulses: Finite-range potentials. Phys. Rev. A 2012, 86, 043417. [Google Scholar] [CrossRef]
- Keldysh, L. Ionization in the field of a strong electromagnetic wave. Sov. Phys. JETP 1965, 20, 1307–1314. [Google Scholar]
- Tibbetts, K.M.; Tarazkar, M.; Bohinski, T.; Romanov, D.A.; Matsika, S.; Levis, R.J. Controlling the dissociation dynamics of acetophenone radical cation through excitation of ground and excited state wavepackets. J. Phys. B At. Mol. Opt. Phys. 2015, 48, 164002. [Google Scholar] [CrossRef] [Green Version]
- Munkerup, K.; Romanov, D.; Bohinski, T.; Stephansen, A.B.; Levis, R.J.; Sølling, T.I. Conserving Coherence and Storing Energy during Internal Conversion: Photoinduced Dynamics of cis- and trans-Azobenzene Radical Cations. J. Phys. Chem. A 2017, 121, 8642–8651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peña, H.L.; Boateng, D.A.; McPherson, S.; Tibbetts, K.M. Using computational chemistry to design pump-probe schemes for measuring radical cation dynamics. Phys. Chem. Chem. Phys. 2021, 23, 13338–13348. [Google Scholar] [CrossRef]
- Ampadu Boateng, D.; Word, M.D.; Gutsev, L.G.; Jena, P.; Tibbetts, K.M. Conserved Vibrational Coherence in the Ultrafast Rearrangement of 2-Nitrotoluene Radical Cation. J. Phys. Chem. A 2019, 123, 1140–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez Pena, H.A.; Shusterman, J.M.; Ampadu Boateng, D.; Lao, K.U.; Tibbetts, K.M. Coherent control of molecular dissociation by selective excitation of nuclear wave packets. Front. Chem. 2022, in press. [Google Scholar] [CrossRef]
- Ampadu Boateng, D.; Word, M.D.; Tibbetts, K.M. Probing Coherent Vibrations of Organic Phosphonate Radical Cations with Femtosecond Time-Resolved Mass Spectrometry. Molecules 2019, 24, 509. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Q.; Fu, Y.C.; Xu, J.Q. Advances in the chemical sensors for the detection of DMMP—A simulant for nerve agent sarin. Procedia Eng. 2010, 7, 179–184. [Google Scholar] [CrossRef] [Green Version]
- Tomchenko, A.A.; Harmer, G.P.; Marquis, B.T. Detection of chemical warfare agents using nanostructured metal oxide sensors. Sens. Actuators B Chem. 2005, 108, 41–55. [Google Scholar] [CrossRef]
- Park, S.; Kim, S.; Kim, M. Observation of conformation-specific pathways in the photodissociation of 1-iodopropane ions. Nature 2002, 415, 306–308. [Google Scholar] [CrossRef] [PubMed]
- Khriachtchev, L.; Pettersson, M.; Rasanen, M. Conformational Memory in Photodissociation of Formic Acid. J. Am. Chem. Soc. 2002, 124, 10994–10995. [Google Scholar] [CrossRef]
- Kim, M.H.; Shen, L.; Tao, H.; Martinez, T.J.; Suits, A.G. Conformationally Controlled Chemistry: Excited-State Dynamics Dictate Ground-State Reaction. Science 2007, 315, 1561. [Google Scholar] [CrossRef]
- Chang, Y.P.; Długołęcki, K.; Küpper, J.; Rösch, D.; Wild, D.; Willitsch, S. Specific Chemical Reactivities of Spatially Separated 3-Aminophenol Conformers with Cold Ca+ Ions. Science 2013, 342, 98–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.; Kim, S.Y.; Lim, J.S.; Kim, J.; Kim, S.K. Conformer Specific Excited-State Structure of 3-Methylthioanisole. J. Phys. Chem. A 2020, 124, 4666–4671. [Google Scholar] [CrossRef]
- Pathak, S.; Obaid, R.; Bhattacharyya, S.; Bürger, J.; Li, X.; Tross, J.; Severt, T.; Davis, B.; Bilodeau, R.C.; Trallero-Herrero, C.A.; et al. Differentiating and Quantifying Gas-Phase Conformational Isomers Using Coulomb Explosion Imaging. J. Phys. Chem. Lett. 2020, 11, 10205–10211. [Google Scholar] [CrossRef] [PubMed]
