4.1. General
All the solvents, starting materials, and chemical reagents were purchased from commercial sources and used without further purification, unless otherwise stated. Anhydrous tetrahydrofuran, dichloromethane, and 1,2-dichloroethane (DCE) were obtained by distillation over sodium wire or calcium hydride, respectively. TLC was performed on silica gel plates (GF254) to visualize components by UV light (254 nm). Column chromatography was carried out on silica gel (200–300 mesh). All non-aqueous reactions are carried out under a nitrogen atmosphere, the reagents do not contain water, and all reaction vessels are dried.
1H NMR spectra were obtained on Bruker at 400 MHz.
13C NMR spectra were obtained at 101 MHz (see
Supplementary Materials). High-resolution mass spectra (HRMS) were performed using a Bruker ESI-TOF high-resolution mass spectrometer. NMR chemical shifts were reported in δ (ppm) using the δ 0 signal of tetramethylsilane, or the residual non-deuterated solvent signal (δ 7.26 signal for CDCl
3, δ 3.31 signal for CD
3OD, or δ 2.50 signal for (CD
3)
2SO, or in case of a mixed solvent, the CD
3OD signal, as internal standards). The following multiplicity abbreviations are used: singlet (s), doublet (d), triplet (t), quartet (q), multiplet (m), and broads (br s). High-resolution mass spectra (HRMS) were performed using a Bruker ESI-TOF high-resolution mass spectrometer. The purity of all final compounds (≥95%) was established by high-performance liquid chromatography (HPLC), which was carried out on a Waters HPLC system using InertSustain-C18 column (5 µm, 250 × 4.6 mm), with a column temperature of 30 °C, detection wavelength at 254 and 280 nm, flow rate = 1 mL/min, and gradient of 5–95% CH
3CN in water (containing 0.5 vol % of HCOOH) in 18 min.
General procedure for the preparation of ethyl (Z)-3-amino-3-(4-methoxyphenyl) acrylate from ethyl 3-(4-methoxyphenyl)-3-oxopropanoate (method A)
A commercially available mixture of 10 (1.0 mmol), ammonium formate (5.0 mmol), and molecular sieves (4 Å, 0.2 g) in 10 mL ethanol was refluxed for 8 h under N2 and then cooled to room temperature. The reaction mixture was filtered through celite. The filtrate was evaporated. The residue was extracted with ethyl acetate (3 × 20 mL), washed with NaCl aqueous solution, the combined organic layer was dried with anhydrous Na2SO4, and the residues were purified by flash chromatography (petroleum ether: ethyl acetate = 20:1) to give 11.
General procedure for the preparation of indole construction (method B)
To a solution of 11 (1.0 mmol) and ZnBr2 (1.0 mmol) in 3 mL, anhydrous THF has added a solution of benzoquinone (1.0 mmol) in THF (2 mL) under N2. After stirring for 6 h at room temperature. The reaction mixture was quenched with saturated NH4Cl aqueous solution and extracted with EtOAc (3 × 10mL). The combined organic phases were dried over anhydrous Na2SO4 and evaporated. The residue was purified by flash chromatography (petroleum ether: ethyl acetate = 10:1) to give 12.
General procedure for the Mannich reaction of 5-hydroxy indole derivatives with amine and formaldehyde (method C)
To a solution of indole analogues (1 mmol) in ethanol (3 mL) were added formaldehyde (37% in water, 4 mmol) and the appropriate amine (4 mmol) at room temperature under N2. The reaction mixture was allowed to reflux for 8–12 h and then cooled to rt. The reaction mixture was evaporated, and the residue was purified by flash chromatography (dichloromethane: methanol = 60:1).
General procedure for the One-pot reaction of 5-hydroxy indole derivatives (method D)
Under the catalysis of montmorillonite (5 mmol), the corresponding amine (1 mmol) and ethyl acetoacetate (1 mmol) was dissolved in refluxing anhydrous DCE under N2 for 30 min and then to it was added benzoquinone (1 mmol) DCE solution dropwise. After reacting for 8 h, it was cooled to room temperature. The reaction mixture was filtered through celite; the filtrate was evaporated; the residue was purified by flash chromatography (petroleum ether: ethyl acetate = 5:1) to give the product.
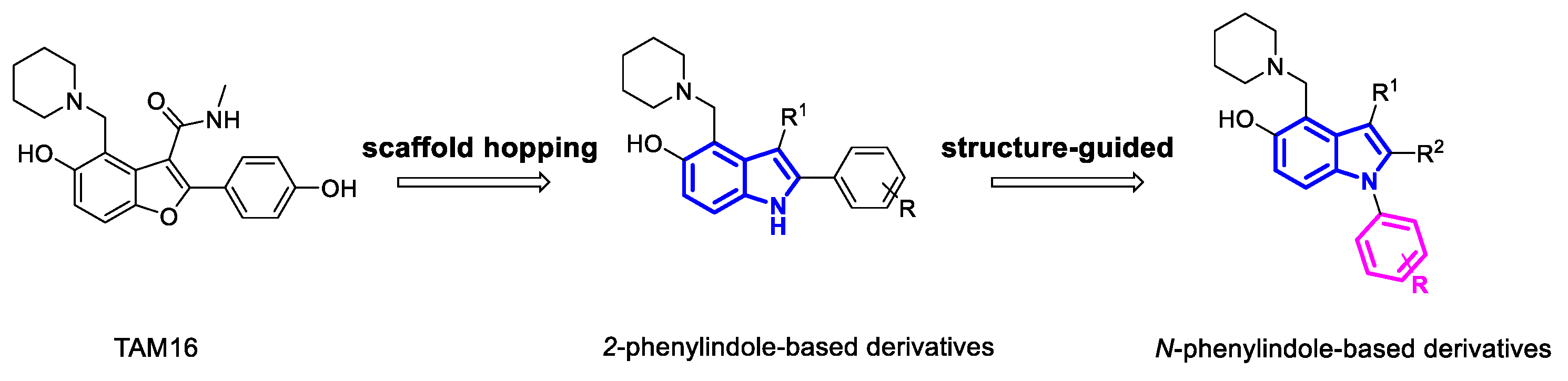
Ethyl (Z)-3-amino-3-(4-methoxyphenyl) acrylate (11).
This compound was obtained from ethyl 3-(4-methoxyphenyl)-3-oxopropanoate 10 by employing Method A. Yield 92%; yellow oil.1H NMR (400 MHz, CDCl3) δ 7.49 (d, J = 8.0 Hz, 2H), 6.91 (d, J = 8.0 Hz, 2H), 4.93 (s, 1H), 4.20 (q, J = 7.0 Hz, 2H), 3.93 (s, 1H), 3.87 (s, 2H), 3.83 (s, 3H), and 1.25 (q, J = 7.0 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 191.1, 131.1, 127.6, 114.3, 114.1, 83.8, 61.6, 59.0, 55.7, 55.5, and 46.0.
Ethyl 5-hydroxy-2-(4-methoxyphenyl)-1H-indole-3-carboxylate (12).
This compound was obtained from Compound 11 by employing Method B. Yield 41%; pale yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 11.70 (s, 1H), 8.93 (s, 1H), 7.60 (d, J = 8.2 Hz, 2H), 7.41 (s, 1H), 7.20 (d, J = 8.6 Hz, 1H), 7.03 (d, J = 8.2 Hz, 2H), 6.68 (d, J = 8.4 Hz, 1H), 4.17 (q, J = 7.0 Hz, 2H), 3.82 (s, 3H), and 1.24 (t, J = 7.0 Hz, 3H).
Ethyl 5-hydroxy-2-(4-methoxyphenyl)-4-(piperidin-1-ylmethyl)-1H-indole-3-carboxylate (13).
