
Experimental and Computational Structural Studies of 2,3,5-Trisubstituted and 1,2,3,5-Tetrasubstituted Indoles as Non-Competitive Antagonists of GluK1/GluK2 Receptors

 , , , and
, , , and
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Chemistry
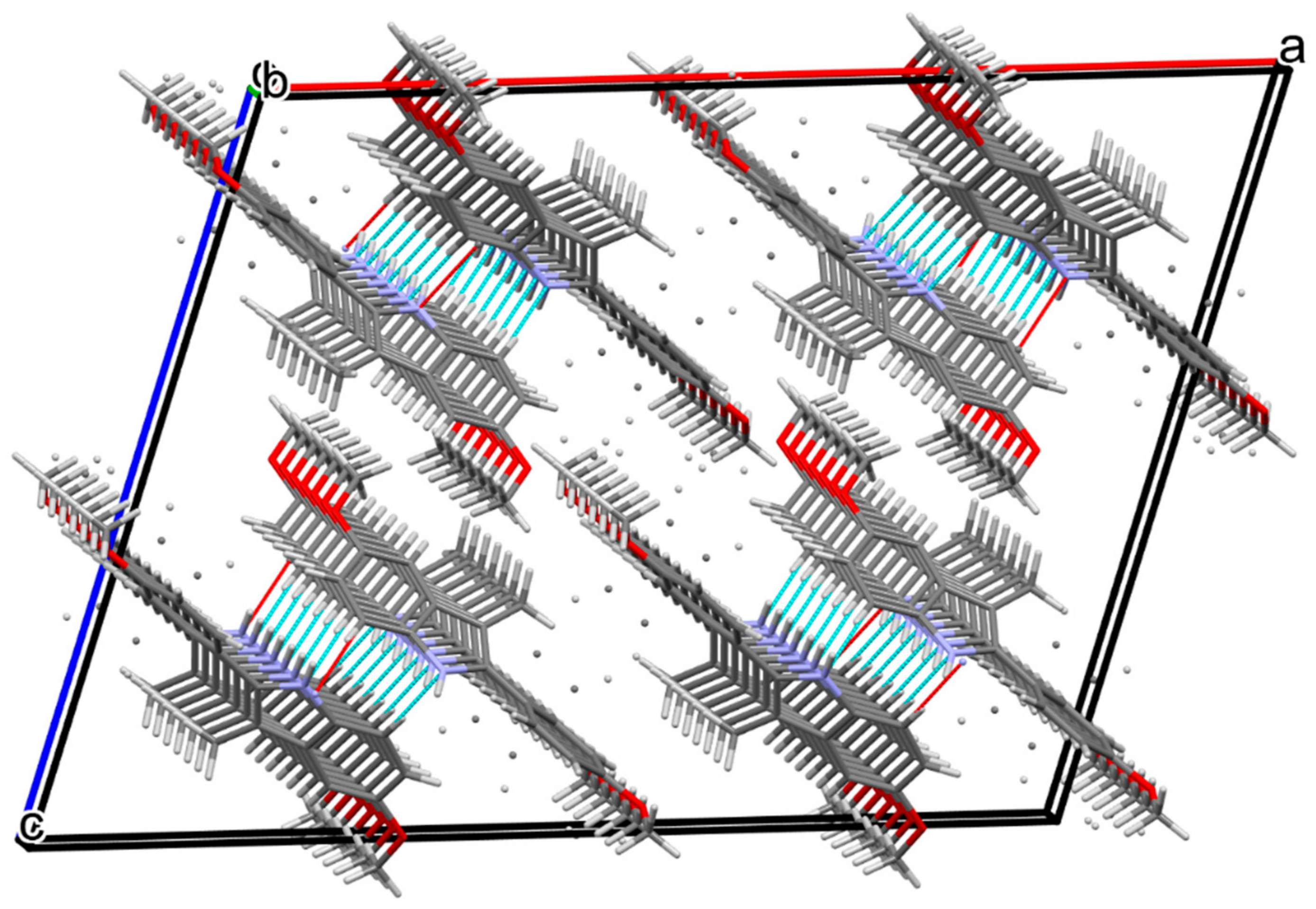
2.2. X-ray Studies
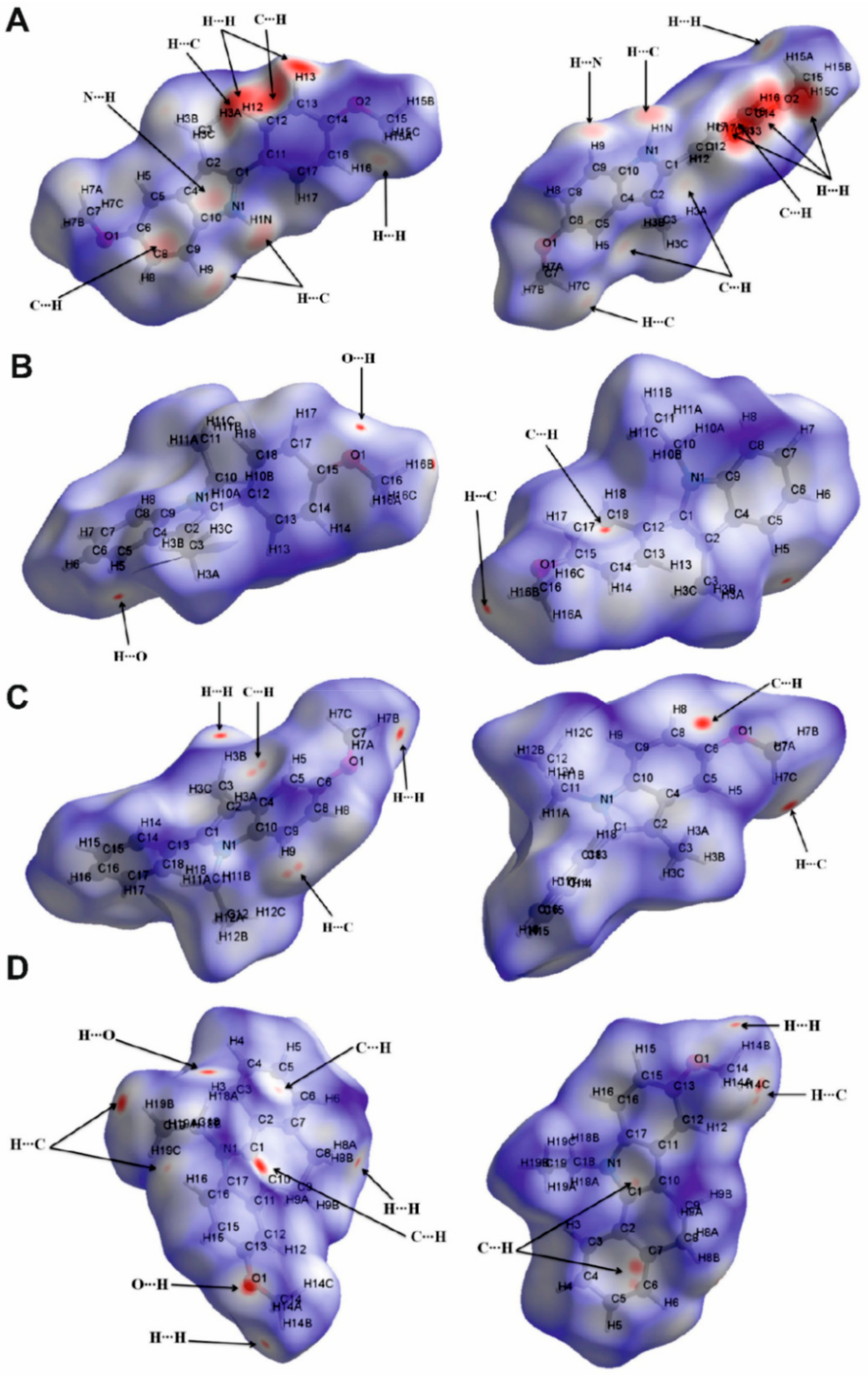
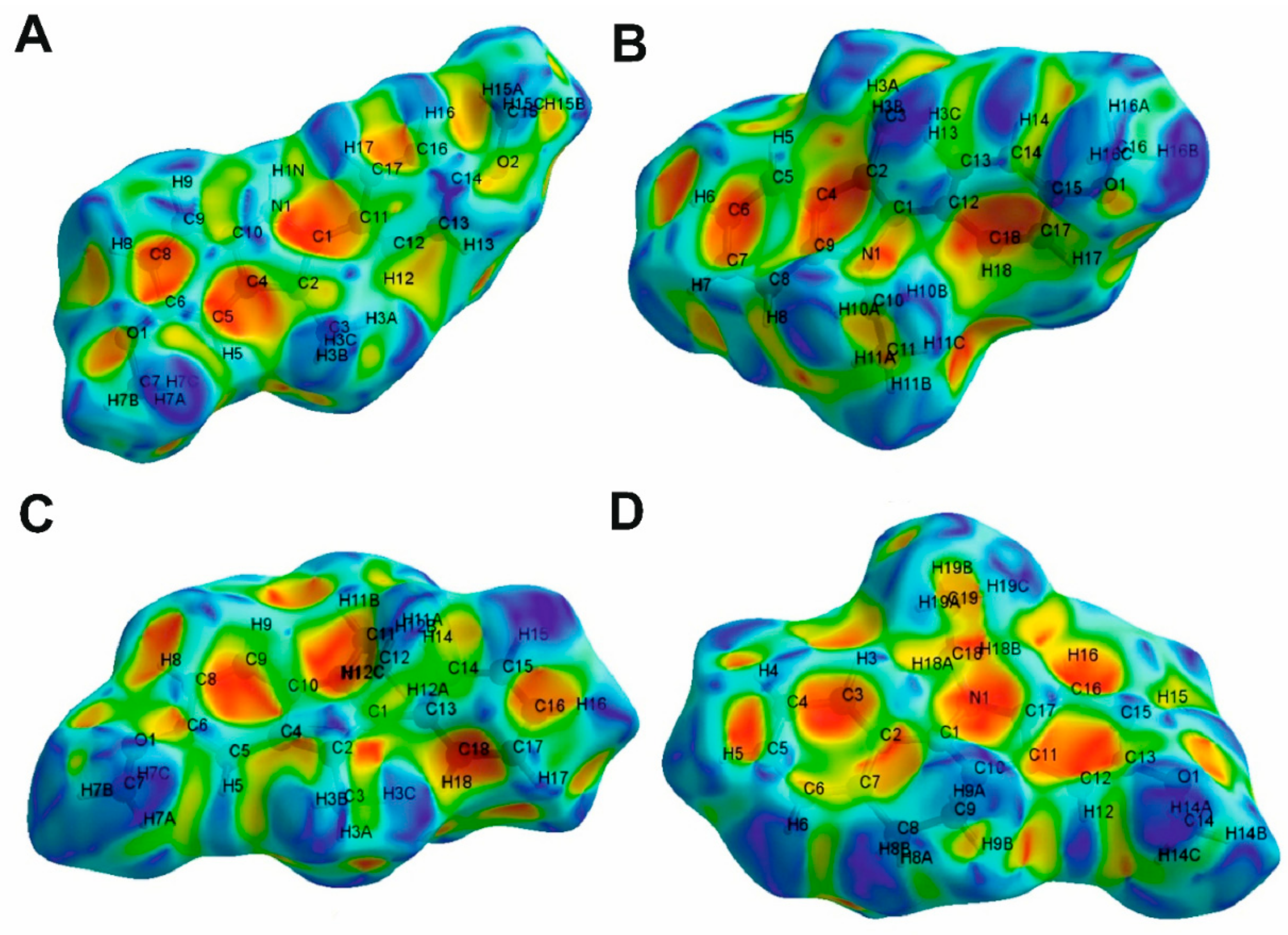
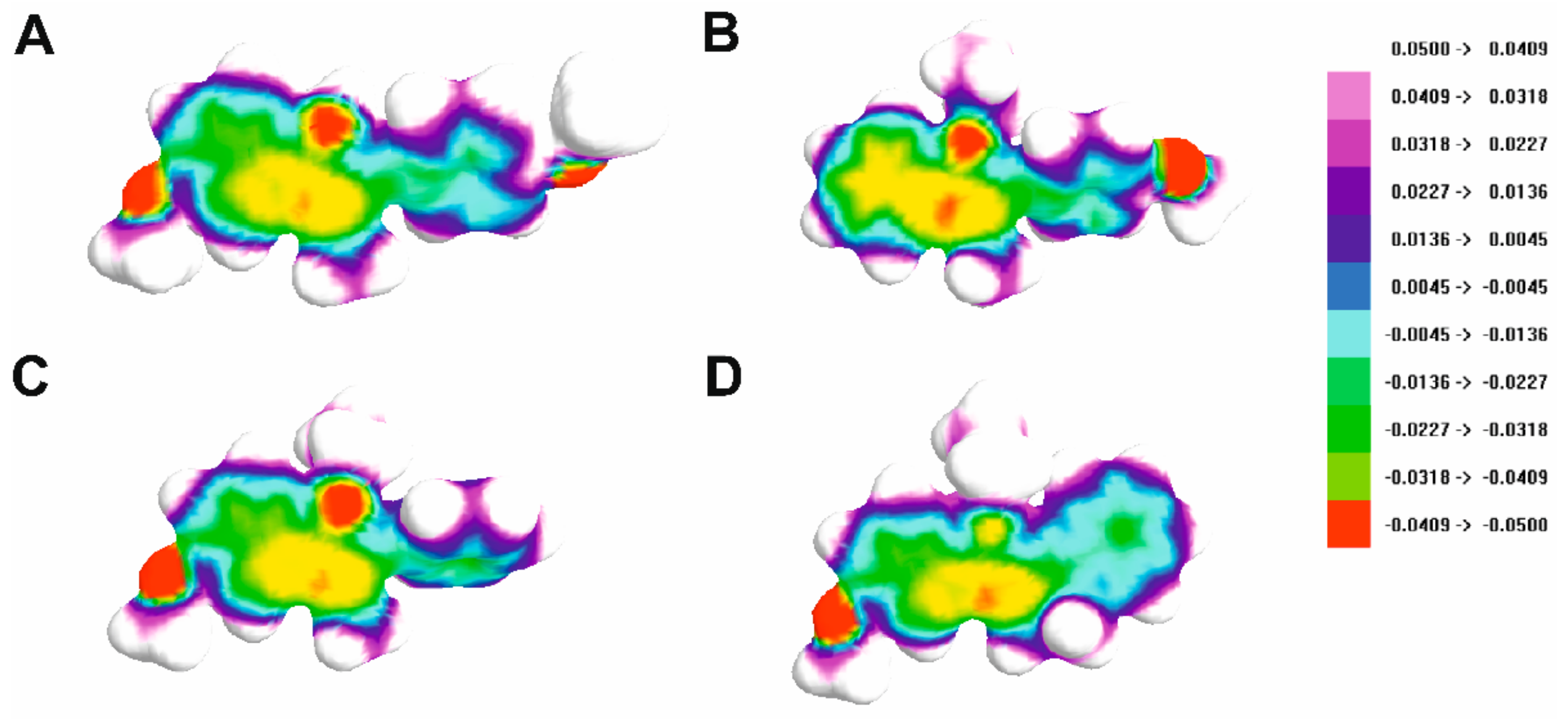
2.3. Hirshfeld Surface Analysis
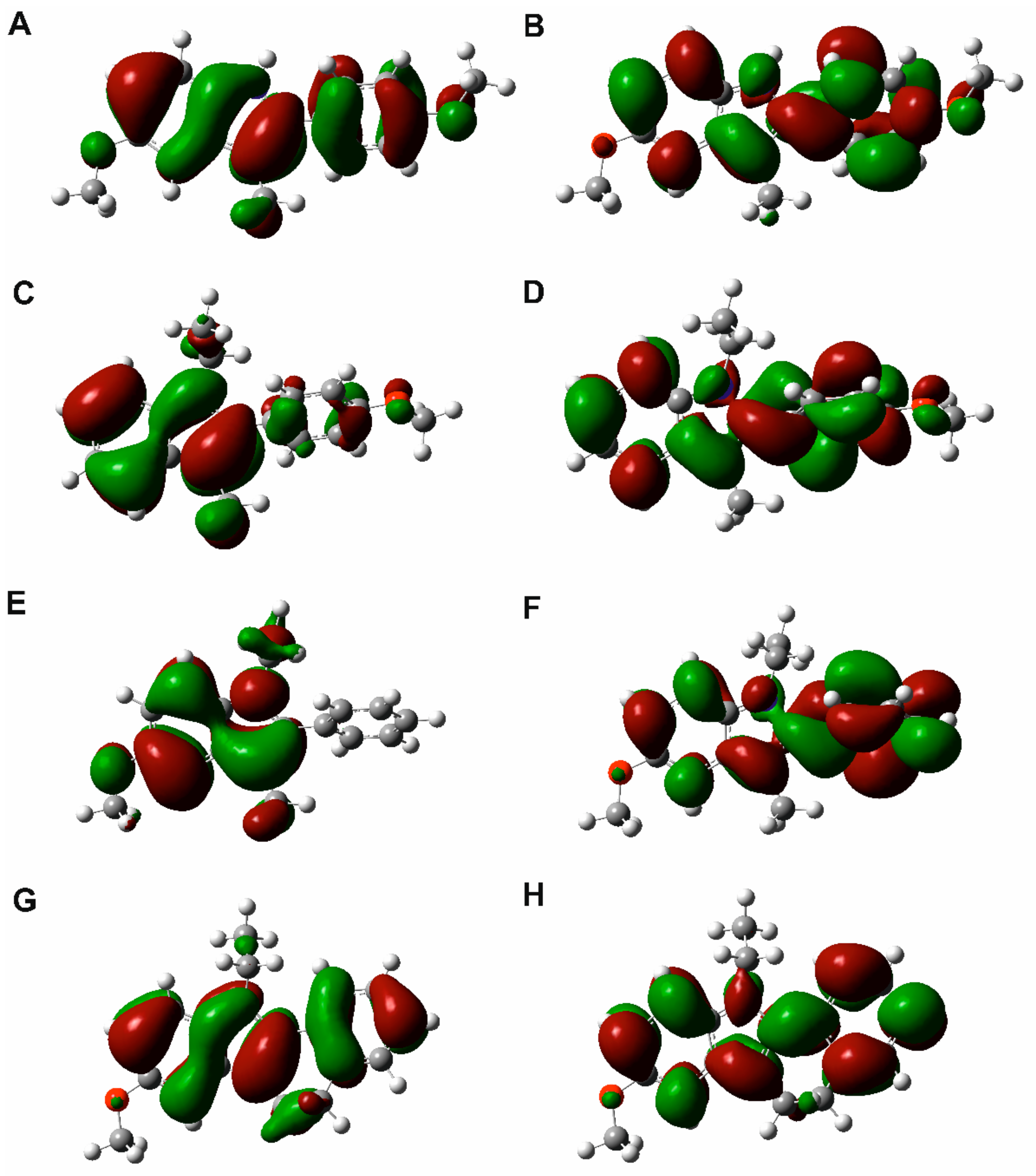
2.4. Quantum Chemical Calculations and NCI Analysis
3. Materials and Methods
3.1. Chemistry
3.2. X-ray Studies
3.3. Hirshfeld Surface and Fingerprint Analysis
3.4. Quantum Chemical Calculations and NCI Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Pollok, S.; Reiner, A. Subunit-Selective IGluR Antagonists Can Potentiate Heteromeric Receptor Responses by Blocking Desensitization. Proc. Natl. Acad. Sci. USA 2020, 117, 25851–25858. [Google Scholar] [CrossRef] [PubMed]
- Larsen, A.M.; Bunch, L. Medicinal Chemistry of Competitive Kainate Receptor Antagonists. ACS Chem. Neurosci. 2010, 2, 60–74. [Google Scholar] [CrossRef] [PubMed]