- Wilkin, K.J.; Parrish, R.M.; Yang, J.; Wolf, T.J.A.; Nunes, J.P.F.; Guehr, M.; Li, R.; Shen, X.; Zheng, Q.; Wang, X.; et al. Diffractive imaging of dissociation and ground-state dynamics in a complex molecule. Phys. Rev. A 2019, 100, 023402. [Google Scholar] [CrossRef] [Green Version]
- Champenois, E.G.; Sanchez, D.M.; Yang, J.; Nunes, J.P.F.; Attar, A.; Centurion, M.; Forbes, R.; Gühr, M.; Hegazy, K.; Ji, F.; et al. Conformer-specific photochemistry imaged in real space and time. Science 2021, 374, 178–182. [Google Scholar] [CrossRef]
- Ziegler, T. Approximate density functional theory as a practical tool in molecular energetics and dynamics. Chem. Rev. 1991, 91, 651–667. [Google Scholar] [CrossRef]
- Kohn, W.; Becke, A.D.; Parr, R.G. Density functional theory of electronic structure. J. Phys. Chem. 1996, 100, 12974–12980. [Google Scholar] [CrossRef] [Green Version]
- Becke, A.D. Becke’s three parameter hybrid method using the LYP correlation functional. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A At. Mol. Opt. Phys. 1988, 38, 3098. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B Condens. Matter Mater. Phys. 1988, 37, 785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef] [Green Version]
- Clark, T.; Chandrasekhar, J.; Spitznagel, G.W.; Schleyer, P.V.R. Efficient diffuse function-augmented basis sets for anion calculations. III. The 3-21+G basis set for first-row elements, Li-F. J. Comput. Chem. 1983, 4, 294–301. [Google Scholar] [CrossRef]
- Francl, M.M.; Pietro, W.J.; Hehre, W.J.; Binkley, J.S.; Gordon, M.S.; DeFrees, D.J.; Pople, J.A. Self-consistent molecular orbital methods. XXIII. A polarization-type basis set for second-row elements. J. Chem. Phys. 1982, 77, 3654–3665. [Google Scholar] [CrossRef] [Green Version]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z=11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Spitznagel, G.W.; Clark, T.; Schleyer, P.v.R.; Hehre, W.J. An evaluation of the performance of diffuse function-augmented basis sets for second row elements, Na-Cl. J. Comput. Chem. 1987, 8, 1109–1116. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; V. Barone, G.A.P.; Nakatsuji, H.; Li, X.; et al. Gaussian 09, Revision B. 01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Yang, L.; Shroll, R.M.; Zhang, J.; Lourderaj, U.; Hase, W.L. Theoretical investigation of mechanisms for the gas-phase unimolecular decomposition of DMMP. J. Phys. Chem. A 2009, 113, 13762–13771. [Google Scholar] [CrossRef]
- Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Woon, D.E.; Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. III. The atoms aluminum through argon. J. Chem. Phys. 1993, 98, 1358–1371. [Google Scholar] [CrossRef] [Green Version]
- Roos, B.O.; Taylor, P.R.; Sigbahn, P.E. A complete active space SCF method (CASSCF) using a density matrix formulated super-CI approach. Chem. Phys. 1980, 48, 157–173. [Google Scholar] [CrossRef]
- Lischka, H.; Shepard, R.; Pitzer, R.M.; Shavitt, I.; Dallos, M.; Müller, T.; Szalay, P.G.; Seth, M.; Kedziora, G.S.; Yabushita, S.; et al. High-Level Multireference Methods in the Quantum-Chemistry Program System COLUMBUS: Analytic MR-CISD and MR-AQCC Gradients and MR-AQCC-LRT for Excited States, GUGA Spin-Orbit CI and Parallel CI Density. Phys. Chem. Chem. Phys. 2001, 3, 664–673. [Google Scholar] [CrossRef]
- Lischka, H.; Shepard, R.; Müller, T.; Szalay, P.G.; Pitzer, R.M.; Aquino, A.J.A.; Araújo do Nascimento, M.M.; Barbatti, M.; Belcher, L.T.; Blaudeau, J.P.; et al. The generality of the GUGA MRCI approach in COLUMBUS for treating complex quantum chemistry. J. Chem. Phys. 2020, 152, 134110. [Google Scholar] [CrossRef] [Green Version]
