
Direct Biocatalytic Processes for CO2 Capture as a Green Tool to Produce Value-Added Chemicals





1
Departamento de Bioquímica y Biología Molecular B e Inmunología, Facultad de Química, Universidad de Murcia, 30100 Murcia, Spain
2
Department of Biotechnology, Delft University of Technology, 2629 HZ Delft, The Netherlands
3
Departamento de Química Inorgánica, Facultad de Química, Universidad de Murcia, 30100 Murcia, Spain
*
Authors to whom correspondence should be addressed.
Molecules 2023, 28(14), 5520; https://0-doi-org.brum.beds.ac.uk/10.3390/molecules28145520
Submission received: 31 May 2023
/
Revised: 14 July 2023
/
Accepted: 16 July 2023
/
Published: 19 July 2023
(This article belongs to the Special Issue Biocatalysis and Enzymes in Pharmaceutical and Biotechnological Processes)
Abstract
:Direct biocatalytic processes for CO2 capture and transformation in value-added chemicals may be considered a useful tool for reducing the concentration of this greenhouse gas in the atmosphere. Among the other enzymes, carbonic anhydrase (CA) and formate dehydrogenase (FDH) are two key biocatalysts suitable for this challenge, facilitating the uptake of carbon dioxide from the atmosphere in complementary ways. Carbonic anhydrases accelerate CO2 uptake by promoting its solubility in water in the form of hydrogen carbonate as the first step in converting the gas into a species widely used in carbon capture storage and its utilization processes (CCSU), particularly in carbonation and mineralization methods. On the other hand, formate dehydrogenases represent the biocatalytic machinery evolved by certain organisms to convert CO2 into enriched, reduced, and easily transportable hydrogen species, such as formic acid, via enzymatic cascade systems that obtain energy from chemical species, electrochemical sources, or light. Formic acid is the basis for fixing C1-carbon species to other, more reduced molecules. In this review, the state-of-the-art of both methods of CO2 uptake is assessed, highlighting the biotechnological approaches that have been developed using both enzymes.
1. Reducing Carbon Dioxide from the Air: The Challenge
One of the main challenges faced by humanity in the 21st century is climate change. Over the last two centuries, the temperature of the Earth’s crust has risen progressively and, since 1980, alarmingly, at a rate of 0.18 °C per decade. Indeed, last year’s average temperature was 1.04 °C higher than the median temperature in the period prior to 1880 [1]. Temperature elevation drives an increase in extreme weather evidenced by a series of well-known events (draughts, floodings, torrential downpours, etc.), the melting of large extensions of frozen water, with the subsequent ascent of the sea, changes in ecosystems with undefined outcomes and, in this sense, uncertainty on how these changes will affect our way of living and welfare, with estimations that are clearly detrimental [2]. Moreover, the acidification of seas and oceans is also a problem, with coral reef weakening already having been detected, as well as the low level of oxygen present in marine life [3,4].
At the end of the nineteenth century, S.A. Arrhenius quantified the contribution of “carbonic acid” (nowadays, carbon dioxide) to the greenhouse effect and was the first in indicating that “The production of carbonic acid by the combustion of coal would therefore suffice to cover the loss of carbonic acid by weathering and by peat formation seven times over. Those are the two chief factors deciding the consumption of carbonic acid, and we thus recognize that the percentage of carbonic acid in the air must be increasing at a constant rate as long as the consumption of coal, petroleum, etc., is maintained at its present figure, and at a still more rapid rate if this consumption should continue to increase as it does now”. He also concluded that this would lead to an increase in the temperature of Earth’s atmosphere [5,6]. Since then, a huge amount of evidence correlating both air CO2 concentration and global warming has accumulated [2,7]. Moreover, there is a direct relationship between human activity, carbon dioxide concentration, and climate change, that is, the anthropogenic origin of global warming is well established. Atmospheric CO2 concentration has increased from 280 ppm (year 1750) to 415 ppm (2021), this value being the highest concentration reached in the last three million years [8]. This carbon dioxide increase is essentially related to the emissions of this gas to the atmosphere as a consequence of the use of fossil fuels by humans [9]. Other gases, such as methane and nitrous oxide, also contribute to global warming, albeit to a minor extent (11 and 7%, respectively) [10]. The objective reached at the Paris Climate Agreement in 2015 to maintain an increase in overall temperature below 2.0 °C with respect to preindustrial levels has recently been revised in the sense that such increments should not exceed 1.5 °C [11].
In this scenario, any scientific strategy that allows for reducing the concentration of CO2 in the atmosphere is an object of interest, although the most relevant solution is to avoid burning fossil energy sources that release CO2, and to substitute them with other sustainable ones. Although more efficient and responsible use of fossil energy sources by society is also essential to contributing to decreasing CO2 emissions, the responsibility of capturing the excess CO2 already emitted is also inescapable.
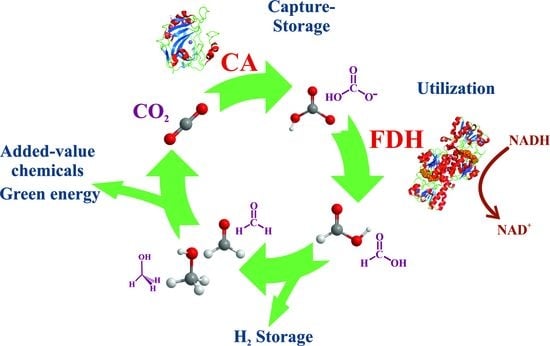
Among other approaches, biocatalysts are useful tools for reducing the accumulation of atmospheric CO2 through either carbon capture and storage (CCS) or carbon capture and its utilization (CCU, Figure 1A). Both approaches are often applied together, known as carbon capture storage and utilization (CCSU) [12]. These methodologies require the passing of CO2 from gas to carbon solid and/or chemically reduced forms, a task that directly implies chemistry in all its fields.
Research in this field has exponentially increased in the last decade. Indeed, Figure 2A shows the number of articles published, directly or indirectly, that relate either to carbon storage or carbon utilization per year, while Figure 2B,C display the percentages of these articles classified by research area. As observed, research on carbon capture is especially intense in areas such as catalysts, synthesis, electrochemistry, and energy and fuels. Nowadays, there are two main approaches for CO2 transformation, namely biological and chemical transformations [13]. In turn, biological CO2 fixation can be photosynthetic or not photosynthetic, while the chemical uptake can be divided into hydrogenation, carboxylation, mineralization, chemical reduction, and photochemical reduction. Although hydrogenation is a well-established technology, nowadays, it is still mostly not green, as it is obtained from the cracking of fossil fuels. The sustainable synthesis and transport of hydrogen is indeed one of the main challenges related to energy sources. The main target of hydrogenation is the production of methanol (see Section 7.2), although other reduced molecules can also be obtained.
Carboxylation is the process of directly converting CO2 into organic value-added compounds. Organic carbonates and polymers are obtained through this process. An example is the production of non-isocyanate polyurethanes (NIPUs) from glycerol carbonates derivatives obtained through CO2 cycloaddition to glycidol moieties [14]. In this case, the authors designed a sustainable chemoenzymatic protocol for the synthesis of glycerol carbonate acrylate (GCA) and glycerol carbonate methacrylate (GCMA) from glycidol and CO2 by using ionic liquid (IL) technologies and enzymes, providing conversions of up to 100% under low-pressure values (1–10 bar). These methods are still in their first stages of development and hence a relatively novel field to work in.
Mineralization is described in Section 6.3 and is used in construction, for instance, for generating cement. This technique allows for the uptake of large quantities of CO2 for obtaining sustainable materials, although it is a highly consuming energy at a high scale. Chemical, electrochemical and photochemical reduction combined with enzymes is described below (see Section 7.2, Section 7.3 and Section 7.4).
The genuine physical and chemical properties of CO2 make it a molecule that cannot be easily captured or retained. Thus, CO2 is a nonpolar molecule that resides in a gaseous state under P and T standard conditions because of the weak van der Waals interactions established between the molecules themselves. In addition, due to its null polarity, CO2 has a low diffusion coefficient in polar solvents (1.26 × 10−5 cm2/s in water under standard conditions), while its solubility follows Henry’s law (76.5 mM in water at 0 °C and partial pressure of 1 atm) [15]. More importantly, its kinetics of capture by water, while increasing with pH, is extremely low at neutral or acidic pH values, behaving as a Brönsted acid according to Equations (1) and (2) (Scheme 1). The Kh value (Equation (2)) indicates that only a minimal fraction of CO2(aq) is present in an aqueous solution of carbonic acid (ca. 1/600 of the molecules at neutral pH values). Both hydrated carbon dioxide and carbonic acid behave as weak Brönsted acids because of the low deprotonation constants (see Equations (3) and (4)) [16].
The equilibrium shown in Equation (3) is essential for the role of CO2 as a buffer between blood and cells, where the interconversion between CO2 and HCO3− (hydro carbonate or bicarbonate) forms should be fast in living beings. Under neutral or weak basic conditions, the formation of bicarbonate anions is slow (the first order kinetic constant, k1, is of the order 10−2 s−1), while it increases in basic media [17]. Consequently, living organisms must have a biocatalyst that allows for fast exchange between acid and basic species. Carbon dioxide gas can be fixed either in aqueous soluble (hydrogen carbonate) or much more insoluble (carbonate) forms using only alkalization or using other chemical processes that incorporate carbon into value-added compounds. In nature, carbonic anhydrase (CA) catalyzes the CO2/hydrogen carbonate reaction (Equation (3)), enabling CO2 capture within reasonable times [17,18]. Thus, CA is used as a tool for incorporating CO2 as a hydrogen carbonate anion in soluble species in a multitude of chemical approaches [19,20]. In contrast, carbonates are usually highly insoluble. It follows that the use of alkaline reactants (Equation (4)) is the easiest and, consequently, main method for sequestering CO2 by forming the corresponding salts or their derivatives.
Alternatively, CO2 can be fixed to hydrogen-enriched energy forms via several natural mechanisms [21,22]. Although the most extended and productive mechanism in nature (green plants and algae) is photosynthesis, where ribulose-1,5-bisphosphate carboxylase-oxygenase (RuBisCo) converts CO2 and water into C3-carbohydrates by taking energy from light (Figure 1B) [22,23,24], here, the focus is on nonphotosynthetic enzymes. The conversion of CO2 into formate anion (C1 species, Equation (5), Scheme 1) is the simplest way in which nature captures CO2 into reduced, highly energetic molecules, and formate dehydrogenases (FDH) are the main actors in this performance (Figure 1C) [25,26,27,28,29].
Formic acid, or its basic form formate, is a simple molecule for efficient hydrogen transport. Moreover, formate synthesis is the first step for obtaining other more complex and energetically enriched molecules [22,30]. Carbon dioxide and formic acid have similar formation energies (Figure 3A), with the most similar energetic states for C1-carbon forms. This converts formic acid as the easiest starting point for obtaining other C1 more reduced carbon species. Nevertheless, reaction 5, in the forward sense, is highly endergonic under physiological conditions. Indeed, the redox potential at pH 7 for CO2 reduction to formate is −430 mV [31], while that of NADH is only −320 mV (Figure 3B). More importantly, for the reasons discussed below, this first step is kinetically, energetically, and economically expensive, and it becomes the bottleneck for obtaining value-added products in CO2 regeneration. Several approaches for circumventing this issue are discussed below. However, other no less relevant problems arise when this reaction is performed for biotechnological purposes. FDHs from different organisms can solve these problems and incorporate formate anions into their biosynthetic routes. Thus, nature provides solutions to the previous difficulties. It is a researcher’s task to adjust these solutions at laboratory and industrial levels for the benefit of society.
In the following pages, the enzymes that participate in both processes of CO2 capture using CA (CCUS processes) and FDH (formate synthesis) are described. How these natural systems circumvent the thermodynamics and kinetics problems derived from reactions 3 and 5 are also discussed, paying special attention to the state-of-the-art concerning the biotechnological applications of these enzymes for CCS and CCU technologies, as well as for reducing CO2 to formic acid.
2. Carbonic Anhydrases: Efficient Devices for CO2 Uptake
2.1. Classification and Structure of Carbonic Anhydrases
Carbonic anhydrases (EC 4.2.1.1) are found in all living kingdoms [18,34,35,36]. They catalyze the reaction of interconversion between CO2 and HCO3− (Equation (3)) in nature and are generally monomeric proteins with molecular weights roughly comprised between 30–50 kDa, depending on their class. CAs have been classified into: α-CAs (found in animal cells, algae, and eubacteria), β-CAs [37] (found in higher plants, microalgae, eubacteria, archaebacteria, and fungi), γ-CAs [37] (algae), δ-CA [38,39,40] (found in the marine diatom Thalassiosira weissflogii), ε-CAs [41] (found, for example, in the carboxysomal shell of Halothiobacillus neapolitanus), ζ-CA [42] (found in Thalassiosira weissflogii), η-CA [43] (found in Plasmodium falciparum), θ-CA [44], and ι-CA [45,46,47] (found in Burkholderia territorii and Phaeodactylum tricornutum, among other bacteria). CAs are zinc(II) enzymes, although some of them can contain other metal ions active in physiological roles: Cd(II) has been found in δ-CA, although its role is disputed [48,49]; Fe(II) is present in some γ-CAs growing under anaerobic conditions [50]; Co(II) has also been replaced in many α-CAs with excellent activities [51], although its original role has not been proven. Remarkably, a group of proteins called “COG4337” has been described as ι-CAs, and strikingly, those from the cyanobacterium Anabaena sp. PCC7120 and the chlorarachniophyte alga Bigelowiella natans display activity without any metal involved [52]. Table 1 lists the main features of some representative CAs.