This compound was obtained from Compound 12 by employing Method C. Yield 82%; pale yellow solid. 1H NMR (400 MHz, CDCl3) δ 10.93 (s, 1H), 7.47–7.37 (m, 3H), 7.07 (d, J = 8.5 Hz, 1H), 6.85 (d, J = 7.8 Hz, 2H), 4.65 (s, 2H), 4.10 (q, J = 6.9 Hz, 2H), 3.79 (s, 3H), 3.54–3.22 (m, 2H), 3.04–2.72 (m, 2H), 1.89–1.77 (m, 4H), 1.53–1.24 (m, 2H), and 1.01 (t, J = 6.9 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 168.8, 160.1, 153.3, 146.2, 131.4, 131.0, 126.8, 125.1, 115.5, 115.3, 113.4, 106.9, 103.5, 60.8, 55.5, 53.9, 52.7, 23.9, 22.4, and 13.9. HRMS (ESI) m/z: Calcd for C24H29N2O4 (M + H)+, 409.2122, found 409.2141.
Ethyl 5-hydroxy-2-(4-hydroxyphenyl)-4-(piperidin-1-ylmethyl)-1H-indole-3-carboxylate (14).
To a solution of Compound 13 (1.0 mmol) in anhydrous CH2Cl2 (4 mL), BBr3 (1 M in CH2Cl2, 4.0 mmol) was added at room temperature under N2. After being stirred overnight, the reaction mixture was quenched with EtOH. The residue was purified by flash chromatography (dichloromethane: methanol = 50:1). Yield 89%; pale yellow solid. 1H NMR (400 MHz, CD3OD:CDCl3 = 1:2) δ 7.33 (d, J = 8.7 Hz, 1H), 7.28 (d, J = 7.8 Hz, 2H), 6.89–6.79 (m, 3H), 4.56 (s, 2H), 4.09 (q, J = 7.0 Hz, 2H), 3.57–3.33 (m, 2H), 3.21–2.86 (m, 2H), 2.06–1.47 (m, 6H), and 0.99 (t, J = 7.0 Hz, 3H). 13C NMR (101 MHz, CD3OD:CDCl3 = 1:2) δ 169.7, 158.6, 153.7, 147.9, 131.6, 131.4, 127.6, 124.6, 115.5, 115.3, 112.8, 105.9, 104.1, 61.3, 54.0, 53.2, 24.4, 22.6, and 14.0. HRMS (ESI) m/z: Calcd for C23H27N2O4 (M + H)+, 395.1965, found 395.1980.
Ethyl 5-hydroxy-2-(4-hydroxyphenyl)-1-methyl-4-(piperidin-1-ylmethyl)-1H-indole-3-carboxylate (15).
This compound was obtained from Compound 10, methylamine, according to the methodology described for 14. Overall yield 22%; yellow solid. 1H NMR (400 MHz, CD3OD:CDCl3 = 1:2) δ 7.38 (d, J = 8.8 Hz, 1H), 7.11 (d, J = 7.7 Hz, 2H), 6.99 (d, J = 8.8 Hz, 1H), 6.91 (d, J = 7.8 Hz, 2H), 4.62 (s, 2H), 3.98 (q, J = 7.0 Hz, 2H), 3.50 (s, 4H), 3.50–3.43 (m, 2H), 3.15–2.99 (m, 2H), 2.09–1.52 (m, 6H), and 0.83 (t, J = 7.0 Hz, 3H). 13C NMR (101 MHz, CD3OD:CDCl3 = 1:2) δ 169.2, 158.6, 154.2, 149.6, 132.6, 131.7, 126.8, 123.3, 115.5, 114.3, 113.0, 105.8, 105.5, 61.2, 53.6, 53.0, 31.5, 24.2, 22.4, and 13.7. HRMS (ESI) m/z: Calcd for C24H29N2O4 (M + H)+, 409.2122, found 409.2153.
Ethyl 5-hydroxy-2-(4-hydroxyphenyl)-1-isopropyl-4-(piperidin-1-ylmethyl)-1H-indole-3-carboxylate (16).
This compound was obtained from Compound 10, isopropylamine, according to the methodology described for 14. Overall yield 21%; pale yellow solid. 1H NMR (400 MHz, CD3OD:CDCl3 = 1:3) δ 7.55 (d, J = 9.0 Hz, 1H), 7.02 (d, J = 8.0 Hz, 2H), 6.95 (d, J = 9.0 Hz, 1H), 6.88 (d, J = 8.0 Hz, 2H), 4.50 (s, 2H), 4.40–4.37 (m, 1H), 3.88 (q, J = 7.0 Hz, 2H), 3.51–3.42 (m, 2H), 3.10–2.98 (m, 2H), 2.06–1.55 (m, 6H), 1.44 (d, J = 7.0 Hz, 6H), and 0.73 (t, J = 7.0 Hz, 3H). 13C NMR (101 MHz, CD3OD:CDCl3 = 1:3) δ 169.3, 158.3, 153.5, 148.9, 130.9, 129.7, 127.5, 123.6, 116.5, 115.5, 112.4, 105.4, 105.2, 61.1, 53.3, 52.8, 49.1, 24.1, 22.2, 21.4, and 13.5. HRMS (ESI) m/z: Calcd for C26H33N2O4 (M + H)+, 437.2435, found 437.2459.
Ethyl 5-hydroxy-2-(4-methoxyphenyl)-1-phenyl-4-(piperidin-1-ylmethyl)-1H-indole-3-carboxylate (17).
This compound was obtained from Compound 10, aniline, by employing Methods A, B, and C. Overall yield 19%; pale yellow solid. 1H NMR (400 MHz, CDCl3) δ 7.35–7.28 (m, 3H), 7.14–7.11(m, 4H), 6.96 (d, J = 8.8 Hz, 1H), 6.79 (d, J = 8.8 Hz, 1H), 6.75 (d, J = 8.1 Hz, 2H), 4.17 (s, 2H), 4.14 (q, 7.0 Hz, 2H), 3.76 (s, 3H), 3.44–2.08 (m, 4H), 1.75–1.42 (m, 6H), and 1.05 (t, J = 7.0 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 167.2, 159.3, 154.6, 143.7, 137.2, 132.4, 131.9, 129.1, 128.4, 127.7, 124.6, 123.7, 114.2, 112.9, 110.9, 110.7, 107.5, 60.3, 58.6, 55.1, 53.8, 25.8, 23.9, and 13.9. HRMS (ESI) m/z: Calcd for C30H33N2O4 (M + H)+, 485.2435, found 485.2447.
Ethyl 5-hydroxy-2-(4-hydroxyphenyl)-1-phenyl-4-(piperidin-1-ylmethyl)-1H-indole-3-carboxylate (18).
This compound was obtained from Compound 17, according to the methodology described for 13–14. Yield 84%; pale yellow solid. 1H NMR (400 MHz, CD3OD:CDCl3 = 1:3) δ 7.40–7.29 (m, 3H), 7.11 (d, J = 6.6 Hz, 2H), 7.04 (d, J = 8.8 Hz, 1H), 6.99 (d, J = 8.0 Hz, 2H), 6.89 (d, J = 8.8 Hz, 1H), 6.67 (d, J = 8.0 Hz, 2H), 4.69 (s, 2H), 4.05 (q, J = 7.0 Hz, 2H), 3.67–3.48 (m, 2H), 3.25–3.01 (m, 2H), 2.10–1.53 (m, 6H), and 0.88 (t, J = 7.0 Hz, 3H). 13C NMR (101 MHz, CD3OD:CDCl3 = 1:3) δ 169.4, 158.3, 154.5, 149.2, 137.1, 133.8, 132.5, 130.0, 129.2, 129.1, 126.8, 123.3, 115.4, 115.1, 113.4, 106.9, 105.9, 61.5, 53.6, 53.3, 24.2, 22.6, and 13.8. HRMS (ESI) m/z: Calcd for C29H31N2O4 (M + H)+, 471.2278, found 471.2285.
1-(5-hydroxy-2-(4-methoxyphenyl)-1-phenyl-4-(piperidin-1-ylmethyl)-1H-indol-3-yl) ethan-1-one (19).