- Szénási, G.; Vegh, M.; Szabo, G.; Kertesz, S.; Kapus, G.; Albert, M.; Greff, Z.; Ling, I.; Barkoczy, J.; Simig, G.; et al. 2,3-Benzodiazepine-Type AMPA Receptor Antagonists and Their Neuroprotective Effects. Neurochem. Int. 2008, 52, 166–183. [Google Scholar] [CrossRef]
- Kaczor, A.A.; Kronbach, C.; Unverferth, K.; Pihlaja, K.; Wiinamäki, K.; Sinkkonen, J.; Kijkowska-Murak, U.; Wróbel, T.; Stachal, T.; Matosiuk, D. Novel Non-Competitive Antagonists of Kainate GluK1/GluK2 Receptors. Lett. Drug Design Discov. 2012, 9, 891–898. [Google Scholar] [CrossRef]
- Kaczor, A.A.; Karczmarzyk, Z.; Fruziński, A.; Pihlaja, K.; Sinkkonen, J.; Wiinämaki, K.; Kronbach, C.; Unverferth, K.; Poso, A.; Matosiuk, D. Structural Studies, Homology Modeling and Molecular Docking of Novel Non-Competitive Antagonists of GluK1/GluK2 Receptors. Bioorg. Med. Chem. 2014, 22, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Kaczor, A.A.; Wróbel, T.; Kronbach, C.; Unverferth, K.; Stachal, T.; Matosiuk, D. Synthesis and Molecular