Mammalian CAs belong to the α-CA class. Depending on their location and primary sequences, several isoforms of human carbonic anhydrases (HCA) have been described, with HCAII being the most studied and best-characterized CA. The HCAII tertiary structure (Figure 4A) consists of a unique domain containing ten β-strands that twist to form a β-sheet (eight of them organized in an antiparallel arrangement and the other two in parallel) [68,69]. Surrounding these β-sheets, up to eight other α-helixes are located on the surface of the protein.
The structure of HCA is the most studied of all HCA isoforms, and its active site can be defined as a cone-shaped cleft, 15 Å deep, formed by a hydrophilic region (Tyr7, Asn62, His64, Asn67, Thr199, and Thr200) and a hydrophobic region (Val121, Val143, Leu198, Val207, and Trp209). Although the core of the active site in α-CAs is highly conserved, there is variability in the polarity and hydropathicity of its periphery [70]. The catalytic Zn(II) ion is located in a deep-centered slot that is accessible to the solvent. The Zn(II) ion is coordinated to three histidine residues (Figure 4B). While His94 and 96 are coordinated through their Nε2 imidazole nitrogen to the metal ion, His119 is bound through its Nδ1 imidazole nitrogen (HCAII numeration) [48]. A water molecule completes the tetrahedral coordination of Zn(II). A second coordination sphere is formed by amino acids that are not directly coordinated with the metal ion but are essential in the catalytic process. Through the formation of a network of hydrogen bonds, residues Tyr7, Asn62, His64, Asn67, Glu106, Thr199, and Thr200 stabilize the mediator species in such a way that the reaction can occur.
The HCA family includes a subclass of three noncatalytic isoforms (HCAs VIII, X, and XI) called CA-related proteins (CA-RPs), whose classification is based on their sequence. The noncatalytic behavior is due to the absence of one or more histidines that coordinate the Zn(II) ion of a catalytic HCA isoform. For instance, in HCA-RP VIII, the Zn-coordinating His94 (HCA II numbering) is replaced by an arginine (Arg116, according to HCA-RP VIII numbering). This residue avoids CO2 hydration in the first step of CA catalysis. [71]. Although the biological functions of CA-RPs have not been defined, these isoforms are of high interest in different scientific research fields. Recently, the X-ray crystal structure of only one HCA-RP (HCA-RP VIII) was determined [72]. HCA-RP VIII is expressed in the cerebellum [73] and has been identified as a binding partner for the inositol 1,4,5 triphosphate (IP3) receptor type [74]. It should be mentioned that the stability and structures of other HCAs such as HCA III or extracellular HCAs (i.e., IV, VI, IX and XIV) have not been as extensively studied as HCAs I and II. For instance, the bovine CA III showed a similar unfolding profile to that of HCA II, providing a molten globule intermediate and an unfolded state at a Cm of 2.6 M guanidinium chloride (GuHCl) concentration [75]. On the contrary, the isoforms VIII, X, and XI showed two distinct transitions, and their sensitivity to guanidinium chloride chemical denaturalization was higher than that of HCA II (Cm 0.4 M for HCA-RP VIII and 0.9 M for HCA II) [71].
2.2. CA Mechanism of Action
Carbonic anhydrase accelerates reaction 3 by more than six orders of magnitude (Table 1) with respect to its rate without the biocatalyst [18,76] (from ca. 3.6 × 10−2 M−1 s−1 to 1.0 × 106 M−1 s−1 in the absence and presence of CA) [17], allowing for the reaction to take place under physiological conditions. Otherwise, CO2 cannot be assimilated as bicarbonate by living organisms.
The kinetic parameters of CA can be determined by measuring hydratase activity (Equation (3), Scheme 1). The method basically consists of saturating a buffered water solution (typically Tris.HCl 0.020 M, pH 8.3) kept in an ice bath with CO2 at a fixed high pH value, then adding CA, and measuring the time that the solution takes to reach a low pH value (generally ca. 6.3) because of the conversion of carbon dioxide to hydrogen carbonate anion that takes the capture of protons (Equation (3)) and hence lowers pH. The Wilbur–Anderson hydratase activity unit [77] is calculated as the ratio ((t0 − t)/t0)/(mg enzyme), where t0 and t are the measured times that an indicator present in the solution shifts its color for the control (without CA) and the sample (in the presence of CA), respectively. This method provides a qualitative measurement of CA activity, but it is not strictly transferable from one set of experiments to another [78]. This method depends on the degree of saturation of CO2 (which in turn changes with temperature and the time at which CO2 is bubbled), the nature of the buffer, its concentration, and its initial pH value. Together, these units are roughly—not strictly—comparable. Most classes of CAs also exhibit esterase activity (δ-CA lacks esterase activity), whose measurement is more direct and contrastable [79]. This is generally performed by the hydrolysis of p-nitrophenyl acetate, which releases free p-nitrophenol, with maximum absorption at 400 nm, which is easily measurable [80]. Accurate experimental requirements for the latter experiments (concentration of the reactants, enzyme, and pH) are easily reproducible and should depend only on contrastable and exchangeable conditions.
The mechanism of action of CA has been deciphered [81] and basically consists of two stages (Figure 4C). In the first phase, a CO2 molecule, partially stabilized by interactions with groups of the enzyme active site, reaches the active center, and subsequently, the hydroxide group bound to the Zn(II) ion attacks the CO2 carbon atom (nucleophilic attack). Then, hydrogen carbonate is formed, and a water molecule replaces it through Zn(II) coordination. In the second step, a proton is transferred from the Zn(II)-coordinated water molecule to the solvent. This is the rate-limiting step. His64 is the amino acid responsible for accepting this proton, which is finally transferred to the bulk solvent. Due to this mechanism, at neutral/soft acid pH levels, the rate of CO2/HCO3− conversion is enhanced by more than six orders of magnitude, making life possible. At pH values higher than 9.5, a direct hydroxide attack can form carbonate anions at rates comparable to those performed by CA at neutral pH values.
3. Formate Dehydrogenases: Natural Machines for Reducing CO2
The specific reduction in carbon dioxide converts it into formate/formic acid (Equation (5), Scheme 1). This C1 metabolism reaction occurs in hydrogenotrophic methanogens (Euryarcheota) and autotrophic acetogens (bacteria) and is carried out by the enzyme formate dehydrogenase (EC 1.17.1.9) [24,82,83,84,85]. Energy and high reduction power (i.e., a cofactor in the NAD(P)H form) are required to perform this process. In contrast, formate dehydrogenases also catalyze the backward reaction (5), oxidizing formate anions and obtaining energy from them. FDHs are divided into two main groups: nonmetal- or NAD-dependent FDHs, and metal-dependent FDHs. The structure, nature, and activity of these two FDH sets are completely different. More importantly, the ability of these two groups to reduce CO2 is manifestly diverse, as discussed below.
3.1. Metal-Independent/NAD-Dependent FDHs
Metal-independent FDHs belong to the family of D-specific 2-oxoacyd dehydrogenases. They are present in bacteria, yeast, plants, and mammals, are globular proteins with ca. 350–400 amino acids, depending on the species, and usually form homodimers [25,26,86]. Each FDH monomer contains two domains, one destined to allocate the substrate (the catalytic domain) and another pocket that allows for the binding of the NADH cofactor. The function of metal-independent FDHs is generally associated with mechanisms for obtaining energy from the oxidation of formate anions in methanogenic pathways (backward reaction 5), that is, these FDHs are machines efficient in catalyzing backward reaction, although much less effective in performing CO2 reduction [25,26,84,87]. The cofactor of all known metal-independent FDHs is NAD(P)+/NAD(P)H, and, consequently, they are also called NAD(P)-dependent FDHs. FDH from the methylotropic yeast Candida boidinii (Cb) is the most studied and best characterized nondependent FDH, since it was the first FDH expressed in Escherichia coli, it is commercially available and relatively inexpensive [88,89]. As shown in Figure 5A, the enzyme (364 amino acids) consists of 15 α-helices and 13 β-strands [90]. There is a deep groove between both domains that allows both the substrate and the cofactor to be bound in this cavity with short contacts between them. Figure 5B displays the active site of the enzyme, including both the NAD+ binding site and the most significant amino acids concerning catalytic properties. NAD+ cofactor strongly binds the protein through its adenine, ribose, and phosphate moieties in such a way that it can only be removed by extensive washing with, for instance, 0.2 M sodium chloride. Formate binds closely to the nicotinamide group through the positively charged residue Arg258. The mechanism of formate oxidation (Figure 5C) consists of hydride transfer to the nicotinamide oxidized group and the release of the CO2 formed. The intermediated anion is stabilized by an arginine (Arg258 CbFDH numbering). The groove where the nicotinamide group and the formate anion are located is hydrophobic; hence, the hydride anion cannot interact with the solvent, so the reaction can take place. Both high formate and NAD+ strong binding with positive residues of the enzyme (Gln287, His311) reduce the ability to exchange the products of the reaction. In turn, this is one of the main factors that decrease the efficient recycling of the cofactor, making these FDHs, in general, not excellent biocatalysts for CO2 reduction.
With wild-type CbFDH being not ideal for forward reaction 5 [94,95], specific mutants increase its capability to capture CO2. For instance, the double mutant V120S-N187D was shown to substantially increase CO2 reduction [96]. Other FDHs have kcat and kM values that are higher than wild-type CbFDHs and its mutants (Table 2). Choe et al. studied Thiobacillus sp. KNK65MA FDH (TsFDH) and concluded that it presented 84-fold higher catalytic efficiency for CO2 reduction than CbFDH [67]. NAD-FDH mutants with higher efficiency have been successfully designed [89,96]. Binay and coworkers obtained and purified mutants of Candida methylica (CmFDH) and another four mutants from Chaetamium thermophilum (CtFDH) in amino acid positions close to the cofactor binding [92,93]. The highest activity was found for the Asn120Cys mutant in CtFDH, for which the kcat value increased 6.5-fold, the same increment observed for the kM value, which indicated that the efficiency of CO2 reduction was due to the lower affinity for the substrate, that is, for the ability to release formate. As observed in Figure 5D, Asn120 is located close to the nicotinamide NAD group; specifically, the amide nitrogen of Asn120 is as close as 3.8 Å from the nicotinamide ring. Its mutation by a smaller cysteine residue introduces more space in the active site and, consequently, higher conformational flexibility, which facilitates the release of formate anion [92]. His96 interacts with the hydrogen carbonate anion stabilizing the hydride in the transition state, as confirmed by molecular dynamics performed on the double mutant. Based on kinetic and molecular dynamic studies, the authors concluded that subtle structural changes around the Asn120 position allowed for the location of two molecules of hydrogen carbonate instead of one of formate, favoring the CO2 forward reaction taking place. On the other hand, replacing key residues G93H/I94Y in CmFDH, located in the catalytic pocket of FDH, increased the catalytic efficiency (kcat/kM) of the wild-type protein 5.4-fold for the reduction of HCO3−. Here, KM values do not vary significantly, while kcat/kM does. The authors suggested that there is a reorganization in the active site that enlarges the space and allows for the reactant (carbonate anion) to adopt a better orientation for catalysis, and so it becomes easier for the HCO3− to reach the nicotinamide ring and the reaction is produced in a faster way [93].
3.2. Metal-Dependent FDHs
FDHs containing metals constitute the other large set of FDHs [28,84,116,117]. All metal-dependent FDHs catalyze the interconversion between formate and CO2. The sense of the reaction (Equation (5), Scheme 1) depends on the external conditions. In general, in biological conditions, formate oxidation (backward reaction 5) is favored, and thus, some organisms obtain their energy from this exergonic reaction. However, metal-dependent FDHs can also catalyze CO2 reduction, and most of them do so, although to a different extent. There are a small number of microorganisms (hydrogenotrophic methanogens, Euryarcheota, and autotrophic acetogens, bacteria) that use FDH not for generating energy (i.e., not for oxidizing formate), but for using C1 carbon species as a primary source of their carbon metabolism [116,117]. All these microorganisms have metal-dependent FDHs.
This type of FDH is much more complex than NAD-FDHs. Indeed, they have more than 700 amino acids arranged in different domains that, in turn, contain several cofactors and/or metal centers such as ferredoxins, heme groups, flavin mononucleotides, etc., depending on the species [118]. For instance, FDH N from E. coli comprises three domains (Figure 6A): the α-domain, which contains the Mo cofactor (see below) and one [4Fe-4S] cluster; the β-domain with four [4Fe-4S] centers; and the γ-domain, with two b-hemes (Figure 6B) [119]. These enzymes receive the electrons from these metal clusters and not necessarily from NADH (although, in some cases, NADH can also be the cofactor), and thus, these FDHs are called nondependent NADHs. Excellent reviews describing the three-dimensional structures of these FDHs, their metal centers, their functions, and the biotechnological achievements of these enzymes have been published [28,84,116,117].