This compound was obtained from 1-(4-methoxyphenyl) butane-1,3-dione, aniline, by employing Methods A, B, and C. Overall yield 14%; pale yellow solid. 1H NMR (400 MHz, CDCl3) δ 7.85 (d, J = 7.6 Hz, 2H), 7.61–7.48 (m, 3H), 7.32 (d, J = 7.2 Hz, 2H), 7.08 (d, J = 8.4 Hz, 1H), 6.97 (d, J = 7.6 Hz, 2H), 6.93 (d, J = 8.8 Hz, 1H), 4.21 (s, 2H), 3.89 (s, 3H), 3.29–2.41 (m, 4H), 1.99 (s, 3H), and 1.86–1.39 (m, 6H). 13C NMR (101 MHz, CDCl3) δ 194.8, 163.8, 154.2, 136.6, 133.5, 133.4, 132.2, 130.0, 129.1, 128.3, 126.4, 115.8, 114.8, 114.2, 112.7, 109.1, 55.7, 53.3, 24.4, 22.9, and 14.1. HRMS (ESI) m/z: Calcd for C29H31N2O3 (M + H)+, 455.2329, found 455.2346.
1-(5-hydroxy-2-(4-hydroxyphenyl)-1-phenyl-4-(piperidin-1-ylmethyl)-1H-indol-3-yl) ethan-1-one (20).
This compound was obtained from Compound 19, according to the methodology described for 13–14. Yield 90%; brown solid. 1H NMR (400 MHz, CD3OD:CDCl3 = 1:1) δ 7.78 (d, J = 7.6 Hz, 2H), 7.61–7.52 (m, 3H), 7.32 (d, J = 7.4 Hz, 2H), 6.93 (d, J = 9.0 Hz, 1H), 6.89 (d, J = 7.8 Hz, 2H), 6.79 (d, J = 8.8 Hz, 1H), 4.20 (s, 2H), 3.28–2.36 (m, 4H), 1.95 (s, 3H), and 1.94–1.73 (m, 6H). 13C NMR (101 MHz, CD3OD:CDCl3 = 1:1) δ 196.5, 163.6, 154.1, 136.6, 133.8, 133.3, 131.8, 130.5, 129.8, 128.5, 126.8, 116.2, 115.1, 114.0, 113.0, 53.5, 25.0, 24.8, 22.8, and 14.6. HRMS (ESI) m/z: Calcd for C28H29N2O3 (M + H)+, 441.2173, found 441.2186.
5-hydroxy-2-(4-methoxyphenyl)-N-methyl-1-phenyl-4-(piperidin-1-ylmethyl)-1H-indole-3-carboxamide (22).
This compound was obtained from Compound 10, aniline, employing Methods A–B to give Compound 21. To a solution of Compound 21 (1.0 mmol) in ethanol (5 mL) was added NaOH aqueous solution (1N 10.0 mL) at rt. After stirring at refluxing for 4–6 h, the reaction mixture was acidified with 2 M aqueous HCl to pH 5–6. the residue was purified by flash chromatography (dichloromethane: methanol = 60:1) to give the product. Then, the result (1 mmol) in anhydrous DMF (5 mL) was added EDC·HCl (1.3 mmol), HOBt (1.3 mmol), N, N-diisopropylethylamine (2.1 mmol), and the methylamine hydrochloride (1.2 mmol) under N2 at rt. After stirring overnight at rt, the reaction mixture was quenched with saturated NH4Cl aqueous solution and extracted with EtOAc (2 × 30 mL). The combined organic phases were washed with NaCl aqueous solution, dried over anhydrous Na2SO4, and evaporated. The residue was purified by flash chromatography (dichloromethane: methanol = 40:1) to give the amide product. Next, we used Method C to give Compound 22. Overall yield 12%; white solid. 1H NMR (400 MHz, CD3OD:CDCl3 = 1:2) δ 7.38–7.28 (m, 3H), 7.11–6.99 (m, 5H), 6.84 (d, J = 9.0 Hz, 1H), 6.75 (d, J = 8.2 Hz, 2H), 4.40 (s, 2H), 3.72 (s, 3H), 3.48–3.35 (m, 2H), 3.08–2.90 (m, 2H), 2.66 (s, 3H), and 2.07–1.60 (m, 6H). 13C NMR (101 MHz, CD3OD:CDCl3 = 1:2) δ 170.2, 160.5, 153.5, 142.7, 137.0, 133.3, 132.1, 129.9, 128.9, 128.8, 125.4, 122.2, 114.9, 114.4, 113.3, 110.4, 104.6, 55.5, 52.8, 27.0, 24.3, 22.8, and 22.5. HRMS (ESI) m/z: Calcd for C29H32N3O3 (M + H)+, 470.2438, found 470.2467.
N-ethyl-5-hydroxy-2-(4-methoxyphenyl)-1-phenyl-4-(piperidin-1-ylmethyl)-1H-indole-3-carboxamide (23).
This compound was obtained from Compound 22, ethylamine, according to the methodology described for 13. Overall yield 17%; pale yellow solid. 1H NMR (400 MHz, CD3OD:CDCl3 = 1:2) δ 7.39–7.27 (m, 3H), 7.16–7.08 (m, 4H), 6.95 (d, J = 8.8 Hz, 1H), 6.76 (d, J = 8.6 Hz, 2H), 6.68 (d, J = 8.8 Hz, 1H), 4.08 (s, 2H), 3.74 (s, 3H), 3.24 (q, J = 7.2 Hz, 2H), 2.92–2.41 (m, 4H), 1.76–1.50 (m, 6H), and 0.97 (t, J = 7.2 Hz, 3H). 13C NMR (101 MHz, CD3OD:CDCl3 = 1:2) δ 170.1, 160.4, 153.8, 140.2, 138.3, 133.2, 132.1, 129.9, 129.0, 128.4, 125.4, 123.6, 114.2, 114.0, 112.3, 111.5, 110.4, 57.7, 55.5, 54.2, 35.4, 26.2, 24.3, and 14.4. HRMS (ESI) m/z: Calcd for C30H34N3O3 (M + H)+, 484.2595, found 484.2623.
5-hydroxy-2-(4-hydroxyphenyl)-N-methyl-1-phenyl-4-(piperidin-1-ylmethyl)-1H-indole-3-carboxamide (24).
This compound was obtained from Compound 22, according to the methodology described for 14. Yield 88%; white solid. 1H NMR (400 MHz, CD3OD:CDCl3 = 1:2) δ 7.38–7.27 (m, 3H), 7.08 (d, J = 6.6 Hz, 2H), 7.03 (d, J = 8.8 Hz, 1H), 6.95 (d, J = 8.6 Hz, 2H), 6.81 (d, J = 8.8 Hz, 1H), 6.66 (d, J = 8.6 Hz, 2H), 4.41 (s, 2H), 3.53–3.30 (m, 2H), 3.10–2.88 (m, 2H), 2.67 (s, 3H), and 2.04–1.50 (m, 6H). 13C NMR (101 MHz, CD3OD:CDCl3 = 1:2) δ 170.5, 158.6, 153.6, 143.3, 137.4, 133.5, 132.4, 123.0, 129.0, 128.9, 125.7, 121.3, 115.9, 114.9, 113.2, 110.5, 105.0, 54.1, 53.0, 27.0, 24.5, and 22.7. HRMS (ESI) m/z: Calcd for C28H30N3O3 (M + H)+, 456.2282, found 456.2292.
N-ethyl-5-hydroxy-2-(4-hydroxyphenyl)-1-phenyl-4-(piperidin-1-ylmethyl)-1H-indole-3-carboxamide (25).