Docking of Novel Non-Competitive Antagonists of GluK2 Receptor. Med. Chem. Res. 2015, 24, 810–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaczor, A.A.; Kijkowska-Murak, U.A.; Matosiuk, D. Theoretical Studies on the Structure and Symmetry of the Transmembrane Region of Glutamatergic GluR5 Receptor. J. Med. Chem. 2008, 51, 3765–3776. [Google Scholar] [CrossRef] [PubMed]
- Kaczor, A.A.; Kijkowska-Murak, U.A.; Kronbach, C.; Unverferth, K.; Matosiuk, D. Modeling of Glutamate GluR6 Receptor and Its Interactions with Novel Noncompetitive Antagonists. J. Chem. Inf. Model. 2009, 49, 1094–1104. [Google Scholar] [CrossRef]
- Bartyzel, A.; Kaczor, A.A.; Głuchowska, H.; Pitucha, M.; Wróbel, T.M.; Matosiuk, D. Thermal and Spectroscopic Studies of 2,3,5-Trisubstituted and 1,2,3,5-Tetrasubstituted Indoles as Non-Competitive Antagonists of GluK1/GluK2 Receptors. J. Therm. Anal. Calorim. 2018, 133, 935–944. [Google Scholar] [CrossRef] [Green Version]
- Pidcock, E. Achiral Molecules in Non-Centrosymmetric Space Groups. Chem. Commun. 2005, 27, 3457–3459. [Google Scholar] [CrossRef]