Importantly, despite their heterogeneity, these FDHs share common features concerning the active center. They all contain a molybdenum or a tungsten metal ion bound to two dithiolene atoms provided by two pyranopterin guanidine dinucleotides, a sulfur or selenium donor atom, and a disulfide anion (Figure 6B) [104]. CysSe residue is present in both Mo- and W-FDHs; hence, it is not specific to a determined metal. On the other hand, according to kinetics parameters (kcat and kM) the presence of CysSe instead of the native amino acid cysteine, is not crucial for FDH activity.
Metal-dependent FDHs catalyze Equation (5) (Scheme 1) in both directions; however, unlike wild-type NAD-dependent FDHs, they are much more efficient in catalyzing forward Equation (5) than the latter, clearly evident in Table 2. This table presents the kinetic parameters, as well as the composition of the active site and cofactors, specifically for metal-dependent FDHs. Hence, for biotechnological applications, that is, for reducing CO2, metal-dependent FDHs are much more active and, hence, more attractive than non-metal FDHs.
What are the key factors that enable metal-dependent FDHs to reduce CO2 efficiently? These issues have been extensively studied in the literature [26,27,28,29,84]. Here, some notes on the crucial aspects are commented upon. First, the existence of different redox centers acts as a corridor for efficient electron transfer toward the CO2 molecule. Second, and importantly, there is presence of a sulfido group that accepts a hydride anion (see Figure 6C). It is well known that, in Mo/W enzymes, a sulfido group accepts a hydride. In FDHs, metal oxidation states change from IV to VI; when they are in a reduced state, ligands tend to be protonated, while tending to deprotonate in the Mo/W(VI) oxidation state. Thus, metal sulfido can act as a donor/acceptor hydride. Indeed, spectroscopic studies are consistent with the transfer of a hydride from a sulfur atom [27,84,116,118]. On the other hand, there is no evidence of the direct coordination either of CO2 or formate directly to the metal center. Altogether, this facilitates the acceptance/donation of a hydride directly towards the CO2 carbon, which has an electronic net deficiency, and so is prone to attack by anions. It is also remarkable that the tungsten or molybdenum metal ion are indistinguishable concerning the catalytic activities while the presence of selenium cysteine does not appear to be relevant in CO2 reduction.
Several schemes of the reaction have been proposed for the mechanism of action of these FDHs [120,121]. However, it is robustly supported that CO2, rather than HCO3−, is the substrate of FDHs for forward Equation (5) [104,122]. This reaction takes place by abstracting (or adding, reverse reaction) a hydride anion, without the intervention of any oxygen atom [123]. Concordantly, the mechanism of action should consider these two fundamental facts. Moura et al. proposed a mechanism for the forward reaction in which the reduced CysSH coordinated to the Mo ion attacks the carbonyl atom and hydride transfer takes place (Figure 6C) [27]. The formate anion is then stabilized by the positive charge of Arg446 (FDH N from E. coli numeration), and afterward, when the protein is again reduced by the other cofactors and by the addition of another hydride to the coordinated Cys, the formate is released. In contrast, the reverse reaction is also produced by the opposite hydride attack from the formate anion on the same Cys (in this case, oxidized).
4. Improving CA Performance: Enzyme Immobilization
For industrial and biotechnological applications to be profitable, enzymes must be as stable and reusable as possible. CA and FDH in solution, like all soluble proteins, behave as solutes with full mobility in the solvent. Although an aqueous medium is, in general, the most suitable for enzyme action, the stability and activity of enzymes in this medium usually decrease rapidly. In addition, the use of enzymes in solution is always constrained by strict pH and temperature conditions. More importantly, enzymes in aqueous solutions can only be applied in the cycle of a specific reaction; hence, their applicability at the industrial level is highly limited. In contrast, enzymes immobilized on solid or gel supports extraordinarily increase their stability, amplifying the range of action of the biocatalyst conditions and the possibility of using more drastic, usually more efficient, reaction conditions (for instance, increasing temperature) [124,125,126]. This immobilization allows for their easy separation from reactants and products, and, consequently, enzymes in this form can be reused for posterior cycles [127,128]. This drastically reduces the cost of the whole process, regardless of the biotechnological industrial reaction. Both CA and FDH have been immobilized on different supports. Based on the immobilization method, the following approaches can be considered: physical adsorption, covalent binding, entrapment, encapsulation, and crosslinking.
Excellent reviews on CA and FDH immobilization have recently been published [129,130,131,132,133,134,135,136,137,138,139]. Here, some illustrative cases regarding the relevance of immobilization in enzyme stabilization are highlighted according to the immobilization method (Figure 7). Because all the examples provided in Section 6 are related to CO2 reduction by immobilized FDH, here, the focus is on CA immobilization. Table 3 summarizes relevant studies on CA immobilization using these methods.
4.1. Physical Adsorption
Physical adsorption was the first method used to immobilize enzymes [161]. It consists of affixing the protein onto a solid matrix utilizing hydrophobic (van der Waals), electrostatic (ionic), or hydrogen bonding interactions [162]. Physical adsorption can involve partial conformational changes and/or denaturation of the enzyme; thus, special attention must be paid to avoid these events and confirm that the whole activity of the enzyme is retained after immobilization. The types of functional groups on the surface that produce the adhesion of the enzyme to the matrix is one of the crucial aspects of this immobilization method [124]. These groups can contain hydroxyl, carboxyl, amino, sulfhydryl, or imidazole groups, among others, and produce interactions with the rest of the amino acids of the protein. Weak interactions are optimal since strong ones could result in enzyme conformational changes or even denaturation. Indeed, weak interactions are nonspecific and reversible; therefore, proteins can be easily recovered. For instance, if the interactions are electrostatic, free protein can be released into the medium by simply increasing the ionic strength of the solution.
Pore structure can affect enzyme accessibility and, consequently, both the quantity and activity of the immobilized enzyme. Finally, the surface area is also a critical factor: the higher the surface area, the larger number of adsorption sites, increasing both the quantity of immobilized enzyme and the global activity of the carrier. Physical adsorption can increase the stability of the enzyme against changes in pH, temperature, or organic solvents; the enzymes can be easily separated from the reaction mixture, making reuse easy. Moreover, in some cases, adsorption can increase the activity of the enzyme due to stabilization of the active conformation of the enzyme. On the other hand, physical adsorption can sometimes result in loss of enzyme activity if the microenvironment is not adequate or in a decrease in activity due to diffusion limitations of the reactants towards the enzyme active center that can reduce the rate of substrate conversion. Finally, the cost of the enzyme immobilization process can be high when applied, in particular, on an industrial scale.
Mesoporous silica and aluminosilicates are excellent candidates for enzyme immobilization using adsorption methods [163]. Here, the size and structure of the pores are crucial. Mesoporous silica with larger pores can allow for higher enzyme accessibility to the adsorption sites, which typically increases the quantity of the immobilized enzyme and, consequently, its activity. Wanjari et al. immobilized CA in an ordered mesoporous synthesized aluminosilicate, obtaining acceptable kinetic values for the biocatalyst compared to the free enzyme, remaining stable for more than 25 days [140]. Yu et al. immobilized CA in silica functionalized with carboxylate groups, which provided a very high degree of enzyme uptake, and, although the enzyme slightly changes its conformation with respect to the free enzyme, their activities were almost equivalent (95.6% that of the free enzyme versus that of the immobilized enzyme) [141]. Vinoba and coworkers adsorbed bovine carbonic anhydrase (BCA) inside octa(aminophenyl)-silsesquioxane silica nanoparticles modified with silver or gold, which continued to be active after 20 recycling runs [142]. This adsorption approach has been developed extensively, a recent example being the fixation of CA together with FDH to produce formate in silica nanoparticles modified by polydopamine and polyethylamine [143]. Here, the production of formate was expedited up to 30-fold with respect to the free enzyme and activity was retained at 86.7% after 10 cycles.
Colloids are another type of support used to adsorb CA [138]. Crummblis et al. immobilized CA in gold sols, obtaining an enzyme with levels of activity comparable to that of the native one [164]. Curiously, denatured CA was also immobilized in modified Sepharose and subsequent enzyme renaturation using a cycle of heating and cooling, resulting in an active enzyme with elevated activity [144]. CA immobilization via electrostatic adsorption has been studied with nanoparticles using different charges [145]. Positively charged nanoparticles do not adsorb human CA II, while negatively charged ones do, showing kinetic activity that depends on the degree of hydration of both the enzyme and the particle surface. More recently, CA was fixed onto two different types of membrane via layer-by-layer assembly: the first with a porous membrane and the second without [146]. The carbonation rate of the porous membrane was three times higher than that of the enzyme alone. On the contrary, the nonporous membrane was less active (70–90%) than the native non-immobilized enzyme. The adhesive properties of the polysaccharide chitosan modified with different compounds have also been employed to immobilize CA [153,165]. Matrixes of chitosan with different coating methods and a given textile package have been shown to adsorb CA in a “drop-in-ready” method, with high efficiency for CO2 scrubbing. The physical properties of these matrixes for CO2 capture were maintained for more than 31 days, with high efficiency (>80%) at moderate temperatures.
4.2. Entrapment and Encapsulation
Immobilization by entrapment occurs when a polymer, gel, or metal organic framework (MOF) is generated in the presence of an enzyme [132,166]. In such cases, the protein can remain trapped within the hollows of the polymer. The nature of these interactions is not chemical in origin, but rather physical. The proteins have free movement at a local level, but the motion is highly restricted to the confined hollows, and most of the molecules are isolated and interact only with the matrix. Drozdov’s group immobilized CA into the pores of different sol–gel magnetite with singular magnetic properties using this method [147]. They studied the physical properties of the new material as well as the overall structure of the enzyme, mainly using infrared spectroscopy, concluding that the protein maintains its 3D arrangement in the generated nanoparticles. The immobilized enzyme was stable and catalytically active at 90 °C, which is the temperature at which the native free enzyme is completely denatured.
Encapsulation is similar to entrapment in the sense that molecules are also free in solution and their movements are restricted; however, molecules are captured in higher bags where they can interact with each other. Sol–gel matrices have also been employed for the encapsulation of CA with excellent results. Polyurethane foam has also been employed to entrap not only the enzyme itself but also E. coli cells expressing CA [148]. Indeed, whole-cell catalyst CO2 hydration activity was measured by comparing both sole and whole-cell immobilized enzymes with respect to the free enzyme. The efficiency of hydratase activity (Equation (4)) was 16-fold higher for the whole-cell immobilized enzyme than for the free enzyme. Interestingly, the activity of the whole cell trapped in the PUF was approximately 100% for at least nine cycles. MOFs are structurally ordered materials formed from inorganic complexes bridged by organic ligands that are projected in three dimensions [167]. Hollows of defined sizes are arranged monotonously in MOFs. MOFs are employed in a multitude of applications, with the immobilization of proteins being one of the most promising [168,169]. CA has been encapsulated in different MOFs with different features, most of them being zeolites constituted by imidazolates, with acceptable or excellent results [149,154,170]. The enzyme encapsulation within MOFs generates enzyme diffusion through windows that have a smaller size than the cavity. Whether or not the term encapsulation can be properly applied to the immobilization of enzymes in MOFs depends on the ratio between the pore and the enzyme size. In any case, an MOF based on Ni(II) showed a high degree of reusability for CA, retaining more than 65% of its activity after eight cycles [149]. Zinc has also been used as a base for MOFs to immobilize CA. In this case, the Zn-OH groups of the hollow imitate the active site of the enzyme, which permits high CO2 capture efficiency [171,172]. MOFs containing several lanthanides have also been employed. In this framework, the existence of a high level of electrostatic interactions substantially increases capacity for CO2 uptake [173]. Here, taking advantage of the lanthanide contraction, the specific dimensions of the hollows can be modulated, with the Eu(III) derivative having the highest affinity towards carbon dioxide. In all these examples, infrared spectroscopy is one of the key techniques for characterizing the degree of CO2 capture, as well as the distortions of the framework.
Ionic liquids (ILs) have also been employed to immobilize CA, although to a lesser extent [150,174,175,176,177]. While CO2 is nonpolar, owing to the difference in electronegativity of the carbon and oxygen atoms, the charges of ILs can absorb CO2 to a high degree; thus, this is a field fertile for exploitation. Recently, CA was immobilized in poly(ionic liquids) (PILs) by mixing the monomer hydrophobic IL 1-vinyl-3-hexylimidazolium bis(trifluoromethylsulfonyl)imide with an ethylene glycol derivative that had previously been polymerized using crosslinking [133]. After generating the PIL, CA was entrapped within the hollows of the polymer. The yield of the resulting CA was highly dependent on the size of the porous material and the degree of humidity (the dry PIL was less efficient). The authors also tuned the degree of particle size using previous sonication and studied the kinetic parameters of CA-PIL. These values were comparable to that of the free enzyme, although the entrapped CA was stable for a month without detectable loss of activity, while the free enzyme decreased its activity by more than 30%. The CA-PIL was reused for five cycles with 60% activity.