This compound was obtained from Compound 23, according to the methodology described for 14. Yield 83%; pale yellow solid. 1H NMR (400 MHz, CD3OD:CDCl3 = 1:3) δ 7.41–7.32 (m, 3H), 7.12 (d, J = 6.5 Hz, 2H), 7.07 (d, J = 8.9 Hz, 1H), 7.01 (d, J = 7.9 Hz, 2H), 6.86 (d, J = 8.9 Hz, 1H), 6.76–6.68 (m, 2H), 4.44 (s, 2H), 3.53–3.34 (m, 2H), 3.21 (q, J = 7.1 Hz, 2H), 3.07–2.96 (m, 2H), 2.15–1.71 (m, 6H), and 0.87 (t, J = 7.2 Hz, 3H). 13C NMR (101 MHz, CD3OD:CDCl3 = 1:3) δ 169.3, 158.6, 153.5, 143.3, 137.0, 133.3, 132.3, 131.3 129.8, 128.8, 125.5, 121.0, 115.9, 114.8, 113.2, 110.4, 104.8, 52.9, 52.8, 35.3, 24.4, 22.5, and 14.1. HRMS (ESI) m/z: Calcd for C29H32N3O3 (M + H)+, 470.2438, found 470.2458.
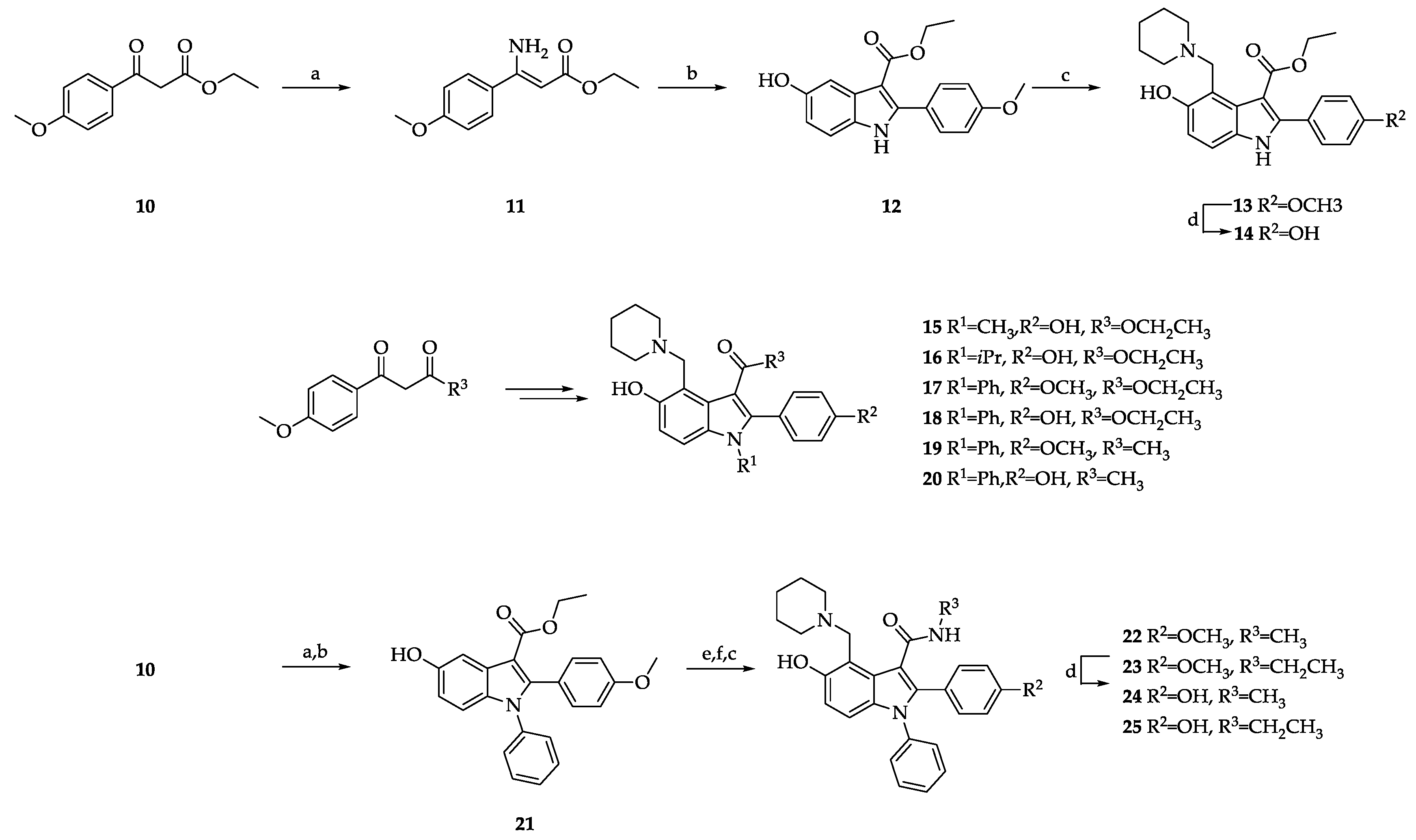
Ethyl 5-hydroxy-2-methyl-1-phenyl-1H-indole-3-carboxylate (27).
This compound was obtained from ethyl acetoacetate (26), aniline, employing Method D. Yield 47%; dark brown solid. 1H NMR (400 MHz, DMSO-d6) δ 9.29 (s, 1H), 7.67–7.54 (m, 3H), 7.48–7.41 (m, 2H), 7.19 (s, 1H), 6.90 (d, J = 7.8 Hz, 1H), 6.65 (d, J = 7.8 Hz, 1H), 4.31 (q, J = 6.8 Hz, 2H), 2.48 (s, 3H), and 1.38 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 165.1, 153.1, 145.4 144.8, 143.0, 136.1, 129.9, 128.9, 127.9, 115.6, 110.7, 105.5, 103.6, 58.9, 14.6, and 12.9.
Ethyl 5-hydroxy-2-methyl-1-phenyl-4-(piperidin-1-ylmethyl)-1H-indole-3-carboxylate (28).
This compound was obtained from Compound 27, employing Method C. Yield 88%; brown solid. 1H NMR (400 MHz, CDCl3) δ 7.62–7.51 (m, 3H), 7.29–7.24 (m, 2H), 7.18 (d, J = 8.8 Hz, 1H), 6.87 (d, J = 8.8 Hz, 1H), 4.98 (s, 2H), 4.41 (q, J = 7.1 Hz, 2H), 3.60–3.31 (m, 2H), 3.16–2.86 (m, 2H), 2.46 (s, 3H), 2.16–1.54 (m, 6H), and 1.43 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 167.7, 154.0, 145.9, 136.2, 133.7, 130.0, 129.4, 128.5, 126.4, 116.5, 113.8, 108.9, 105.5, 60.8, 53.9, 52.7, 23.6, 22.5, 14.7, and 14.7. HRMS (ESI) m/z: Calcd for C24H29N2O3 (M + H)+, 393.2173, found 393.2190.
5-hydroxy-N,2-dimethyl-1-phenyl-4-(piperidin-1-ylmethyl)-1H-indole-3-carboxamide (29).
This compound was obtained from N-methyl-3-oxobutanamide and aniline, employing Methods D and C. Overall yield 41%; pale brown solid. 1H NMR (400 MHz, CD3OD:CDCl3 = 1:1) δ 7.60–7.51 (m, 3H), 7.29 (d, J = 7.4 Hz, 2H), 6.91 (d, J = 8.8 Hz, 1H), 6.76 (d, J = 8.8 Hz, 1H), 4.38 (s, 2H), 3.49–3.35 (m, 2H), 3.12–3.04 (m, 2H), 2.99 (s, 3H), 2.33 (s, 3H), and 2.03–1.72 (m, 6H). 13C NMR (101 MHz, CD3OD:CDCl3 = 1:1) δ 170.5, 153.2, 140.1, 136.8, 133.3, 130.4, 129.6, 128.6, 125.6, 114.3, 112.3, 110.2, 104.7, 52.9, 52.8, 27.0, 24.3, 23.0, and 13.0. HRMS (ESI) m/z: Calcd for C23H28N3O2 (M + H)+, 378.2176, found 378.2199.
5-hydroxy-1-(4-methoxyphenyl)-N,2-dimethyl-4-(piperidin-1-ylmethyl)-1H-indole-3-carboxamide (30).