- Saha, B.K.; Nangia, A.; Nicoud, J. Using Halogen⋯halogen Interactions to Direct Noncentrosymmetric Crystal Packing in Dipolar Organic Molecules. Cryst. Growth Des. 2006, 6, 1278–1281. [Google Scholar] [CrossRef]
- Mayo, R.A.; Sullivan, D.J.; Fillion, T.A.P.; Kycia, S.W.; Soldatov, D.V.; Preuss, K.E. Reversible Crystal-to-Crystal Chiral Resolution: Making/Breaking Non-Bonding S⋯O Interactions. Chem. Commun. 2017, 53, 3964–3966. [Google Scholar] [CrossRef] [PubMed]
- Allen, F.H.; Watson, D.G.; Brammer, L.; Orpen, A.G.; Taylor, R. Typical Interatomic Distances: Organic Compounds in International Tables for Crystallography; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2006; Chapter 9.5. [Google Scholar]
- Li, B.; Ju, Z.; Zhou, M.; Su, K.; Yuan, D. A Reusable MOF-Supported Single-Site Zinc(II) Catalyst for Efficient Intramolecular Hydroamination of o-Alkynylanilines. Angew. Chem. Int. Ed. 2019, 58, 7687–7691. [Google Scholar] [CrossRef]
- Kerr, J.R.; Trembleau, L.; Storey, J.M.D.; Wardell, J.L.; Harrison, W.T.A. Crystal Structures of Four Indole Derivatives as Possible Cannabinoid Allosteric Antagonists. Acta Crystallogr. E Crystallogr. Commun. 2015, 71, 654–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondej, M.; Bartyzel, A.; Pitucha, M.; Wróbel, T.M.; Silva, A.G.; Matosiuk, D.; Castro, M.; Kaczor, A.A. Synthesis, Structural and Thermal Studies of 3-(1-Benzyl-1,2,3,6-Tetrahydropyridin-4-Yl)-5-Ethoxy-1H-Indole (D2AAK1_3) as Dopamine D2 Receptor Ligand. Molecules 2018, 23, 2249. [Google Scholar] [CrossRef] [Green Version]
- Bartyzel, A.; Kondej, M.; Stępnicki, P.; Wróbel, T.M.; Kaczor, A.A. Experimental and Computational Structural Studies of 5-Substituted-3-(1-Arylmethyl-1,2,3,6-Tetrahydropyridin-4-Yl)-1H-Indoles. J. Mol. Struct. 2021, 1245, 130998. [Google Scholar] [CrossRef]
- Wolff, S.K.; Grimwood, D.J.; McKinnon, J.J.; Turner, M.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer; University of Western Australia: Crawley, Australia, 2012. [Google Scholar]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld Surface Analysis. Cryst. Eng. Commun. 