4.3. Covalent Binding and Crosslinking
Covalent binding implies the formation of bonds between the groups of adequately functionalized supports and an enzyme. This is, by far, the most extended approach for immobilizing enzymes, particularly for CA [152,155,178,179]. Several protein functional groups can be used for this purpose. CA has been covalently bound to different supports by its amine groups by reaction with glutaraldehyde [156,180]. Generally, mesoporous supports containing hollows of controlled sizes are grafted with amine groups to obtain solid materials that are prone to covalently binding to enzymes using glutaraldehyde. This was carried out with the support SBA-15, in which three different amine compounds were inserted, followed by covalent immobilization of HCA [142,152,181]. The resulting material was morphologically characterized, and its activity, thermal stability, and reusability were also determined, obtaining better results than those of the free enzyme. Kimmel et al. immobilized CA on the surface of propylene fiber membranes. These membranes, commercially available, were coated with a siloxane layer and functionalized with amine groups. Posteriorly, CA was attached to these fibers via glutaraldehyde crosslinking under two conditions: with and without chitosan tethering. Then, the authors applied these fibers to CO2 removal, finding enhancements of 115% and 37% versus the buffer and the blood controls, respectively [157]. Moreover, carboxylic groups activated by carbodiimide and N-hydroxysuccinimide agents were used to covalently immobilize CA in microtubes [182]. The resulting immobilized enzyme enhanced its ability to sequester CO2 with respect to the free enzyme.
Finally, the crosslinking method is actually a special way of covalent binding. The proteins are bound to other large proteins to form high molecular weight complexes without any solid support or, properly, the enzymes themselves being a solid support. Typically, the protein is precipitated with the appropriate agent and then crosslinked, which can be performed with a purified protein or an extract of a still unpurified enzyme. This method confers high stability and a high degree of enzyme recovery. CA has been immobilized via crosslinking in numerous studies. Recently, Xu et al. encapsulated crosslinked CA in alginate beads and confirmed that this crosslinked CA enhanced the growth of microalgae cultures [183]. The crosslinked CA was stable during 10-cycle assays. Magnetic nanoparticles aggregated with CA were obtained by crosslinking the enzyme with glutaraldehyde, improving the yield of absorbing CO2 up to 3.4-fold with respect to the free enzyme and retaining 95% activity after five cycles of reuse [158]. These magnetic nanoparticles are amply used because they allow for very simple separation and recovery of the biocatalyst by using an external magnetic field. They are considered excellent carriers and supporting matrices for enzyme immobilization, providing several advantages for the design of biocatalytic processes (i.e., large surface area, large surface-to-volume ratio, high mass transference, etc.). More recently, Chang et al. crosslinked CA and geopolymer microspheres with glutaraldehyde and performed a detailed study on the morphology, stability, and activity of the immobilized support [159]. After 60 days of storage at 25 °C, immobilized CA still presented 28.9% activity, whereas free enzyme activity was less than 10%. CA and FDH have also been crosslinked to reduce CO2. Zhang et al. used microbial transglutaminase (MTG) as the crosslinking medium for CA and FDH labeled with peptide tags and previously expressed in E. coli [160]. MTG catalyzes the formation of an isopeptide bond between the ε-amino group of lysine and a glutamine. The authors studied the activity and reusability of several crosslinked particles with different tags at CA/FDH ratios of 1:1, 1:2, and 1:3. Because CA is much more active than FDH, it is expected that the lower the CA/FDH ratio, the higher the formate yield obtained. However, the optimal found CA/FDH ratio was 1:2. The authors attributed the lower yields obtained for a 1:3 ratio to FDH steric hindrances in the crosslinked aggregates.
5. Carbon Capture Storage and Utilization: State-of-the-Art, Costs, and Perspectives
Methods for CO2 capture are classified into precombustion, postcombustion, and oxy-combustion processes [12,184]. The precombustion approach is related to hydrogen gas production. This is obtained in numerous industrial processes such as electric power generation, ammonia or fertilizer synthesis, and petroleum refinement. The precombustion process refers to the conversion of the primary solid fuel (coal or biomass) by reforming it into a mixture of CO and H2 gas (syngas). This gas reacts with the water stream at high temperatures and pressures to produce CO2 and more H2 (water–gas shift reaction). Finally, CO2 is captured using several methods. Postcombustion CCS methodology denotes all the processes employed to capture CO2 from exhaust gas (its major component being nitrogen) resulting from industrial chemical processes. CO2 gas is emitted at relatively low temperature and pressure. Oxy-fuel combustion consists of the oxidation of fuel using pure oxygen instead of air, obtaining an almost pure CO2 atmosphere without nitrogen gas. Most CCS methods have been developed for postcombustion gases and are referred to here, except where otherwise indicated. In many cases, the methodology can be the same for both post- and precombustion approaches, although with different designs depending on the P/T conditions of the exhaust gases. Physical adsorption and absorption [185] (geological storage [186] probably being the most relevant among absorption approaches) and cryogenic distillation [187] are the main methods used for CO2 CCS.
Global CO2 emissions from combustion processes grew by 0.9% in 2022, reaching a total of 36.8 Gt [188]. Energy used in industry, agriculture or land use, buildings, transport, direct industrial processes, waste, and others with 37.8%, 18.4%, 17.5%, 16.2%, 5.2%, 3.2%, and 1.7%, respectively, are the contributions to CO2 emissions by the different sectors [189]. Only ca. 40 million Tm, i.e., 0.1%, was removed from the atmosphere using CCS methods in 2019. Thus, we are still very far from being efficient in eliminating the CO2 expulsed into the atmosphere.
The estimated present costs of CO2 Tm removal vary nowadays from 40 to 80 USD depending on the method [190]; however, the net contribution to CO2 elimination from the atmosphere is difficult to calculate, since net contributions in the whole process have to be taken into account. Hepburn and coworkers analyzed the perspectives, including cost, for different methods of CO2 utilization [190]. They analyzed ten different methods of CO2 utilization. For instance, chemical production, particularly the generation of urea, on one hand, and the production of polycarbonate polyols, on the other, are two fields in which CO2 capture can be exploited. They estimated that CO2 utilization in chemicals in 2050 could be around 0.3–0.6 Gt CO2/yr with costs ranging from −80 to 320 USD per Tm of CO2 (a negative value would indicate an additional economic benefit, while a positive value indicates that the cost of capturing and utilizing CO2 would be higher than the value generated from it). Fuels, that is, CO2-methanol plants, were also considered in their study, although they stated that many different scenarios can vary their prospects from 1 to 4.2 Gt/yr in 2050 and, in terms of cost, from 0 to 670 USD per Tm of CO2.
6. Carbonic Anhydrase in Carbon Capture Storage
Although CA is not used in all previous technologies, its use is extended or has a good perspective in others, mainly chemical adsorption and mineralization, whose description is commented on below. Table 4 describes relevant studies performed with CA in CCS research.
6.1. Chemical Absorption
Chemical absorption has been the most used CCS method for decades [203]. This procedure involves the scrubbing of exhaust gas at low pressures and temperatures with alkaline solutions typically containing amines and/or carbonates or hydroxide solutions [203]. Amines are weak bases that can capture protons from Brönsted acid CO2. The reactions of primary/secondary or tertiary amines produce carbamates or bicarbonate anions, respectively, according to the reactions described below.
The amines typically used for these purposes are alkanolamines. An alcohol group increases water solubility and decreases vapor pressure compared to analogous amines. The main chemical solvent used as an absorber is monoethanolamine (MEA). The solutions typically consist of an aqueous solution of 20–30 wt% MEA. CO2 is captured at low pressure (ca. 1 bar) and in a mixed gas containing other gases, such as N2, SOx, and NOx. Secondary amine diethanolamine (DEA) and tertiary amine N-methyl diethanolamine (MDEA) are also amply used amines.
Several points should be considered when choosing a suitable solvent for CO2 sequestration. First, the enthalpy of the reaction: the higher the enthalpy of the reaction, the higher the cost of solvent regeneration. This is a crucial point because it is estimated that 60–80% of the costs of these processes arise from solvent regeneration [204]. The enthalpy of (exothermic) reactions 6 and 7 increases from tertiary to secondary and primary amines; thus, primary amines improve both energetic and economic costs. Moreover, the power of corrosion also follows the same order (primary amines are the most corrosive). Another decisive issue is their ability to load CO2. Tertiary amines have the highest capabilities in this regard [205]. According to these thermodynamic aspects, tertiary amines are the best ones to use. However, another crucial point is the kinetics of CO2 sequestration; indeed, tertiary amines have low reaction rates and are kinetically much more inert than primary amines [206,207]. Owing to the slow kinetics of tertiary amines, primary amines (or secondary amines) are currently preferred. However, the high costs of cooperation and maintenance due to the ease of amine degradation and the formation of highly corrosive salts are drawbacks when operating with amines.
Consequently, for kinetic reasons, primary amines, specifically MEA, are by far the most widely used alkanolamines in the industry. Numerous plants have been developed using MEA solutions. These systems are typically coupled with industrial processes and CO2 is captured with postcombustion gases. For instance, their use is extended to the iron and steel industries (responsible for approximately 31% of all industrial CO2 emissions). These plants can recover 85–95% of the CO2 in gas. As an example, a steel production plant recently established by Emirate Steel Industries has a yield plant using CCS based on amine absorption that captures 0.8 Mt CO2/year [208].
In the last decade, CA has been revealed as a tool for accelerating CO2 uptake in chemical absorption processes. Gundersen et al. studied CA stability and activity for a long time (150 days) as a function of pH, temperature, and the solvent, combining MEA and MDEA solutions, among others [191]. They concluded that CA was suitable for these purposes; the biocatalyst was stable and active between pH 7 and 11, with maximum activity at 40 °C. In addition, the enzyme preserved its activity between 12 and 91% of the original activity depending on the solvent employed. The absorption of CA was also accomplished in MOF ZIF-L-1 in the presence of MDEA [198]. In ZIF-L (a zeolitic imidazolate framework), the imidazolate groups enhance CA immobilization, and CO2 uptake is hence greatly increased. The authors highlighted that this new MOF obtained excellent CO2 absorption rates at 40 °C and a CO2 partial pressure of 15 kPa, while the activity was maintained for six reuse cycles [192]. Additionally, a pilot-scale plant was set up with CA in solution in the presence of MDEA. The authors observed an enhancement in CO2 capture in the presence of the enzyme and demonstrated the possibility of translating the laboratory results to higher scales [193]. However, because the free enzyme is damaged by amines, immobilization is necessary. Kim et al. also studied the effect of CA on CO2 absorption rates in the presence of MEA and MDEA, although they used a membrane contactor with hydrophobic and hydrophilic supports [194]. This system allows an expanded contact surface to enhance CO2 absorption.
6.2. Chemical Carbonation
Chemical carbonation is probably the most efficient method for capturing CO2. This is performed when CO2 is bubbled through an alkaline solution, typically consisting of dissolved KOH or Ca(OH)2, where potassium or calcium carbonates precipitate. The limiting step for capturing CO2 in postcombustion processes is Equation (3). However, due to Equation (4), CO2 capture is much faster in alkaline media since carbonates are formed, and so hydrogen carbonate concentration decreases, and Equation (3) is shifted towards the consumption of CO2. Indeed, once bicarbonate anion (soluble) is formed, reactions such as Equations (6) and (7) (amine formation, Scheme 2) occur much faster in alkaline media. Even so, the limiting step, for kinetic reasons, continues to be the CO2 gas uptake, as commented previously. Thus, the main challenge in applying alkaline solutions, either amines or carbonates, to the CCS approach is speeding up CO2 conversion to bicarbonate [195]. Consequently, numerous studies on CA to increase the mass transfer of CO2 capture have been proven not only at the laboratory level, and its feasibility has been demonstrated on an industrial scale [15,205]. Novozymes NS81239 CA (NCA) at 2 μM increased the absorption rate of CO2 into potassium carbonate by ca. 30%, augmenting this uptake at temperatures in the range of 40–60 °C [195,196]. Power et al. demonstrated that bovine CA accelerated the carbonation rate of brucite Mg(OH)2 from CO2 gas by up to 240% [209]. In these studies, CA was supplied as a free enzyme; therefore, its regeneration was not studied. Biological tools have also been used to enhance carbonation. Jin et al. accelerated calcium carbonate precipitation by employing Bacillus mucilaginosus on steel slag powder [197], increasing the carbonation degree from 66.34 to 86.25%. Moreover, the mechanical properties and durability of the treated steel slag were enhanced. The CA immobilization, as described in the previous section, strongly improves the reusability of the enzyme as well as the chemical carbonation. For instance, Jo and coworkers proved the suitability of CA encapsulated in a biosilica matrix, obtaining good yields for carbonation compared to the free enzyme [198].
6.3. Mineralization
Biomineralization is a very slow and exothermic process by which carbonate minerals are formed from silicates and CO2 under basic conditions [210]. The starting silicates usually contain divalent metals such as Ca(II) and Mg(II) or trivalent metals such as Fe(III). This event occurs in nature on a regular basis and is responsible for the formation of inorganic structures in living organisms such as exoskeletons in protozoa, algae or invertebrates, and shells or plant mineral structures. It is also responsible for the presence of large amounts of limestone on the Earth’s surface [211,212]. When trying to emulate biomineralization, which takes place over very large timescales, the main drawback is speeding up the process. Artificial mineralization mimics nature, although in short periods. It involves the injection of CO2 directly into geological formations to promote a carbonate-forming reaction with alkaline minerals [213]. This mineral sequestration would be a viable alternative for subsequent storage because the carbonate products formed would not require monitoring owing to their high stability and safety. On the other hand, in-ground or ex situ mineralization is based on the exposure of crushed rock material in a processing plant where CO2 is introduced, facilitating the formation of carbonate minerals. Natural minerals or alkaline solid waste can be used [213,214]. The use of natural silicates requires a large amount of material, which implies a very large operational size and an unfeasible economic mineral impact. Ex situ mineralization can also be carried out using alkaline wastes containing divalent metals such as ash originating from the coal or metallurgical industry, cement and concrete wastes, or iron and steel slag [215,216]. This method would reduce not only environmental CO2 but also the accumulation of waste from industrial activities, although a major disadvantage in that its capacity is much smaller than that of CO2 mineralization from silicates. At laboratory scale, this mineralization has been satisfactorily performed by directly extracting CO2 from the air, and its direct extraction by passing the air through cooling towers using NaOH solutions has also been proposed for larger scales [217]. However, the same authors pointed out the elevated costs of this approach on a large scale.