This compound was obtained from N-methyl-3-oxobutanamide and 4-methoxyaniline, employing Methods D and C. Overall yield 35%; pale brown solid. 1H NMR (400 MHz, CD3OD:CDCl3 = 1:1) δ 7.15 (d, J = 8.2 Hz, 2H), 6.99 (d, J = 8.2 Hz, 2H), 6.82 (d, J = 8.8 Hz, 1H), 6.75 (d, J = 8.8 Hz, 1H), 4.27 (s, 2H), 3.82 (s, 3H), 3.42–3.30 (m, 2H), 2.94 (s, 3H), 2.93–2.83 (m, 2H), 2.29 (s, 3H), and 1.98–1.48 (m, 6H). 13C NMR (101 MHz, CD3OD:CDCl3 = 1:1) δ 170.3, 160.1, 152.8, 140.3, 133.2, 129.5, 129.1, 125.2, 115.2, 114.0, 112.1, 109.5, 104.2, 55.8, 52.6, 52.4, 26.8, 24.0, 22.3, and 13.0. HRMS (ESI) m/z: Calcd for C24H30N3O3 (M + H)+, 408.2282, found 408.2311.
Ethyl 5-hydroxy-2-methyl-4-(piperidin-1-ylmethyl)-1-(p-tolyl)-1H-indole-3-carboxylate (31).
This compound was obtained from ethyl acetoacetate (26) and 4-methylaniline, employing Methods D and C. Overall yield 41%; pale yellow solid. 1H NMR (400 MHz, CDCl3) δ 7.34 (d, J = 7.6 Hz, 2H), 7.16 (d, J = 8.8 Hz, 1H), 7.11 (d, J = 7.6 Hz, 2H), 6.84 (d, J = 8.8 Hz, 1H), 4.93 (s, 2H), 4.38 (q, J = 7.0 Hz, 2H), 3.55–3.29 (m, 2H), 3.10–2.86 (m, 2H), 2.45 (s, 3H), 2.43 (s, 3H), 2.04–1.88 (br, 6H), and 1.41 (t, J = 7.0 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 167.8, 154.0, 146.0, 139.5, 133.7, 133.5, 130.6, 128.1, 126.3, 116.2, 113.8, 108.6, 105.2, 60.7, 53.9, 52.6, 23.7, 22.5, 21.4, and 14.6. HRMS (ESI) m/z: Calcd for C25H31N2O3 (M + H)+, 407.2329, found 407.2303.
Ethyl 5-hydroxy-2-methyl-4-(piperidin-1-ylmethyl)-1-(m-tolyl)-1H-indole-3-carboxylate (32).
This compound was obtained from ethyl acetoacetate (26) and 3-methylaniline, employing Methods D and C. Overall yield 35%; pale yellow solid. 1H NMR (400 MHz, CDCl3) δ 7.45 (t, J = 7.6 Hz, 1H), 7.34 (d, J = 7.4 Hz, 1H), 7.15 (d, J = 8.8 Hz, 1H), 7.09–7.03 (m, 2H), 6.88 (d, J = 8.8 Hz, 1H), 5.00 (s, 2H), 4.39 (q, J = 7.0 Hz, 2H), 3.52–3.33 (m, 2H), 3.11–2.92 (m, 2H), 2.46 (s, 3H), 2.44 (s, 3H), 2.09–1.80 (m, 6H), and 1.43 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 167.7, 153.9, 146.0, 140.3, 136.2, 133.8, 130.2, 129.8, 129.0, 126.4, 125.4, 116.9, 114.0, 109.3, 105.3, 60.7, 52.7, 31.6, 23.6, 22.5, 21.5 14.8, and 14.7. HRMS (ESI) m/z: Calcd for C25H31N2O3 (M + H)+, 407.2329, found 407.2351.
Ethyl 5-hydroxy-2-methyl-4-(piperidin-1-ylmethyl)-1-(o-tolyl)-1H-indole-3-carboxylate (33).
This compound was obtained from ethyl acetoacetate (26) and 2-methylaniline, employing Methods D and C. Overall yield 33%; yellow solid. 1H NMR (400 MHz, CDCl3) δ 7.48–7.39 (m, 2H), 7.36 (t, J = 7.4 Hz, 1H), 7.16–7.09 (m, 2H), 6.66 (d, J = 8.8 Hz, 1H), 4.96–4.92 (m, 2H), 4.39 (q, J = 7.0 Hz, 2H), 3.53–3.26 (m, 2H), 3.11–2.81 (m, 2H), 2.36 (s, 3H), 2.07–1.65 (m, 9H), and 1.42 (t, J = 7.0 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 167.6, 154.0, 145.9, 137.0, 135.1, 132.9, 131.6, 129.9, 129.0, 127.6, 126.3, 116.5, 113.4, 109.4, 105.1, 60.7, 54.4, 52.8, 29.8, 23.8, 22.6, 17.3, 14.7, and 14.2. HRMS (ESI) m/z: Calcd for C25H31N2O3 (M + H)+, 407.2329, found 407.2355.
Ethyl 5-hydroxy-1-(4-methoxyphenyl)-2-methyl-4-(piperidin-1-ylmethyl)-1H-indole-3-carboxylate (34).
This compound was obtained from ethyl acetoacetate (26) and 4-methoxyaniline, employing Methods D and C. Overall yield 41%; yellow solid. 1H NMR (400 MHz, CD3OD:CDCl3 = 1:2) δ 7.17 (d, J = 8.5 Hz, 2H), 7.08 (d, J = 8.5 Hz, 2H), 6.88 (d, J = 8.8 Hz, 1H), 6.82 (d, J = 8.8 Hz, 1H), 4.67 (s, 2H), 4.41 (q, J = 7.1 Hz, 2H), 3.89 (s, 3H), 3.63–3.45 (m, 2H), 3.19–3.00 (m, 2H), 2.46 (s, 3H), 2.10–1.53 (m, 6H), and 1.42 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, CD3OD:CDCl3 = 1:2) δ 169.2, 160.8, 154.3, 147.5, 134.0, 129.8, 128.8, 126.8, 115.7, 114.9, 112.5, 105.5, 105.3, 61.6, 55.9, 53.6, 53.0, 24.2, 22.4, 14.8, and 14.6. HRMS (ESI) m/z: Calcd for C25H31N2O4 (M + H)+, 423.2278, found 423.2300.
Ethyl 5-hydroxy-1-(3-methoxyphenyl)-2-methyl-4-(piperidin-1-ylmethyl)-1H-indole-3-carboxylate (35).
This compound was obtained from ethyl acetoacetate (26) and 3-methoxyaniline, employing Methods D and C. Overall yield 47%; yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.46 (t, J = 7.8 Hz, 1H), 7.13 (d, J = 8.8 Hz, 1H), 7.06 (d, J = 8.6 Hz, 1H), 6.90 (d, J = 8.8 Hz, 1H), 6.84 (d, J = 7.7 Hz, 1H), 6.77 (s, 1H), 4.92 (s, 2H), 4.39 (q, J = 7.0 Hz, 2H), 3.83 (s, 3H), 3.56–3.22 (m, 2H), 3.13–2.76 (m, 2H), 2.46 (s, 3H), 2.06–1.53 (m, 6H), and 1.42 (t, J = 7.0 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 167.6, 160.8, 154.0, 145.7, 137.3, 133.6, 130.7, 126.3, 120.6, 116.5, 115.1, 114.1, 113.7, 109.3, 105.5, 60.7, 55.7, 54.3, 52.8, 23.8, 22.6, 14.7, and 14.6. HRMS (ESI) m/z: Calcd for C25H31N2O4 (M + H)+, 423.2278, found 423.2308.
Ethyl 5-hydroxy-1-(2-methoxyphenyl)-2-methyl-4-(piperidin-1-ylmethyl)-1H-indole-3-carboxylate (36).