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Grabowsky, S.; Dean, P.M.; Skelton, B.W.; Sobolev, A.N.; Spackman, M.A.; White, A.H. Crystal Packing in the 2-R,4-Oxo-[1,3-a/b]-Naphthodioxanes—Hirshfeld Surface Analysis and Melting Point Correlation. Cryst. Eng. Commun. 2012, 14, 1083–1093. [Google Scholar] [CrossRef]
- Miar, M.; Shiroudi, A.; Pourshamsian, K.; Oliaey, A.R.; Hatamjafari, F. Theoretical Investigations on the HOMO–LUMO Gap and Global Reactivity Descriptor Studies, Natural Bond Orbital, and Nucleus-Independent Chemical Shifts Analyses of 3-Phenylbenzo[d]Thiazole-2(3H)-Imine and Its Para-Substituted Derivatives: Solvent and Substituent Effects. J. Chem. Res. 2021, 45, 147–158. [Google Scholar] [CrossRef]
- Rathi, P.C.; Ludlow, R.F.; Verdonk, M.L. Practical High-Quality Electrostatic Potential Surfaces for Drug Discovery Using a Graph-Convolutional Deep Neural Network. J. Med. Chem. 2020, 63, 8778–8790. [Google Scholar] [CrossRef] [Green Version]
- Contreras-García, J.; Johnson, E.R.; Keinan, S.; Chaudret, R.; Piquemal, J.-P.; Beratan, D.N.; Yang, W. NCIPLOT: A Program for Plotting Non-Covalent Interaction Regions. J. Chem. Theory Comput. 2011, 7, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Sagaama, A.; Issaoui, N.; Al-Dossary, O.; Kazachenko, A.S.; Wojcik, M.J. Non Covalent Interactions and Molecular Docking Studies on Morphine Compound. J. King Saud Univ. Sci. 2021, 33, 101606. [Google Scholar] [CrossRef]
- Agilent Technologies Ltd. CrysAlis Pro; Agilent Technologies Ltd.: Oxfordshire, UK, 2014. [Google Scholar]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Cryst. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX and ORTEP for Windows: An Update. J. Appl. Cryst. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From Visualization to Analysis, Design and Prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [Google Scholar] [CrossRef] [Green Version]
- Spek, A.L. Single-Crystal Structure Validation with the Program PLATON. J. Appl. Cryst. 2003, 36, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision E.01; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Available online: www.Arguslab.com (accessed on 1 February 2022).