The efficiency of the biomineralization process can also be accelerated by modifying certain parameters such as increasing the temperature, pressure, or retention time. The biomineralization process is also favored by the presence of purines, NaCl, or CA. The presence of CA accelerates the rate of hydration of CO2 dissolved in water; therefore, possible modifications to CA to support high pH and temperature conditions without losing its advantageous functionality have been studied [218,219]. On the other hand, some varieties of carbonic anhydrase are inhibited in the presence of high concentrations of hydrogen carbonate, which becomes a problem for its use in industry. However, this can be circumvented, at least partially, by increasing the pH to values equal to or higher than 9.0, conditions under which some CAs are still stable and functional. Immobilization improves the stability of CA at high temperatures or alkaline conditions, as confirmed by Arias et al. when forming calcite in vitro by mineralization using CA immobilized in eggshell membranes [219]. Recombinant CAs have also been used to accelerate mineralization under extreme conditions. For instance, CA from the alkalistable Aeribacillus pallidus was genetically modified, achieving acceptable yields in the presence of pollutants such as NOx and SOx [200]. Similarly, CA from the thermophilic bacterium Sulfurihydrogenibium azorense was modified, and its half-life was found to be 8 days when the biomineralization process was carried out at 70 °C and 53 days at a reaction temperature of 50 °C [201]. Di Lorenzo et al. studied the effect of CA and a Zr-based MOF in the carbonation process of wollastonite (CaSiO3) to produce calcite (CaCO3) [202]. Although CA accelerated CO2 uptake by the silicate, the total gas absorber quantity was lower than that of the MOF. Jin et al. also took advantage of CA to accelerate the carbonation of γ-dicalcium silicate, which is also present in steel slag. They used a powder containing alkali-resistant CA bacteria, increasing the yield by 19.0% [220].
7. Biotechnological Aspects of CO2 Reduction
7.1. Hindrances to Biochemically Reducing CO2
Forward Equation (5) presents several drawbacks that hinder its application outside the natural living environment, that is, employing it with biotechnological aims. First, owing to the low redox potential of the CO2/HCO2− pair (−430 mV), the reaction is highly endergonic under physiological conditions. Three main strategies have been developed to overcome this problem (Figure 8): coupling thermodynamically favorable reactions in the presence of an excess concentration of the reducing agent [136,221,222,223,224]; coupling the reaction to an electrochemical device that provides electrons from a battery anode (i.e., by supplying electric energy) [225,226,227,228]; and using photosensitive molecules able to absorb light and regenerate redox partners [31,229,230,231,232,233].
Regeneration of the cofactor NADH is crucial for obtaining formate with acceptable yields [234,235,236,237]. As previously mentioned, metal-dependent FDHs do not necessarily accept electrons from NADH (see Figure 6); nevertheless, whatever the cofactor is, either in nature or in biotechnological approaches, it has to be regenerated for adequate progress of the reaction. NADH, the primary source of electrons, is an expensive reactant, and thus economic aspects are also relevant in cofactor regeneration. The approaches to regenerating NADH are similar to those followed to facilitate the thermodynamics of the reaction. Two additional issues related to the oxidation of this cofactor also have to be kept in mind: NAD+ can strongly interact through its negative phosphate charges, with positive charges on the active site of the biocatalyst, inhibiting the biocatalyst, particularly in nonmetal-dependent FDHs [238]. Moreover, it is well known that NAD+ trends to form dimers (see below), which makes cofactor regeneration impossible [221,234]. Carbon dioxide uptake into the reaction medium is another crucial factor. As commented throughout this text, since CO2 is a nonpolar gas, its solubility in water or similar solvents is low and, more importantly, the kinetics of solubilization and mass transfer are very slow [204]. Hence, the employment of systems (specific solvents or solutions) that can incorporate CO2 into their structures is decisive. As discussed above (see Section 6.1 and Section 6.2), amines have been used extensively [204].
Ionic liquids, solvents in which positive cations can interact with the oxygen atoms of a CO2 molecule through their sole pair of electrons, are also excellent media to solve, in high quantities, carbon dioxide gas [214,239,240]. The selection of the appropriate conditions for the reaction is also crucial. Dealing with a gas, high pressures and low temperatures are ideal conditions for solubilizing CO2, while with respect to pH, carbon dioxide solubilization is favored at high pH values (Equations (3) and (4)). However, optimal conditions are determined by FDH stability and activity, and temperatures must thus be moderated (lower than 50 °C), pressures cannot be high, and pH values must be between 6.0 and 7.5, the range at which FDH shows its highest yields. Finally, the use of the adequate enzyme, that is, the FDH of the appropriate organism, is also a key factor in the success of the reaction.
Approaches to circumventing these problems and achieving significant formate yields as a starting point for obtaining value-added chemicals are discussed in the following sections.
7.2. Coupled reactions: Enzymatic Multicascades
Equation (5) is reversible although under physiological conditions is highly shifted towards the backward reaction. The first successful conversion of CO2 from formate using FDH in a laboratory system was reported by Hopner and collaborators [100]. They used the metal-dependent FDH from Pseudomonas oxalaticus as biocatalyst and NADH concentrations in such a way that reaction 5, still being thermodynamically unfavorable, was not completely shifted towards the oxidation of formate. They devised a sealed system containing 14CO2 and measured the radioactivity of the H14COOH formed. In this pioneering study, the kinetic parameters (kcat/KM) of the enzyme and its pH activity profile were determined. However, the cofactor was not regenerated and the rates between the forward and reverse reactions in their working conditions were 1:30, with a turnover number for CO2 reduction as low as 3 s−1, which makes these results insufficient.
The simplest and most straightforward way to increase formic acid generation is to eliminate the products from the reaction media, that is, regenerating the NADH cofactor using a reducing chemical agent present in the medium. This approach was developed in the 1980s with redox biocatalytic systems [241,242]. The reaction desired to take place is coupled with another “inverse” reaction, in which the reactant is added at high concentrations. Thus, the reaction is thermodynamically favored, and the cofactor is regenerated (Figure 9A). Chenault and Whitsides, pioneers in using this technique with formate dehydrogenase, employed CbFDH to regenerate NADH by coupling formate oxidation with the reduction in lactate to pyruvate using D-lactate dehydrogenase and obtained acceptable results, with nicotinamide residual activity of 55% after each run [242]. However, in their reaction, FDH was used for formate oxidation (backward Equation (5)), and the coupled reaction was used to regenerate NAD+. Since then, the reduction of CO2 (forward Equation (5)) using FDHs in a conjugated oxidation–reduction reaction system for regenerating the NADH cofactor has been carried out with several enzymes. Yu et al. cloned the FDH gene from Cupriavidus necator in E. coli and coupled the reduction of CO2 with the oxidation of D-glucose to D-δ-gluconolactone using glucose dehydrogenase (GcDH) to regenerate NADH [243] (Figure 9A). The expressed enzyme, an O2-tolerant, Mo-dependent FDH, was able to effectively reduce formic acid comparable to that of the nonrecombinant protein. Glutamate dehydrogenase (GDH) is also used to regenerate NADH. GDH catalyzes the oxidation of glutamate to α-ketoglutarate and ammonia, thereby reducing NAD+ to NADH. GDH is highly stable over a wide range of pH values and at temperatures as high as 85 °C, as well as being widely commercially available and inexpensive [236,244].
Nonetheless, the most extensive coupled reaction approach is the well-known and widely employed enzymatic cascade reaction that drives from CO2 to formic acid, catalyzed using FDH, from this species to formaldehyde, i.e., formaldehyde dehydrogenase (FalDH), the biocatalyst, and, lately, to reduce this molecule to methanol, by the action of the alcohol dehydrogenase (ADH) (Figure 9B). The oxidases mentioned above are typically used for NADH regeneration.
El-Zahab et al. co-immobilized FDH, FaldDH, and ADH together with GDH into polystyrene particles to reduce CO2 to methanol [245] (Figure 9B). The NADH cofactor was also immobilized, although separately. The results obtained with the immobilized enzymes were similar to those obtained for the free enzymes; however, importantly, the yield of the reaction was maintained at over 80% after 11 cycles. Ji et al. coupled the same reaction cascade [246], but in their study, the four redox enzymes were entrapped in hollow nanofibers together with CA to facilitate CO2 absorption. The methanol yield was 36.17%, retaining ca. 80% of the productivity after 10 reuses, with an accumulative yield of more than of more than 900% for NADH regeneration. In another study using the same cascade, Ren and collaborators encapsulated the same biocatalysts in MOF ZIF-8 and investigated the effect of polyethyleneimine (PEI) on anchoring the NADH cofactor and, hence, on the yield of the reaction [247]. Compared to the free system, the yield of this reactor system increased 4.6-fold and the activity after eight cycles was retained by 50%.
Ionic liquids are known to solubilize CO2 [248,249]. Taking advantage of this, Pinelo’s laboratory immobilized the four mentioned enzymes as well as the cofactor in a series of modified ILs composed of choline (CH) and amino acids (CHGlu, CHPro, CHGLy, and CHHis) [250]. They generated a membrane reactor in which the products were removed in situ and the reaction was displaced towards the desired product. The yield of CHGlu increased up to fivefold compared to that of with the control aqueous system when NADH was regenerated. In another study, immobilization of the four enzymes was performed on superparamagnetic nanoparticles. Here, the yield was low (2.3% of methanol per NADH molecule); however, under CO2 pressure (126 psi), the reaction yield increased 64-fold after 30 min of reaction [250].
Figure 9.
(A) Several enzymes whose oxidation reactions have been coupled to FDH CO2 reduction for regenerating NADH. (B) A classical enzyme multicascade for the reduction of CO2 to methanol; here, GDH is coupled for regenerating NADH. (C) Uses of methanol at the global level in the year 2020 shown in percentages according to their production and demand [251].
Figure 9.
(A) Several enzymes whose oxidation reactions have been coupled to FDH CO2 reduction for regenerating NADH. (B) A classical enzyme multicascade for the reduction of CO2 to methanol; here, GDH is coupled for regenerating NADH. (C) Uses of methanol at the global level in the year 2020 shown in percentages according to their production and demand [251].

Phosphite dehydrogenase (PTDH, Figure 10A) has also been used for NADH regeneration in a multicascade approach to obtain methanol from CO2. Cazelles et al. studied the effect of regenerating NADH using three different coupled reactions, namely phosphite oxidation, catalyzed using phosphite reductase; glycerol oxidation to dihydroxyacetone, performed using glycerol dehydrogenase; and a natural photosystem (chloroplasts) extracted from spinach leaves that oxidize water to molecular oxygen [252]. They encapsulated the three enzymes in phospholipid–silica nanocapsules and obtained excellent activities with PTDH with respect to the free enzymes in solution (55 times higher activities) under 5 bar of CO2 pressure for 3 h, although the other two systems were not so efficient. Singh et al. also employed PTDH for regenerating NADH [253]. They expressed recombinant proteins (FDH, FalDH from different bacteria, and ADH from yeast) in E. coli and performed assays with free enzymes in water solution and in the presence of many different cosolvents. IL 1-ethyl-3-methylimidazolium acetate (EMIM-Ac) was found to be the most effective in increasing methanol production. Indeed, the yield was enhanced more than twofold (from 3.28 mM of methanol to 7.86 mM, 6 h of reaction) in the presence of 1% EMIM-Ac because of the ability of EMIM cations to interact with CO2, increasing solubility. Finally, lactate dehydrogenase (LDH) was also used to regenerate NADH in a multicascade reaction [254]. This enzyme was immobilized in a sol–gel matrix, and CO2 reduction was acceptable after 1 h of reaction, as indicated by the authors.
Using CO2 as a substrate for the generation of methanol is an attractive process because of its potential use as a fuel and the multitude of products obtained from it at the industrial level. Figure 9C shows the chemicals produced from methanol at a global level in 2020 [251]. Methanol is a precursor of numerous compounds such as olefines (25.9% of the demand for methanol, the year 2020), formaldehyde (25.0%), gasoline blending (13.1%), biodiesel, and so on. Therefore, it is of great commercial interest due to its potential application both in the energy industry as a fuel and in environmental CO2 sequestration to mitigate high atmospheric CO2 levels. The methanol production in the year 2023 was 98.9 × 106 Tm and it is projected that by the year 2027, more than 8 × 106 Tm will be obtained from e-methanol (produced from captured carbon dioxide and hydrogen produced from renewable electricity, ca 5 × 106 Tm) and biomethanol (produced from sustainable biomass, ca 3 × 106 Tm). For example, currently, 4000 Tm of methanol is produced annually using biocatalysts based on copper and zinc oxide in Iceland, recycling some 5500 tons of CO2 annually. Other approaches to achieving the same effect have also been undertaken in Germany and China [222]. The use of enzymes is an advantage for the conversion of CO2 to methanol because of the high selectivity of the catalyzed reaction. This reduction of CO2 to methanol is considered a green chemical process and occurs at atmospheric temperature and pressure [253].