This compound was obtained from ethyl acetoacetate (26) and 2-methoxyaniline, employing Methods D and C. Overall yield 43%; yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.47 (t, J = 7.8 Hz, 1H), 7.22 (d, J = 7.7 Hz, 1H), 7.13–7.05 (m, 2H), 6.67 (q, J = 8.7 Hz, 2H), 4.39 (q, J = 7.0 Hz, 2H), 4.32–4.17 (m, 2H), 3.72 (s, 3H), 3.10–1.82 (m, 7H), 1.65 (br, 6H), and 1.42 (t, J = 7.0 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 167.0, 156.0, 154.8, 144.7, 132.5, 130.5, 130.3, 125.3, 125.0, 121.1, 113.2, 112.4, 111.4, 110.1, 105.6, 60.1, 59.4, 55.8, 53.9, 26.1, 24.2, 14.8, and 13.2. HRMS (ESI) m/z: Calcd for C25H31N2O4 (M + H)+, 423.2278, found 423.2305.
Ethyl 1-(4-fluorophenyl)-5-hydroxy-2-methyl-4-(piperidin-1-ylmethyl)-1H-indole-3-carboxylate (37).
This compound was obtained from ethyl acetoacetate (26) and 4-fluoroaniline, employing Methods D and C. Overall yield 47%; pale brown solid. 1H NMR (400 MHz, CDCl3) δ 7.28–7.25 (m, 4H), 7.08 (d, J = 8.6 Hz, 1H), 6.83 (d, J = 8.8 Hz, 1H), 4.84 (s, 2H), 4.40 (q, J = 7.0 Hz, 2H), 3.49–3.16 (m, 2H), 3.02–2.59 (m, 2H), 2.44 (s, 3H), 1.96–1.57 (m, 6H), and 1.43 (t, J = 7.0 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 167.4, 162.8 (d, JC-F = 251.0 Hz), 154.3, 145.3, 133.6, 132.4 (d, JC-F = 3.0 Hz), 130.3 (d, JC-F = 8.0 Hz), 126.1, 117.1 (d, JC-F = 23.0 Hz), 116.4, 112.9, 110.0, 105.8, 60.7, 55.1, 53.0, 24.1, 22.9, 14.7, and 14.4. HRMS (ESI) m/z: Calcd for C24H28FN2O3 (M + H)+, 411.2078, found 411.2102.
Ethyl 1-(3-fluorophenyl)-5-hydroxy-2-methyl-4-(piperidin-1-ylmethyl)-1H-indole-3-carboxylate (38).
This compound was obtained from ethyl acetoacetate (26) and 3-fluoroaniline, employing Methods D and C. Overall yield 36%; brown oil. 1H NMR (400 MHz, CDCl3) δ 7.52 (q, J = 7.3 Hz, 1H), 7.21 (t, J = 8.3 Hz, 1H), 7.10 (d, J = 7.8 Hz, 1H), 7.03 (d, J = 9.0 Hz, 1H), 6.79 (d, J = 8.7 Hz, 1H), 6.72 (d, J = 8.7 Hz, 1H), 4.40 (q, J = 6.9 Hz, 2H), 4.19 (s, 2H), 3.17–1.89 (m, 7H), 1.66 (br, 6H), and 1.43 (t, J = 7.0 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 166.8, 163.0 (d, JC-F = 250.6 Hz), 155.0, 142.7, 138.3 (d, JC-F = 9.6 Hz), 132.3, 130.9 (d, JC-F = 9.2 Hz), 124.8, 124.3 (d, JC-F = 2.6 Hz), 116.0 (d, JC-F = 2.6 Hz), 115.8, 113.7, 111.3, 110.0, 106.5, 60.2, 59.2, 53.8, 25.9, 24.0, 14.6, and 13.3. HRMS (ESI) m/z: Calcd for C24H28FN2O3 (M + H)+, 411.2078, found 411.2091.
Ethyl 1-(2-fluorophenyl)-5-hydroxy-2-methyl-4-(piperidin-1-ylmethyl)-1H-indole-3-carboxylate (39).
This compound was obtained from ethyl acetoacetate (26) and 2-fluoroaniline, employing Methods D and C. Overall yield 39%; brown oil. 1H NMR (400 MHz, CDCl3) δ 7.50 (q, J = 6.1 Hz, 1H), 7.38–7.27 (m, 3H), 6.74–6.68 (m, 2H), 4.40 (q, J = 7.0 Hz, 2H), 4.28–4.14 (m, 2H), 3.18–1.82 (m, 7H), 1.65 (br, 6H), and 1.43 (t, J = 7.0 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 166.9, 158.6 (d, JC-F = 253.6 Hz), 155.1, 143.8, 132.2, 130.9 (d, JC-F = 7.8 Hz), 130.7, 125.1 (d, JC-F = 4.2 Hz), 125.1, 124.5 (d, JC-F = 13.2 Hz), 117.3 (d, JC-F = 19.2 Hz), 113.7, 111.6, 109.9, 106.7, 60.3, 59.3, 53.9, 26.1, 24.2, 14.7, and 13.1. HRMS (ESI) m/z: Calcd for C24H28FN2O3 (M + H)+, 411.2078, found 411.2108.
(Z)-4-((4-bromophenyl)amino)pent-3-en-2-one (42).
This compound was obtained from pentane-2,4-dione (40) and 4-bromoaniline (41), employing Method A. Overall yield 72%; yellow oil. 1H NMR (400 MHz, CDCl3) δ 12.43 (s, 1H), 7.45 (d, J = 8.6 Hz, 2H), 6.98 (d, J = 8.6Hz, 2H), 5.21 (s, 1H), 2.10 (s, 3H), and 1.99 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 191.3, 156.1, 137.5, 132.2, 126.2, 118.8, 98.3, 29.3, and 19.7.
1-(1-(4-bromophenyl)-5-hydroxy-2-methyl-1H-indol-3-yl)ethan-1-one(43).
This compound was obtained from Compound 42, employing Method B. Overall yield 67%; brown solid.1H NMR (400 MHz, DMSO-d6) δ 9.12 (s, 1H), 7.83 (d, J = 8.2 Hz, 2H), 7.50 (s, 1H), 7.42 (d, J = 8.2 Hz, 2H), 6.80 (d, J = 8.8 Hz, 1H), 6.65 (d, J = 8.8 Hz, 1H), 2.55 (s, 3H), and 2.49 (s, 4H). 13C NMR (101 MHz, DMSO-d6) δ 193.4, 153.5, 144.2, 135.3, 133.0, 131.3, 130.4, 127.1, 122.0, 114.4, 112.1, 110.9, 105.7, 39.9, 39.7, 39.5, 39.3, 39.1, 31.3, and 13.8.
1-(1-([1,1′-biphenyl]-4-yl)-5-hydroxy-2-methyl-1H-indol-3-yl)ethan-1-one (44).
To a solution of Compound 43 (1mmol) in toluene: EtOH: H2O (3:2:1) (12 mL) under N2, were added 3 M aqueous solution of K2CO3 (18.88 mmol), phenylboronic acid (11.62 mmol), and Pd(PPh3)4 (0.29 mmol). The mixture was stirred at 100 °C for 4 h. After cooling to r.t. The resulting suspension was diluted with EtOAc (20 mL), the aqueous layer was extracted with EtOAc (30mL), and the combined organic layers washed with NaCl aqueous solution. The residue was purified by flash chromatography (dichloromethane: methanol = 40:1) to afford 44. Yield 42%; pale yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 9.11 (s, 1H), 7.92 (d, J = 7.6 Hz, 2H), 7.79 (d, J = 7.6 Hz, 2H), 7.67–7.59 (m, 3H), 7.57–7.49 (m, 6H), 7.43 (t, J = 7.2 Hz, 1H), 6.85 (d, J = 8.8 Hz, 1H), 6.66 (d, J = 8.8 Hz, 1H), 2.56 (s, 3H), and 2.54 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 193.0, 153.0, 144.0, 140.1, 138.6, 134.7, 131.7, 131.1, 128.7, 128.3, 128.2, 127.7, 126.5, 115.2, 113.8, 111.6, 105.2, 30.8, and 13.5.
1-(1-([1,1′-biphenyl]-4-yl)-5-hydroxy-2-methyl-4-(piperidin-1-ylmethyl)-1H-indol-3-yl)ethan-1-one (45).