- Lischka, H.; Shepard, R.; Shavitt, I.; Pitzer, R.M.; Dallos, M.; Müller, T.; Szalay, P.G.; Brown, F.B.; Ahlrichs, R.; Böhm, H.J.; et al. COLUMBUS, an ab Initio Electronic Structure Program, Release 7.0 (2017). Phys. Chem. Chem. Phys. 2017, 19, 5888–5894. [Google Scholar]
- Shao, Y.; Gan, Z.; Epifanovsky, E.; Gilbert, A.T.; Wormit, M.; Kussmann, J.; Lange, A.W.; Behn, A.; Deng, J.; Feng, X.; et al. Advances in molecular quantum chemistry contained in the Q-Chem 4 program package. Mol. Phys. 2015, 113, 184–215. [Google Scholar] [CrossRef] [Green Version]
- Werner, H.J.; Knowles, P.J.; Manby, F.R.; Black, J.A.; Doll, K.; Heßelmann, A.; Kats, D.; Köhn, A.; Korona, T.; Kreplin, D.A.; et al. The Molpro quantum chemistry package. J. Chem. Phys. 2020, 152, 144107. [Google Scholar] [CrossRef] [Green Version]
- Barbatti, M. Nonadiabatic dynamics with trajectory surface hopping method. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 620–633. [Google Scholar] [CrossRef]
- Tully, J.C. Perspective: Nonadiabatic dynamics theory. J. Chem. Phys. 2012, 137, 22A301. [Google Scholar] [CrossRef] [PubMed]
- Barbatti, M.; Granucci, G.; Ruckenbauer, M.; Plasser, F.; Crespo-Otero, R.; Pittner, J.; Persico, M.; Lischka, H. NEWTON-X: A Package for Newtonian Dynamics Close to the Crossing Seam. Version 2. Available online: www.newtonx.org (accessed on 22 February 2022).
- Tully, J.C. Molecular dynamics with electronic transitions. J. Chem. Phys. 1990, 93, 1061–1071. [Google Scholar] [CrossRef]
- Barbatti, M.; Granucci, G.; Ruckenbauer, M.; Plasser, F.; Crespo-Otero, R.; Pittner, J.; Persico, M.; Lischka, H. NEWTON-X: A surface-hopping program for nonadiabatic molecular dynamics. WIREs Comp. Mol. Sci. 2014, 4, 96. [Google Scholar] [CrossRef]
- Granucci, G.; Persico, M. Critical appraisal of the fewest switches algorithm for surface hopping. J. Chem. Phys. 2007, 126, 134114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, C.; Nangia, S.; Jasper, A.W.; Truhlar, D.G. Coherent switching with decay of mixing: An improved treatment of electronic coherence for non-Born—Oppenheimer trajectories. J. Chem. Phys. 2004, 121, 7658–7670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boateng, D.A.; Tibbetts, K.M. Measurement of ultrafast vibrational coherences in polyatomic radical cations with strong-field adiabatic ionization. J. Vis. Exp. JoVE 2018, 138, e58263. [Google Scholar]
- Holtzclaw, J.R.; Wyatt, J.R.; Campana, J.E. Structure and Fragmentation of Dimethyl Methylphosphonate and Trimethyl Phosphite. Org. Mass Spectrom. 1985, 20, 90–97. [Google Scholar] [CrossRef]
- Gutsev, G.L.; Ampadu Boateng, D.; Jena, P.; Tibbetts, K.M. A Theoretical and Mass Spectrometry Study of Dimethyl Methylphosphonate: New Isomers and Cation Decay Channels in an Intense Femtosecond Laser Field. J. Phys. Chem. A 2017, 121, 8414–8424. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, V.; López Peña, H.A.; Shusterman, J.M.; Vindel-Zandbergen, P.; Tibbetts, K.M.; Matsika, S. Conformer-Specific Dissociation Dynamics in Dimethyl Methylphosphonate Radical Cation. Molecules 2022, 27, 2269. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27072269
Singh V, López Peña HA, Shusterman JM, Vindel-Zandbergen P, Tibbetts KM, Matsika S. Conformer-Specific Dissociation Dynamics in Dimethyl Methylphosphonate Radical Cation. Molecules. 2022; 27(7):2269. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27072269
Chicago/Turabian StyleSingh, Vaibhav, Hugo A. López Peña, Jacob M. Shusterman, Patricia Vindel-Zandbergen, Katharine Moore Tibbetts, and Spiridoula Matsika. 2022. "Conformer-Specific Dissociation Dynamics in Dimethyl Methylphosphonate Radical Cation" Molecules 27, no. 7: 2269. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27072269