7.3. Electrochemical Regeneration of NADH Cofactor
NADH can be regenerated at the cathode of an electrolytic cell by applying adequate electric voltage (Figure 10A). This method can be extensively applied and permits the easy separation of products [225,226,227,228]. In principle, as the supported potential difference can be as high as desired within technical constraints, the cofactor regeneration under appropriate conditions could be high, although the complete energy cycle is probably not. If a cell is supported by renewable energy, this approach can also be considered green. In the electrolytic cell, the cofactor can be the primary acceptor of electrons from the electrode (direct electrochemical regeneration, Figure 10B) or, in contrast, other molecules can accept the electrons, the cofactor being reduced by these mediators (indirect mode Figure 10B).
The direct mode has two intrinsic disadvantages that are very difficult to overcome: the formation of (NAD)2-inactive dimers and the necessity of using high overpotentials. The electrochemical reduction of NAD+ molecule proceeds in two stages (Figure 11). In the first step, NAD+ captures an electron and an NAD* radical is formed. In the second step, an additional electron and a proton are accepted by the NAD* radical. However, NAD* can dimerize and, furthermore, due to the adsorption of NAD+ onto the electrode, this dimerization process is favored over the uptake of the second electron. The need to use high overpotentials for direct NADH reduction is another limitation of this method. Several studies modifying the electrode nature concluded that mass transfer between the cathode and the cofactor was a crucial step for favoring reduction versus dimer formation. These problems can be partially circumvented by selecting an appropriate electrode [255]. For instance, Ag or Pt electrodes coated on Cu foams were successfully employed for NADH regeneration [256]. The existence of a mediator on the electrode surface is another key point for avoiding dimer formation. Mediators such as (2,2-bipyridyl) (pentamethylcyclopentadienyl)rhodium, [Cp*Rh(bpy)], and methylviologen (MV) have been shown to decrease the overpotential for NADH regeneration, facilitating electron transfer and NADH recovery [221].
In an elegant experiment, Song et al. generated a cysteine residue in FDH from Thiobacillus sp. KNK65MA and attached the mutated enzyme to copper nanoparticles (CuNPs) deposited on the electrode surface [257]. Polyethylene glycol (PEG) was then used to crosslink the FDH-NAD+ system so that the cofactor could swing from CuNP to the enzyme and vice versa. No mediator was used. This system produced 8.5 mM formate, several times that achieved with a free enzyme.
The enzyme cascade for methanol production from CO2 (Figure 10B) has also been widely performed using an electrochemical approach. Addo et al. coupled the multienzyme cascade for producing methanol from CO2 to a polyneutral red electrode to regenerate NADH [258]. Electroenzymatic reduction in CO2 was also achieved with maximum Faradaic efficiency (99 ± 5%) by the immobilization of Mo-dependent FDH from E. coli at the surface of a carbon electrode [259]. Here, reduction was achieved through the mediator cobaltocene being covalently bound to the linear polymer poly(allylamine), which transferred electrons from the cathode to the enzyme. In another interesting study, CO2 reduction was electrochemically achieved by using copper deposited in a glassy carbon electrode, the Rh(III) complex [Cp*Rh(bpy)Cl]+ (Cp* = pentamethylcyclopentadienyl; bipy = bipyridine) as a mediator and CbFDH as a biocatalyst, with yields threefold higher than those of previous analogous works with copper foil electrodes [260]. Chen et al. also used an Rh(III) complex as a mediator for the electrochemical regeneration of NADH [261]. MOF NU-1006 containing CbFDH was deposited on the electrode surface of fluorine-dopped tin oxide glass. NU-1006 is a mesoporous material with a channel size that can accommodate CbFDH (6 × 4 × 11 nm). The electrode was obtained using a multilayer system in which the entrapped enzyme was the latter and was in contact with the solution containing CO2. Regeneration of the cofactor was optimum because of the modified electrode, with a formate generation rate of 79 ± 3.4 mM h−1. All these experiments have the advantage of easy regeneration of NADH, due to the possibility of creating a sufficient negative redox potential. However, its translation towards more complex systems, such as a multicascade system coupled to other oxidoreductases, no longer seems biotechnologically accessible.
Barin et al. immobilized CbFDH on polystyrene nanofibers and NADH in a copper foam electrode [262]. Although the activity of this system was inferior to that of the free enzyme, the immobilized enzyme was stable for a long period (41% of the initial yield after 20 days) and had acceptable reusability after eight cycles (53% of the initial activity). The authors also observed an inhibitory effect of NADH at concentrations higher than 0.51 mM. Indeed, several studies have demonstrated the inhibition of FDH by the cofactor at concentrations higher than millimolar. On the other hand, Zhang et al. encapsulated FDH, FaldDH, and ADH in MOF ZIF-8 and used the Rh complex (Cp*Rh(2,2′-bipyridyl-5,5′-dicarboxylic acid)Cl2) grafted on the cathode as a mediator to perform NADH regeneration [263]. The concentration of methanol obtained was fivefold (from 0.061 to 0.320 mM) with the encapsulated enzyme compared to the free enzyme. Moreover, when NADH was electrochemically regenerated, an increase of 0.742 mM was observed. This again demonstrated that electrochemical NADH regeneration is probably the best approach to achieving this goal.
7.4. Photochemical NADH Regeneration
Photochemical reactions are another suitable approach to reducing CO2 [31,229,230,231,232]. The energy arises from light, which is an inexpensive, renewable, and clean source. This requires a supply of electrons and a photosensitizer mediator scheme (Figure 12). The electron donor, D, also called the sacrificial agent, is typically a stable solute present in large concentrations that can be easily oxidized using a photosynthesizer in its excited state [223]. Amines such as triethanolamine (TEOA), triethylamine (TEA), and ethylenediaminetetraacetic acid (EDTA) are the most commonly employed, although many other species, such as 3-(N-morpholino) propanesulfonic acid, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, 2-(N-morpholino)ethanesulfonic acid mercaptoethanol, phosphite, propanol, and even molecular hydrogen gas have also been used [30]. The redox potentials of these systems must be low enough to be oxidized using the photosynthesizer so that a pool of electrons is always present in the solution. This electron donor supplier must be present at high concentrations, typically not lower than 100 mM, for the system to be effective. A water molecule has also been proposed, imitating natural photosynthesis, as a sacrificial agent; however, the redox potential of H2/H2O is too low to produce an efficient system.
The key to these reactions is photosensitizers, molecules that are excited by light [31,232]. These species act as electron mediators, capturing electrons from the sacrificial agent and transferring them to the cofactor (Figure 12). Then, the cofactor is regenerated, and CO2 can be reduced using FDH. The mediator is a molecule that is stable in its oxidized state and can be photoactivated, passing from the ground to an excited state in which this molecule has a much higher redox potential, being able to oxidize the sacrificial agent and becoming reduced. In this reduced state, the mediator is highly reducing and immediately relinquishes the electrons to the cofactor. This transfer proceeds with a proton cession, such that the cofactor in its oxidation state (NAD+ or others) is reduced (NADH or analogous). The photosensitizers must fulfill three requirements [30,264]: First, the band gap between the HOMO and LUMO must be low enough to accept an electron from visible light (see Figure 3). This is normally satisfied in extended π–π-conjugated systems or semiconductors. Second, in the excited state, its redox potential must be sufficiently high to oxidize the sacrificial agent under solution conditions. Finally, the ground state redox potential must be sufficiently low to reduce the cofactor. Other chemical (stability), economical (inexpensive), and environmental (clean) requirements must also be satisfied.
Various photosensitizers have also been used. Rh(III) and Ru(III) bipyridyl complexes are well known for their photosensitivity; consequently, their use has been extended [264,265,266,267,268,269]. Guo et al. employed Cp*Rh(bpy)(H2O)]2+ (Cp = cyclopentadienyl; bpy = 2,2′-bipyridyl) as a synthesizer in a system where FDH and FalDH were immobilized on polyethylene membranes doped with the widely used semiconductor TiO2 [270]. A comparison between the results using water or EDTA as a sacrificial agent and as a function of pH was described, the latter being much more efficient for formaldehyde production. The optimal FDH:FalDH ratio was 1:0.3, reaching up to 6.5% formaldehyde production after 4 h of reaction. Photosynthesizers composed of xanthene dyes have also been used. Kim and col. combined eosin Y with cobaloxime complexes for regenerating NADH using TEOA as the sacrificial electron donor [271]. The system exhibited an acceptable turnover number for formate generation (ca. 1.6) and an optimal NADH production (0.038 mM/h). In situ changes in eosin Y infrared and UV-visible spectroscopy properties eosin Y was also used to follow in situ formic acid generation using infrared and UV-visible absorption spectroscopies [272]. Interestingly, EDTA was used not only as an electron sacrificial agent but also as a source of CO2, without any additional electron carriers.
Nanomaterials, which act as porphyrin-based photosensitizers, are another set of well-developed approaches for acting in these photochemical reactions. Ji and coworkers designed a biomimetic chlorosome by combining porphyrin, eosin Y, and [Cp*RhCl2]2 to generate supramolecular assemblies [273]. Using TEOA as the ultimate electron donor agent, an enzyme cascade was coupled, obtaining 38 μM methanol from CO2 after 2 h of reaction. MOFs have revolutionized many technochemical applications [157]. The MOFs used for CO2 reduction are also basic pivots in this respect. MOFs contain molecules and holes with different chemical and physical features (hydrophilicity, hydrophobicity, acid-base properties, photochemical features, etc.) that make them ideal for catalysis in general, and photobiocatalysis in particular. An excellent recent example is Xing’s work [229], where a porphyrin ligand was covalently bound to a Zr-based MOF and, posteriorly, the complex Cp*Rh(bpydc)Cl2 (bpydc = 2,2′-bipyridine-5,5′-dicarboxylic acid) was incorporated into the organic frame, generating a system that can be activated by light owing to the porphyrin, the Rh(III) complex, and original aromatic ligands of the MOF. FDH was then immobilized via electrostatic entrapment. Using TEOA, up to 244 μg/mL of formic acid was formed after 4 h of reaction. A similar approach employing an MOF based on pyrane skeleton linkers, NU-1006, has also been developed [261]. In this case, the Rh(III) complex attached to the MOF was used as an electron mediator, reducing NADH and being reduced by a pyrene photosynthesizer. FDH was also trapped in the MOF and the reaction took place with a yield of formic acid production of 0.144 ± 0.003 mM after 24 h, while the cofactor was regenerated at a rate of 28 mMh−1. In another recent study, MOF Mil-125-NH2 was functionalized with the Rh complex in such a way that it was covalently fixed to the secondary sphere of the MOF and FDH was subsequently immobilized [274]. The system obtained a formic acid yield of 9.5 mM in 24 h, whereas the NADH regeneration was 64%.
Graphitic carbon nitride (g-C3N4)-based materials are other fascinating agents with extremely interesting properties, such as photocatalysts [275]. These materials have also been used in biocatalysis and to generate formate from CO2. Zeng et al. used g-C3N4 doped with the metal dichalcogenide WS2, which provides g-C3N4 with specific semiconductor properties that can be used as photosensitizers [276]. Using [Cp*Rh(phen)H2O]2+ (phen = 1,10-phenanthroline) as a mediator and TEOA as the sacrificial agent, a multicascade system for obtaining methanol from CO2 was designed with acceptable results (methanol productivity 372.1 μmol h−1 gcat−1). In a similar approach, Meng and col. designed nanospheres with thiophene incorporated into hollows in a double shell that acted as the photosynthesizer [277]. This nanomaterial was coupled to [Cp*Rh(bpy)H2O]2+, which in turn was coupled to the cofactor to reduce CO2. Optimal NADH yield regeneration was obtained (74%). Silver nanoclusters combined with TiO2 and g-C3N4 have also been employed in efficient devices for formate production [278]. These nanoclusters are good light sensors, making it possible to obtain good yields in CO2 uptake with the metal-dependent FDH from Clostridium ljungdahliia. Carbon nitride has also been used in microbial electrosynthesis. Here, the photoanode chamber was formed using an activated carbon fiber (ACF) supported by NiCoWO4 in g-C3N4 [279]. The oxidation of water by light in this chamber provides the electrons for reducing CO2 in the cathode chamber, formed by a g-C3N4/ACF (without NiCoWO4), in which a culture of E. coli was adhered as a biofilm. FDH from E. coli produced the reduction of CO2 due to the electrons arriving from the biocathode and the protons, also produced in the cathode by the photochemical decomposition of water, which crossed a cation exchange membrane. The witty system, first applied in CO2 reduction, provided highly efficient formate synthesis (12.8 mM per day).
When dealing with photochemical reactions, a critical point is the reactive oxygen species (ROS) that can be generated by photoexcitation processes. To avoid this, systems mimicking chloroplasts have been developed, creating divided compartments where reactions occur separately. The photoactivation process was achieved by combining a Rh(III) complex conjugated onto g-C3N4 previously modified using thiophene, with triethanolamine used as the electron sacrificial agent [280]. The key to this approach arises from the encapsulation of FDH into a MOF (MAF-7), which protects the enzyme from photoactivation reactions. NADH shuttled electrons from the reduction compartment towards FDH, obtaining 16.75 mM of formic acid after 9 h of illumination. Immobilization of FDH together with CA in the TOF ZIF-8 was also carried out by Yu et al. in another system imitating photosynthesis and with g-C3N4 as the photosynthesizer [281]. CA accelerated the interconversion of CO2/HCO3−, allowing for an effective mass transfer rate between the gas and liquid phases. The authors reported production of 243 μM formic acid with excellent system stability since the yield production was above 80% after 10 batches.