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Pitucha, M.; Sobotka-Polska, K.; Keller, R.; Pachuta-Stec, A.; Mendyk, E.; Kaczor, A.A. Synthesis and Structure of New 1-Cyanoacetyl-4-Arylsemicarbazide Derivatives with Potential Anticancer Activity. J. Mol. Struct. 2016, 1104, 24–32. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| D-H···A | d (D-H) | d (H···A) | d (D···A) | ∠ DHA |
|---|---|---|---|---|
| 1 | ||||
| C(9)-H(9)∙∙∙N(1) 1a | 0.93 | 2.52 | 3.449(2) | 174 |
| 4 | ||||
| C(18)-H(18A)∙∙∙O(1) 4a | 0.99 | 2.56 | 3.430(3) | 146 |
| Compound | EHOMO (eV) | ELUMO (eV) | HOMO-LUMO Gap (eV) |
|---|---|---|---|
| 1 | −5.30 | −0.91 | 4.39 |
| 2 | −5.31 | −0.75 | 4.56 |
| 3 | −5.36 | −0.99 | 4.37 |
| 4 | −5.26 | −1.21 | 4.05 |
| Compound | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| Temperature K | 298(2) | 298(2) | 100(2) | 100(2) |
| Empirical formula | C17H17NO2 | C18H19NO | C18H19NO | C19H19NO |
| Formula weight | 267.31 | 265.34 | 265.34 | 277.35 |
| Crystal system | monoclinic | monoclinic | orthorhombic | orthorhombic |
| Space group | C2/c | P21/n | P212121 | Pbc21 |
| a (Å) | 27.464(3) | 9.2916(7) | 7.6910(3) | 17.2868(5) |
| b (Å) | 4.9385(4) | 8.7458(5) | 7.8087(3) | 8.7543(3) |
| c (Å) | 20.956(2) | 18.6641(13) | 23.5793(10) | 9.4312(2) |
| α (°) | 90.00 | 90 | 90 | |
| β (°) | 108.564(11) | 102.484(7) | 90 | 90 |
| γ (°) | 90.00 | 90 | 90 | |
| Volume (Å3) | 2694.4(5) | 1480.83(18) | 1416.10(10) | 1427.26(7) |
| Z | 8 | 4 | 4 | 4 |
| μ (mm−1) | 0.086 | 0.073 | 0.077 | 0.079 |
| Absorption correction | multi-scan | multi-scan | multi-scan | multi-scan |
| F(000) | 1136 | 568 | 568 | 592 |
| Crystal size (mm) | 0.10 × 0.15 × 0.40 | 0.13 × 0.27 × 0.50 | 0.40 × 0.40 × 0.50 | 0.28 × 0.30 × 0.30 |
| θ range (°) | 2.95 to 26.02 | 2.58 to 27.10 | 2.75 to 26.01 | 3.20 to 27.09 |
| Reflections collected/unique | 5674/2615 | 10931/3261 | 8686/2778 | 9669/3008 |
| Rint | 0.0289 | 0.0427 | 0.0361 | 0.0314 |
| Data/restraints/parameters | 2615/0/235 | 3261/0/184 | 2778/0/184 | 3008/1/192 |
| GooF on F2 | 1.039 | 1.018 | 1.070 | 1.074 |
| Final R indices [I > 2σ(I)] | R1 = 0.0410, wR2 = 0.0912 | R1 = 0.0585, wR2 = 0.1320 | R1 = 0.0356, wR2 = 0.0743 | R1 = 0.0347, wR2 = 0.0756 |
| R indices (all data) | R1 = 0.0578, wR2 = 0.0998 | R1 = 0.1012, wR2 = 0.1582 | R1 = 0.0429, wR2 = 0.0781 | R1 = 0.0399, wR2 = 0.0787 |
| Largest diff. peak/hole, e Å−3 | 0.255/−0.157 | 0.370/−0.236 | 0.171/−0.153 | 0.245/−0.139 |
| CCDC number | 2153868 | 2153866 | 2153867 | 2153869 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bartyzel, A.; Kaczor, A.A.; Mahmoudi, G.; Masoudiasl, A.; Wróbel, T.M.; Pitucha, M.; Matosiuk, D. Experimental and Computational Structural Studies of 2,3,5-Trisubstituted and 1,2,3,5-Tetrasubstituted Indoles as Non-Competitive Antagonists of GluK1/GluK2 Receptors. Molecules 2022, 27, 2479. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27082479
Bartyzel A, Kaczor AA, Mahmoudi G, Masoudiasl A, Wróbel TM, Pitucha M, Matosiuk D. Experimental and Computational Structural Studies of 2,3,5-Trisubstituted and 1,2,3,5-Tetrasubstituted Indoles as Non-Competitive Antagonists of GluK1/GluK2 Receptors. Molecules. 2022; 27(8):2479. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27082479
Chicago/Turabian StyleBartyzel, Agata, Agnieszka A. Kaczor, Ghodrat Mahmoudi, Ardavan Masoudiasl, Tomasz M. Wróbel, Monika Pitucha, and Dariusz Matosiuk. 2022. "Experimental and Computational Structural Studies of 2,3,5-Trisubstituted and 1,2,3,5-Tetrasubstituted Indoles as Non-Competitive Antagonists of GluK1/GluK2 Receptors" Molecules 27, no. 8: 2479. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27082479