This compound was obtained from Compound 44, employing Method C. Overall yield 83%; pale yellow solid. 1H NMR (400 MHz, CD3OD:CDCl3 = 1:2) δ 7.82 (d, J = 7.8 Hz, 2H), 7.65 (d, J = 7.6 Hz, 2H), 7.50–7.47 (m, 3H), 7.35 (d, J = 7.8 Hz, 2H), 6.95 (d, J = 8.9 Hz, 1H), 6.84 (d, J = 8.7 Hz, 1H), 4.48 (s, 2H), 3.68–3.50 (m, 2H), 3.18–3.03 (m, 2H), 2.68 (s, 3H), 2.58 (s, 3H), 2.14–2.01 (m, 2H), 1.87–1.73 (m, 2H), and 1.68–1.51 (m, 2H). 13C NMR (101 MHz, CD3OD:CDCl3 = 1:2) δ 199.8, 154.8, 146.7, 143.5, 139.9, 135.0, 134.0, 129.5, 129.3, 129.1, 128.7, 127.6, 126.2, 117.7, 115.1, 113.3, 106.4, 53.5, 53.1, 31.9, 24.4, 22.4, and 15.6. HRMS (ESI) m/z: Calcd for C29H31N2O2 (M + H)+, 439.2380, found 439.2386.
4-(piperidin-1-yl) aniline (47).
To a solution of indole P-nitroaniline (1 mol) in DMSO (30 mL) was added K2CO3 and piperidine (4 mol) at room temperature under N2. The reaction mixture was allowed to 90 °C for 8–12 h and then cooled to rt. The reaction mixture was extracted with ethyl acetate (3 × 20 mL), washed with NaCl aqueous solution, and the combined organic layer was dried with anhydrous Na2SO4. The residue was evaporated and purified by flash chromatography (petroleum ether: ethyl acetate = 20:1). Then, performed catalytic hydrogenation without further purification. Overall yield 90%; yellow solid. 1H NMR (400 MHz, CDCl3) δ 6.83 (d, J = 8.0 Hz, 2H), 6.64 (d, J = 8.0 Hz, 2H), 3.32 (s, 2H), 3.01–2.95 (m, 4H), 1.77–1.65 (m, 4H), and 1.58–1.47 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 146.0, 139.9, 119.3, 116.3, 52.7, 26.3, and 24.3.
1-(5-hydroxy-2-methyl-1-(4-(piperidin-1-yl) phenyl)-4-(piperidin-1-ylmethyl)-1H-indol-3-yl) ethan-1-one (48).
This compound was obtained from pentane-2,4-dione (40) and Compound 47, employing Methods A, B, and C. Overall yield 41%; brown solid. 1H NMR (400 MHz, CDCl3) δ 7.30 (d, J = 8.7 Hz, 1H), 7.13–6.95 (m, 4H), 6.79 (d, J = 8.7 Hz, 1H), 4.63 (s, 2H), 3.56–3.41 (m, 2H), 3.35–3.20 (m, 4H), 3.14–2.95 (m, 2H), 2.60 (s, 3H), 2.45 (s, 3H), and 2.00–1.55 (m, 12H). 13C NMR (101 MHz, DMSO-d6) δ 196.2, 152.8, 144.9, 132.2, 128.1, 125.3, 116.2, 115.3, 113.0, 111.7, 110.4, 110.1, 106.3, 51.7, 51.6, 48.2, 31.3, 24.5, 23.2, 21.9, 20.7, and 14.2. HRMS (ESI) m/z: Calcd for C28H36N3O2 (M + H)+, 446.2802, found 446.2835.
Ethyl 1-(4-(dimethylamino)phenyl)-5-hydroxy-2-methyl-4-(piperidin-1-ylmethyl)-1H-indole-3-carboxylate (49).
This compound was obtained from ethyl acetoacetate (26) and N,N-dimethyl-1,4-phenylenediamine, employing Methods A, B, and C. Overall yield 33%; brown solid. 1H NMR (400 MHz, CD3OD:CDCl3 = 1:3) δ 6.98 (d, J = 8.0 Hz, 2H), 6.85–6.80 (m, 2H), 6.73 (d, J = 8.1 Hz, 2H), 4.58 (s, 2H), 4.32 (q, J = 7.0 Hz, 2H), 3.40 (s, 2H), 3.11–2.87 (m, 8H), 2.38 (s, 3H), 2.04–1.56 (m, 6H), and 1.35 (t, J = 7.0 Hz, 3H). 13C NMR (101 MHz, CD3OD:CDCl3 = 1:3) δ 168.9, 153.8, 150.9, 147.4, 133.8, 128.8, 126.1, 124.0, 114.5, 112.7, 112.5, 105.4, 104.4, 61.1, 53.8, 52.6, 40.5, 24.0, 22.2, and 14.5. HRMS (ESI) m/z: Calcd for C26H34N3O3 (M + H)+, 436.2595, found 436.2625.
1-(1-(4-(dimethylamino)phenyl)-5-hydroxy-2-methyl-4-(piperidin-1-ylmethyl)-1H-indol-3-yl)ethan-1-one (50).
This compound was obtained from pentane-2,4-dione (40) and N,N-dimethyl-1,4-phenylenediamine, employing Methods A, B, and C. Overall yield 37%; pale brown solid. 1H NMR (400 MHz, CD3OD:CDCl3 = 1:1) δ 7.06 (d, J = 8.0 Hz, 2H), 6.80 (d, J = 8.0 Hz, 2H), 6.74 (d, J = 8.6 Hz, 1H), 6.64 (d, J = 8.6 Hz, 1H), 4.05 (s, 2H), 3.01 (s, 6H), 2.81–2.53 (m, 4H), 2.37 (s, 3H), and 1.72–1.45 (m, 6H). 13C NMR (101 MHz, CD3OD:CDCl3 = 1:1) δ 199.2, 155.0, 151.3, 143.9, 134.1, 129.3, 125.3, 125.1, 117.4, 113.8, 113.2, 111.7, 111.3, 59.4, 54.0, 40.7, 26.2, 24.2, and 14.3. HRMS (ESI) m/z: Calcd for C25H32N3O2 (M + H)+, 406.2489, found 406.2523.
1-(4-((dimethylamino)methyl)-1-(4-(dimethylamino)phenyl)-5-hydroxy-2-methyl-1H-indol-3-yl)ethan-1-one (51).
This compound was obtained from pentane-2,4-dione (40), N,N-dimethyl-1,4-phenylenediamine, and N, N-dimethylamine, employing Methods A, B, and C. Overall yield 34%; dark brown solid. 1H NMR (400 MHz, CD3OD:CDCl3 = 1:2) δ 7.05 (d, J = 8.3 Hz, 2H), 6.87 (d, J = 8.8 Hz, 1H), 6.83–6.77 (m, 3H), 4.62 (s, 2H), 3.03 (s, 6H), 2.92 (s, 6H), 2.61 (s, 3H), and 2.49 (s, 3H). 13C NMR (101 MHz, CD3OD:CDCl3 = 1:2) δ 198.0, 154.1, 151.2, 147.0, 134.3, 128.9, 125.9, 123.7, 116.8, 114.9, 112.9, 112.8, 107.1, 54.8, 42.71, 40.5, 31.8, and 15.4. HRMS (ESI) m/z: Calcd for C22H28N3O2 (M + H)+, 366.2176, found 366.2199.
1-(1-(4-(dimethylamino)phenyl)-5-hydroxy-2-methyl-4-(pyrrolidin-1-ylmethyl)-1H-indol-3-yl)ethan-1-one (52).