Graphene has also been employed as a photocatalyst in systems that imitate photosynthesis and produce formic acid from CO2 [210]. Ji’s group conceived and developed a nanofiber polyurethane system as a support where the cationic electrolyte, a polyalanine, was deposited onto graphene oxide, while the cascade of proteins FDH, FalDH, and ADH was entrapped in the hollows of the nanofiber to generate methanol [246,282,283]. Finally, polymers that mimic photosystems in CO2 reduction have also been candidates for biotechnological applications. Kim et al. reported a photosystem based on polydiacetylenes and covalently attached (phen)Ru(bpy)2 as the mediator [284]. The system exhibited acceptable regeneration values for NADH.
It should also be noted that semiartificial photocatalysis centers have been also designed to reduce CO2. Sokol et al. generated a sophisticated electrocell in which the photoanode was the photosystem II from Thermosynechococcus elongatus embedded in a redox polymer composed of the complex [Os(bipy)2Cl]Cl bound to a polymer of a derivative of allylamine was, in turn, deposited on TiO2 [285]. Here, light (680 nm) oxidized water to O2, generating electrons that were shuttled towards the cathode, where FDH from Desulfovibrio vulgaris had adhered to TiO2 but, in this case, coated with a fluorine tin oxide. Excellent Faraday efficiencies (>70%) and acceptable yields (0.185 μmol/cm2) were obtained, although the system exhibited the progressive photodegradation of PSII.
Due to its high substrate and product selectivity as a biocatalyst, FDH has been used in recent years to obtain chemical products from reduced forms of CO2, such as formic acid. In addition to its own applicability, this acid is used as a springboard product to obtain a wide variety of derivatives. For example, CO2 can be converted to oxalic acid through the coupling of two formate molecules [286]. This can occur via a metal–formate intermediate (typically sodium or potassium alkaline metals), obtained from the electro- or photocatalytic reduction of CO2 coupled to FDH. After obtaining this metal–formate, two molecules are coupled to give oxalate, which, after acidification, gives rise to oxalic acid. Oxalic acid produced through this coupling route can be used in various industries, including pharmaceuticals, as a component of some antibiotics, or textiles and in other industries such as food (beer or wine production) or chemicals [286]. In addition, from the reduction of this oxalic acid, a wide variety of derivatives are obtained. The first oxalic acid reduction product is glyoxylic acid, which, after reduction, produces glycolic acid and also ethylene glycol. These chemicals are the starting points in the production of agrochemicals, flavors, cosmetics, and polymers [287].
7.5. CO2 Reduction by Whole-Cell Bacteria
Finally, it should be pointed out that recent studies focused on CO2 reduction encompass an even more open overview of what nature provides us in this area. Indeed, whole cells can also be used for fixing CO2, using H2 as the reductant agent [288,289,290], electrochemical reduction [291], or even a combination of H2 and photocatalysis [290]. Whole-cell biocatalysis is a promising method developed in the last few years that shows high efficacy and selectivity and can be used in soft conditions. Moreover, since there is no requirement for purifying enzymes, the most expensive step in biocatalytic processes, the whole-cell approach is a hopeful methodology that will probably have the highest efficiency/cost ratio. It is also relatively easy to adjust the experimental conditions to be developed if the suitable organism or correct molecular biology tools in them are adequately employed. Table 5 reports the most relevant results obtained in the CO2 reduction to formate using the whole-cell approach. The culture of these microorganisms supplied by H2 gas reduces CO2 obtaining extraordinarily high concentrations of formate. Methylobacteria species are satisfactory in this aspect [290].
Leo et al. genetically modified E. coli cells to express the hydrogen-dependent CO2 reductase from Acetobacterium woodie [294]. In this bacterial culture, increasing the cell density up to 30 mg/mL resulted in a formate production yield of 6 mM/h, whereas the addition of 2.5 mM HCO3− increased the formate generation rate fourfold. In this way, the authors confirmed that E. coli CA also played an important role in providing the correct substrate to FDH. Müller’s laboratory took advantage of the metabolic machinery of the acetogenic bacteria Acetobacterium woodii and Thermoanaerobacter kivui to convert H2 and CO2 from syngas into formic acid, obtaining production rates of 234 mmol g−1protein h−1 [288]. E. coli has been also used for formate generation with and without overexpression of exogenous FDH genes. Indeed, as commented previously (see Section 3.2), E. coli has two metal-dependent FDHs capable of reducing CO2 to formate. This bacterium has been used in the whole-cell strategy with extraordinarily high formate generation. Indeed, more than 0.5 molL−1 of formate concentration was achieved when E. coli cells were grown under CO2 and H2 gas at 10 bar pressure for 23 h [293]. E. coli JM109(DE3) strain has also been used as a host bacterium where to overexpress FDHs from Clostridium carboxidivorans, Pyrococcus furiosus (Pf) and Methanobacterium thermosformicicum, also obtaining excellent yields [292]. Indeed, the highest formate generation yield of this study, obtained with PfFDH, was more than 1 gL−1h−1.
Electrochemical reduction is another approach for obtaining high yields of formate within the whole-cell frame. An excellent and very recently released review describes in detail this methodology [291]. Table 5 highlights the remarkable features of the—up to now—few articles published in this area concerning formic acid generation. In these studies, yields are as good as for those using the H2 reduction strategy. For instance, Le et al. achieved to obtain 3.8 mM h−1 g−1wet-cell of formic acid in the cathode electrode at 72 h when Shewanella oneidensis MR-1 was grown in LB media supplemented with nitrate 1 mM and DL-lactate 20 mM [296].
It is also remarkable that two other different approaches to applying whole-cell have been developed. Guntermann and col. devised a two-phase system where Saccharomyces cerevisiae D-glucose fermentation produces ethanol and CO2 in the water phase. This was coupled to a hydrogen gas source that, in the presence of a Ru catalyst solved in the tetradecane phase, generated formic acid at concentrations of 128 mM after 48 h of reaction [300]. On the other hand, light-driven photocatalytic hydrogenation has been applied to a culture of Shewanella oneidensis MR-1 in anaerobic conditions [301]. Using methyl viologen as a photoactivated molecule and triethanolamine (TEOA) as the sacrificial agent, Rowe et al. obtained formic acid at concentrations higher than 1.5 mmol for 48 h of reaction.
Finally, it is worth remarking that the use of microorganisms that incorporate formic acid into their metabolic routes has also been another object of research in the last years. Formatotrophic organisms (capable of assimilating formate for use as a carbon source) offer a new approach to formate utilization, although they are difficult to cultivate, which partially restricts their applicability [302,303]. One strategy is to biotechnologically adapt microorganisms to assimilate formate by adapting their metabolism to the formatotrophic growth model through metabolic engineering tools. Both methanol and formate can be assimilated in the central metabolism through various metabolic pathways, and the bio-production of other compounds such as ethanol, acetone, isopropanol, or short- and medium-chain fatty acids and alcohols from these compounds is very promising [304,305]. To this end, the introduction of pathways, such as the Calvin cycle, the serine cycle, the acetyl-CoA reductive pathway or the glycine pathway in hosts, or the design of new pathways, is proposed [304,305]. The most commonly used hosts are E. coli, S. cerevisiae and Cupriavidus necator, as they are easy to modify with genetic engineering tools and are industrially applicable [306].
Synthetic pathways, such as the reductive glycine pathway (rGly, pathway for formate assimilation), can support higher biological yields than natural pathways and could therefore be implemented in a variety of microorganisms. The rGly pathway has been introduced into the E. coli host by redesigning its central metabolism to be able to assimilate formate. In terms of strategy, the pathway to be integrated is divided into modules and introduced together into the host bacteria to express the enzymes necessary to form the complete metabolic pathway [304,305]. With this methodology, a strain capable of growing from formate with a doubling time of ~70 h and a growth yield of ~1.5 g cell dry weight (gCDW) per mole of formate was achieved [303]. In this way, products such as lactate or isobutanol, both pyruvate derivatives, were obtained. Cotton et al. used C. necator to modify it with rGly, so that it was able to grow under formate-rich conditions. In this respect, the production of methylketones, isoprenoids and terpenes, isobutanol, alkanes, and alkenes from CO2 using C. necator seems particularly promising. With the same microorganism, the Calvin cycle was also used for the assimilation of formate or methanol, although with low energy efficiencies (20–35%) [307]. On the other hand, Collas et al. very recently devised an intelligent approach in which they engineered a crotonate biosynthetic pathway in C. necator [306]. This mechanism permits the conversion of formic acid in crotonate with extraordinary yields in a continuous process. Indeed, using this approach, they obtained 148 mg/L of product. This is a new starting point for the generation of new value-added chemicals.
Although whole-cell technology is still incipient, new opportunities that open a window to capture CO2 under standard conditions, where nature develops, are enormous.
8. Conclusions and Perspectives
A panoramic overview of the state-of-the-art on carbon capture and its reduction in C1 forms by employing the biocatalysts CA and FDH was presented. CO2 uptake is one of the main challenges that science, in general, and chemistry, in particular, face today. Nature offers appropriate tools and clues for finding approaches to solve this huge problem posed to mankind. CCSU techniques are well developed in the laboratory and, to a lesser extent, at a large scale, although their application is still far from being appropriately adapted for obtaining high yields without great energetic costs. While CA immobilization is currently a reality and it stabilizes the enzyme allowing its reuse, the employment of recombinant CAs with higher resistance to temperature and pressure is a field that still remains to be fully developed. These molecular biology tools will help speed up CO2/HCO3− conversion at an industrial scale in the next few years.
Carbon dioxide reduction is also a reality at the laboratory scale, and research lines are well established, although they are still far from showing high yields. The formic acid molecule is a rich form for transporting hydrogen in an efficient way and is the primary step for producing many more reduced molecules in such a way that carbon is recycled and reused as an energy H2 vector. Mass transfer from the gas to the solution is a limiting step in reaching the substrate to the biocatalyst. On the other hand, improvements in the stability and high performance of FDHs, as well as in their stability and reuse via immobilization, have been developed, with a significant explosion of this research in the last decade, despite still being a relatively virgin area. However, its application to larger scales has not been established.
Measures to mitigate climate change are urgently needed. CAs and FDHs are excellent devices for capturing CO2 and transforming it into fuel, which is an interesting way to reduce two problems into one. According to the exponential progress existing in the research in this area nowadays, surprising approaches and solutions to these problems will probably be found not in the next decades but in years.
Funding
This work was partially supported by MICINN-FEDER-AEI 10.13039/501100011033 (PID2021-124695OB-C21/C22 and PDC2022-133313-C21/C22), MICINN—European Union Next Generation EU-PRTR (TED2021-129626B-C21/C22), and SENECA (21884/PI/22) grants.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- National Center for Environmental Information. Annual 2021 Global Climate Report. Available online: https://www.ncei.noaa.gov/access/monitoring/monthly-report/global/202113 (accessed on 24 February 2023).
- Intergovernmental Panel on Climate Change (IPCC). Climate Change 2022, Mitigation of Climate Change. Available online: https://www.ipcc.ch/report/ar6/wg3/downloads/report/IPCC_AR6_WGIII_FullReport.pdf (accessed on 24 February 2023).
- Fellowes, T.E.; Anggadi, F.; Byrne, M.; Vila-Concejo, A.; Bruce, E.; Baker, E. Stability of Coral Reef Islands and Associated Legal Maritime Zones in a Changing Ocean. Environ. Res. Lett. 2022, 17, 093003. [Google Scholar] [CrossRef]
- Borges, F.O.; Sampaio, E.; Santos, C.P.; Rosa, R. Impacts of Low Oxygen on Marine Life: Neglected, but a Crucial Priority for Research. Biol. Bull. 2022, 243, 104–119. [Google Scholar] [CrossRef] [PubMed]
- Arrhenius, S.; Holden, E.S. On the Influence of Carbonic Acid in the Air Upon the Temperature of the Earth. Publ. Astron. Soc. Pac. 1897, 9, 14–24. [Google Scholar] [CrossRef] [Green Version]
- Arrhenius, S. The Celestial Bodies, In Particular the Earth, as Abodes of Living Beings. In Worlds in the Making: The Evolution of the Universe; Harper: New York, NY, USA, 1908; pp. 39–63. [Google Scholar]
- Friedlingstein, P.; Andrew, R.M.; Rogelj, J.; Peters, G.P.; Canadell, J.G.; Knutti, R.; Luderer, G.; Raupach, M.R.; Schaeffer, M.; van Vuuren, D.P.; et al. Persistent Growth of CO2 Emissions and Implications for Reaching Climate Targets. Nat. Geosci. 2014, 7, 709–715. [Google Scholar] [CrossRef]
- Lindsey, R. Climate Change: Atmospheric Carbon Dioxide. Science and Information for a Climate-Smart Nation. Available online: https://www.climate.gov/news-features/understanding-climate/climate-change-atmospheric-carbon-dioxide (accessed on 24 February 2023).
- IPCC. Climate Change 2007: Synthesis Report. Summary for Policymakers. Valencia (Spain) 2007. Available online: https://www.ctc-n.org/resources/ipcc-fourth-assessment-report-climate-change-2007-synthesis-report-summary-policymakers (accessed on 24 February 2023).