This compound was obtained from pentane-2,4-dione (40), N,N-dimethyl-1,4-phenylenediamine, and pyrrolidine, employing Methods A, B, and C. Overall yield 37%; brown solid. 1H NMR (400 MHz, CDCl3) δ 7.08 (d, J = 8.5 Hz, 2H), 7.00 (d, J = 8.7 Hz, 1H), 6.85–6.77 (m, 3H), 4.66 (s, 2H), 3.23–3.08 (m, 4H), 3.05 (s, 6H), 2.60 (s, 3H), 2.45 (s, 3H), and 2.06–1.97 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 197.5, 154.2, 150.7, 144.3, 134.0, 129.0, 125.0, 124.3, 117.0, 115.2, 112.8, 112.7, 111.1, 53.4, 53.3, 40.6, 32.3, 23.7, and 14.8. HRMS (ESI) m/z: Calcd for C24H30N3O2 (M + H)+, 392.2333, found 392.2361.
1-(1-(4-(dimethylamino)phenyl)-5-hydroxy-2-methyl-4-((4-methylpiperidin-1-yl)methyl)-1H-indol-3-yl)ethan-1-one (53).
This compound was obtained from pentane-2,4-dione (40), N,N-dimethyl-1,4-phenylenediamine, and 4-methylpiperidine, employing Methods A, B, and C. Overall yield 31%; brown solid. 1H NMR (400 MHz, CD3OD:CDCl3 = 1:2) δ 7.08 (d, J = 8.0 Hz, 2H), 6.90–6.82 (m, 3H), 6.77 (d, J = 8.7 Hz, 1H), 4.45 (s, 2H), 3.54–3.42 (m, 2H), 3.14–2.88 (m, 9H), 2.68–2.60 (m, 2H), 2.50 (s, 3H), 2.01–1.90 (m, 2H), 1.81–1.67 (m, 1H), 1.57–1.39 (m, 2H), and 1.02 (d, J = 6.0 Hz, 3H). 13C NMR (101 MHz, CD3OD:CDCl3 = 1:2) δ 199.3, 155.1, 151.4, 143.9, 134.2, 129.4, 125.3, 125.2, 117.6, 113.9, 113.3, 111.7, 111.6, 59.3, 53.6, 40.8, 34.7, 31.1, 31.0, 22.0, and 14.4. HRMS (ESI) m/z: Calcd for C26H34N3O2 (M + H)+, 420.2669, found 420.2646.
1-(5-hydroxy-1-(4-isopropylphenyl)-2-methyl-4-(piperidin-1-ylmethyl)-1H-indol-3-yl)ethan-1-one (54).
This compound was obtained from pentane-2,4-dione (40) and 4-isopropylaniline, employing Methods A, B, and C. Overall yield 41%; brown solid. 1H NMR (400 MHz, CDCl3) δ 7.39 (d, J = 7.8 Hz, 2H), 7.17 (d, J = 7.8 Hz, 2H), 6.94 (d, J = 8.5 Hz, 1H), 6.80 (d, J = 8.7 Hz, 1H), 4.29 (s, 2H), 3.24–2.69 (m, 5H), 2.60 (s, 3H), 2.41 (s, 3H), 1.83–1.56 (m, 6H), and 1.32 (d, J = 6.8 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 198.3, 154.9, 150.1, 142.6, 133.9, 133.2, 128.2, 127.9, 125.1, 117.5, 114.7, 112.1, 109.8, 57.2, 53.3, 34.02, 32.27, 25.12, 24.02, 23.36, and 14.42. HRMS (ESI) m/z: Calcd for C26H33N2O2 (M + H)+, 405.2537, found 405.2564.
1-(1-(4-(tert-butyl)phenyl)-5-hydroxy-2-methyl-4-(piperidin-1-ylmethyl)-1H-indol-3-yl)ethan-1-one (55).
This compound was obtained from pentane-2,4-dione (40) and 4-(tert-butyl)aniline, employing Methods A, B, and C. Overall yield 45%; brown solid. 1H NMR (400 MHz, CDCl3) δ 7.54 (d, J = 8.0 Hz, 2H), 7.20 (d, J = 8.3 Hz, 2H), 6.79 (d, J = 8.8 Hz, 1H), 6.71 (d, J = 8.7 Hz, 1H), 4.00 (s, 2H), 2.83–2.20 (m, 10H), 1.64 (s, 6H), and 1.39 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 198.0, 154.8, 151.7, 140.6, 133.8, 132.6, 127.7, 126.5, 124.6, 117.5, 113.5, 111.1, 110.3, 59.4, 53.5, 34.7, 32.2, 31.3, 25.8, 23.9, and 13.5. HRMS (ESI) m/z: Calcd for C27H35N2O2 (M + H)+, 419.2693, found 419.2714.
1-(1-(4-(tert-butyl)phenyl)-5-hydroxy-2-methyl-4-(pyrrolidin-1-ylmethyl)-1H-indol-3-yl)ethan-1-one (56).
This compound was obtained from pentane-2,4-dione (40), 4-(tert-butyl)aniline, and pyrrolidine, employing Methods A, B and C. Overall yield 42%; brown solid. 1H NMR (400 MHz, CD3OD:CDCl3 = 1:3) δ 7.53 (d, J = 7.6 Hz, 2H), 7.12 (d, J = 7.6 Hz, 2H), 6.84–6.79 (m, 2H), 4.66 (s, 2H), 3.58–3.31 (m, 4H), 2.59 (s, 3H), 2.46 (s, 3H), 2.25–2.02 (m, 4H), and 1.33 (s, 9H). 13C NMR (101 MHz, CD3OD:CDCl3 = 1:3) δ 198.4, 153.8, 153.3, 146.2, 133.8, 133.1, 128.0, 127.3, 125.8, 117.2, 114.6, 113.1, 107.8, 53.6, 51.5, 35.2, 31.9, 31.4, 23.5, and 15.5. HRMS (ESI) m/z: Calcd for C26H33N2O2 (M + H)+, 405.2537, found 405.2563.
Ethyl 1-(4-(tert-butyl)phenyl)-5-hydroxy-2-methyl-4-(piperidin-1-ylmethyl)-1H-indole-3-carboxylate (57).
This compound was obtained from ethyl acetoacetate (26) and 4-(tert-butyl)aniline, employing Methods A, B, and C. Overall yield 33%; brown solid. 1H NMR (400 MHz, CDCl3) δ 7.53 (d, J = 7.8 Hz, 2H), 7.19 (d, J = 7.8 Hz, 2H), 6.79 (d, J = 8.7 Hz, 1H), 6.70 (d, J = 8.7 Hz, 1H), 4.40 (q, J = 7.0 Hz, 2H), 4.22 (s, 2H), 3.01–1.99 (m, 7H), 1.60 (br, 6H), and 1.39 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 167.0, 154.8, 151.8, 143.5, 134.1, 132.7, 127.8, 126.6, 124.8, 113.3, 111.2, 110.4, 105.8, 60.1, 59.3, 53.8, 34.9, 31.4, 26.0, 24.1, 14.7, and 13.5. HRMS (ESI) m/z: Calcd for C28H37N2O3 (M + H)+, 449.2799, found 449.2814.
1-(1-(4-bromophenyl)-5-hydroxy-2-methyl-4-(piperidin-1-ylmethyl)-1H-indol-3-yl)ethan-1-one (58).
This compound was obtained from Compound 43, employing Method C. Overall yield 84%; pale yellow solid. 1H NMR (400 MHz, CD3OD:CDCl3 = 1:1) δ 7.73 (d, J = 7.7 Hz, 2H), 7.17 (d, J = 7.7 Hz, 2H), 6.86–6.81 (m, 2H), 4.41 (s, 2H), 3.68–3.34 (m, 2H), 3.26–2.83 (m, 2H), 2.64 (s, 3H), 2.49 (s, 3H), and 2.08–1.57 (m, 6H). 13C NMR (101 MHz, CD3OD:CDCl3 = 1:1) δ 199.7, 154.8, 145.3, 135.1, 133.8, 133.5, 130.3, 125.9, 124.2, 117.9, 114.1, 113.5, 107.0, 54.2, 53.1, 32.0, 24.5, 22.5, and 15.1. HRMS (ESI) m/z: Calcd for C23H26BrN2O2 (M + H)+, 441.1172, found 441.1194.


 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