- EPA. United States Environmental Protection Agency. Overview of Greenhouse Gases. Available online: https://www.epa.gov/ghgemissions/overview-greenhouse-gases (accessed on 24 February 2023).
- Hoegh-Guldberg, O.; Jacob, D.; Taylor, M.; Bolanos, T.G.; Bindi, M.; Brown, S.; Camilloni, I.A.; Diedhiou, A.; Djalante, R.; Ebi, K.; et al. The Human Imperative of Stabilizing Global Climate Change at 1.5 Degrees C. Science 2019, 365, eaaw6974. [Google Scholar] [CrossRef] [Green Version]
- Rajabloo, T.; Valee, J.; Marenne, Y.; Coppens, L.; De Ceuninck, W. Carbon Capture and Utilization for Industrial Applications. Energy Rep. 2023, 9, 111–116. [Google Scholar] [CrossRef]
- Kamkeng, A.D.N.; Wang, M.; Hu, J.; Du, W.; Qian, F. Transformation technologies for CO2 utilisation: Current status, challenges and future prospects. Chem. Eng. J. 2021, 409, 128138. [Google Scholar] [CrossRef]
- Villa, R.; Porcar, R.; Nieto, S.; Donaire, A.; García-Verdugo, E.; Luis, S.V.; Lozano-Rodríguez, P. Sustainable chemo-enzymatic synthesis of glycerol carbonate (meth)acrylate from glycidol and carbon dioxide enabled by ionic liquid technologies. Green Chem. 2021, 11, 4191–4200. [Google Scholar] [CrossRef]
- Knoche, W. Chemical Reactions of CO2 in Water. In Biophysics and Physiology of Carbon Dioxide, Proceedings in Life Sciences; Bauer, C., Gros, G., Bartels, H., Eds.; Springer: Berlin/Heidelberg, Germany, 1980. [Google Scholar] [CrossRef]
- Greenwood, N.N.; Earnshow, A. Chemistry of the Elements, 2nd ed.; Butterworth-Heinemann, Elsevier: Oxford, UK, 1997. [Google Scholar] [CrossRef]
- Talekar, S.; Jo, B.H.; Dordick, J.S.; Kim, J. Carbonic Anhydrase for CO2 Capture, Conversion and Utilization. Curr. Opin. Biotechnol. 2022, 74, 230–240. [Google Scholar] [CrossRef]
- Lindskog, S. Structure and Mechanism of Carbonic Anhydrase. Pharmacol. Ther. 1997, 74, 1–20. [Google Scholar] [CrossRef]
- De Oliveira Maciel, A.; Christakopoulos, P.; Rova, U.; Antonopoulou, I. Carbonic Anhydrase to Boost CO2 Sequestration: Improving Carbon Capture Utilization and Storage (CCUS). Chemosphere 2022, 299, 134419. [Google Scholar] [CrossRef]
- Yoshimoto, M.; Walde, P. Immobilized Carbonic Anhydrase: Preparation, Characteristics and Biotechnological Applications. World J. Microbiol. Biotechnol. 2018, 34, 151. [Google Scholar] [CrossRef]
- Appel, A.M.; Bercaw, J.E.; Bocarsly, A.B.; Dobbek, H.; DuBois, D.L.; Dupuis, M.; Ferry, J.G.; Fujita, E.; Hille, R.; Kenis, P.J.A.; et al. Frontiers, Opportunities, and Challenges in Biochemical and Chemical Catalysis of CO2 Fixation. Chem. Rev. 2013, 113, 6621–6658. [Google Scholar] [CrossRef] [Green Version]
- Bierbaumer, S.; Nattermann, M.; Schulz, L.; Zschoche, R.; Erb, T.J.; Winkler, C.K.; Tinzl, M.; Glueck, S.M. Enzymatic Conversion of CO2: From Natural to Artificial Utilization. Chem. Rev. 2023, 123, 5702–5754. [Google Scholar] [CrossRef]
- Badger, M.R.; Sharwood, R.E. Rubisco, the Imperfect Winner: It’s All about the Base. J. Exp. Bot. 2023, 74, 562–580. [Google Scholar] [CrossRef]
- Bauwe, H. Photorespiration—Rubisco’s Repair Crew. J. Plant. Physiol. 2023, 280, 153899. [Google Scholar] [CrossRef]
- Alpdagtas, S.; Binay, B. Nadp+-Dependent Formate Dehydrogenase: A Review. Biocatal. Biotransform. 2021, 39, 260–268. [Google Scholar] [CrossRef]
- Alekseeva, A.A.; Savin, S.S.; Tishkov, V.I. NAD+-Dependent Formate Dehydrogenase from Plants. Acta Nat. 2011, 3, 38–54. [Google Scholar] [CrossRef]
- Maia, L.B.; Moura, I.; Moura, J.J.G. Molybdenum and Tungsten-Containing Formate Dehydrogenases: Aiming to Inspire a Catalyst for Carbon Dioxide Utilization. Inorganica Chim. Acta 2017, 455, 350–363. [Google Scholar] [CrossRef]
- Jormakka, M.; Byrne, B.; Iwata, S. Formate Dehydrogenase—A Versatile Enzyme in Changing Environments. Curr. Opin. Struct. Biol. 2003, 13, 418–423. [Google Scholar] [CrossRef]
- Yang, J.Y.; Kerr, T.A.; Wang, X.S.; Barlow, J.M. Reducing CO2 to HCO2- at Mild Potentials: Lessons from Formate Dehydrogenase. J. Am. Chem. Soc. 2020, 142, 19438–19445. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, M.; Zhao, Z.; Liu, W. Effect of Carbonic Anhydrase on Enzymatic Conversion of CO2 to Formic Acid and Optimization of Reaction Conditions. J. Mol. Catal. B Enzym. 2015, 116, 89–94. [Google Scholar] [CrossRef]
- Thauer, R.K.; Jungermann, K.; Decker, K. Energy-Conservation in Chemotropic Anaerobic Bacteria. Bacteriol. Rev. 1977, 41, 100–180. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhao, Y.; Li, R.; Liu, J. Bioinspired NADH Regeneration Based on Conjugated Photocatalytic Systems. Solar RRL 2021, 5, 2000339. [Google Scholar] [CrossRef]
- Lee, S.H.; Choi, D.S.; Kuk, S.K.; Park, C.B. Photobiocatalysis: Activating Redox Enzymes by Direct or Indirect Transfer of Photoinduced Electrons. Angew. Chem. Int. Ed. 2018, 57, 7958–7985. [Google Scholar] [CrossRef]
- Supuran, C.T.; Capasso, C. An Overview of the Bacterial Carbonic Anhydrases. Metabolites 2017, 7, 56. [Google Scholar] [CrossRef] [Green Version]
- DiMario, R.J.; Machingura, M.C.; Waldrop, G.L.; Moroney, J.V. The Many Types of Carbonic Anhydrases in Photosynthetic Organisms. Plant Sci. 2018, 268, 11–17. [Google Scholar] [CrossRef]
- Kupriyanova, E.; Pronina, N.; Los, D. Carbonic Anhydrase—A Universal Enzyme of the Carbon-Based Life. Photosynthetica 2017, 55, 3–19. [Google Scholar] [CrossRef]
- Zimmerman, S.A.; Ferry, J.G. The Beta and Gamma Classes of Carbonic Anhydrase. Curr. Pharm. Des. 2008, 14, 716–721. [Google Scholar] [CrossRef]
- McGinn, P.J.; Morel, F.M.M. Expression and Regulation of Carbonic Anhydrases in the Marine Diatom Thalassiosira pseudonana and in Natural Phytoplankton Assemblages from Great Bay, New Jersey. Physiol. Plant. 2008, 133, 78–91. [Google Scholar] [CrossRef]
- Roberts, S.B.; Lane, T.W.; Morel, F.M.M. Carbonic Anhydrase in the Marine Diatom Thalassiosira weissflogii (Bacillariophyceae). J. Phycol. 1997, 33, 845–850. [Google Scholar] [CrossRef]
- Beauchemin, M.; Morse, D. δ-Carbonic Anhydrases: Structure, Distribution, and Potential Roles. In Carbonic Anhydrases as Biocatalysts: From Theory to Medical and Industrial Applications; Université de Montréal: Montréal, QC, Canada, 2015; pp. 337–349. [Google Scholar] [CrossRef]
- Dou, Z.; Heinhorst, S.; Williams, E.B.; Murin, C.D.; Shively, J.M.; Cannon, G.C. CO2 Fixation Kinetics of Halothiobacillus neapolitanus Mutant Carboxysomes Lacking Carbonic Anhydrase Suggest the Shell Acts as a Diffusional Barrier for CO2. J. Biol. Chem. 2008, 283, 10377–10384. [Google Scholar] [CrossRef] [Green Version]
- Alterio, V.; Langella, E.; Buonanno, M.; Esposito, D.; Nocentini, A.; Berrino, E.; Bua, S.; Polentarutti, M.; Supuran, C.T.; Monti, S.M.; et al. Zeta-Carbonic Anhydrases Show CS2 Hydrolase Activity: A New Metabolic Carbon Acquisition Pathway in Diatoms? Comput. Struct. Biotechnol. J. 2021, 19, 3427–3436. [Google Scholar] [CrossRef]
- Del Prete, S.; Vullo, D.; Fisher, G.M.; Andrews, K.T.; Poulsen, S.-A.; Capasso, C.; Supuran, C.T. Discovery of a New Family of Carbonic Anhydrases in the Malaria Pathogen Plasmodium falciparum—The η-Carbonic Anhydrases. Bioorg. Med. Chem. Lett. 2014, 24, 4389–4396. [Google Scholar] [CrossRef] [Green Version]
- Nawaly, H.; Tanaka, A.; Toyoshima, Y.; Tsuji, Y.; Matsuda, Y. Localization and Characterization Theta Carbonic Anhydrases in Thalassiosira pseudonana. Photosynth. Res. 2023, 156, 217–229. [Google Scholar] [CrossRef]
- Nocentini, A.; Supuran, C.T.; Capasso, C. An Overview on the Recently Discovered Iota-Carbonic Anhydrases. J. Enzym. Inhib. Med. Chem. 2021, 36, 1988–1995. [Google Scholar] [CrossRef]
- Del Prete, S.; Nocentini, A.; Supuran, C.T.; Capasso, C. Bacterial Iota-Carbonic Anhydrase: A New Active Class of Carbonic Anhydrase Identified in the Genome of the Gram-Negative Bacterium Burkholderia Territorii. J. Enzym. Inhib. Med. Chem. 2020, 35, 1060–1068. [Google Scholar] [CrossRef] [Green Version]
- Kikutani, S.; Nakajima, K.; Nagasato, C.; Tsuji, Y.; Miyatake, A.; Matsuda, Y. Thylakoid Luminal Theta-Carbonic Anhydrase Critical for Growth and Photosynthesis in the Marine Diatom Phaeodactylum Tricornutum. Proc. Natl. Acad. Sci. USA 2016, 113, 9828–9833. [Google Scholar] [CrossRef]
- Alterio, V.; Langella, E.; De Simone, G.; Monti, S.M. Cadmium-Containing Carbonic Anhydrase CDCA1 in Marine Diatom Thalassiosira weissflogii. Mar. Drugs 2015, 13, 1688–1697. [Google Scholar] [CrossRef] [Green Version]
- Angeli, A.; Buonanno, M.; Donald, W.A.; Monti, S.M.; Supuran, C.T. The Zinc–but Not Cadmium–Containing ζ-Carbonic from the Diatom Thalassiosira weissflogii Is Potently Activated by Amines and Amino Acids. Bioorg. Chem. 2018, 80, 261–265. [Google Scholar] [CrossRef]
- Nathan, V.K.; Ammini, P. Carbon Dioxide Sequestering Ability of Bacterial Carbonic Anhydrase in a Mangrove Soil Microcosm and Its Bio-Mineralization Properties. Water Air Soil Pollut. 2019, 230, 192. [Google Scholar] [CrossRef]
- Banci, L.; Bertini, I.; Luchinat, C.; Donaire, A.; Martinez, M.-J. The Factors Governing the Coordination Number in the Anion Derivatives of Carbonic Anhydrase. Comments Inorg. Chem. 1990, 9, 245–261. [Google Scholar] [CrossRef]
- Hirakawa, Y.; Senda, M.; Fukuda, K.; Yu, H.Y.; Ishida, M.; Taira, M.; Kinbara, K.; Senda, T. Characterization of a Novel Type of Carbonic Anhydrase That Acts without Metal Cofactors. BMC Biol. 2021, 19, 105. [Google Scholar] [CrossRef] [PubMed]
- Jo, B.H.; Kim, I.G.; Seo, J.H.; Kang, D.G.; Cha, H.J. Engineered Escherichia coli with Periplasmic Carbonic Anhydrase as a Biocatalyst for CO2 Sequestration. Appl. Environ. Microbiol. 2013, 79, 6697–6705. [Google Scholar] [CrossRef] [Green Version]
- Ekinci, D.; Beydemir, S.; Alim, Z. Some Drugs Inhibit in vitro Hydratase and Esterase Activities of Human Carbonic Anhydrase-I and II. Pharmacol. Rep. 2007, 59, 580–587. [Google Scholar] [PubMed]
- Steger, F.; Reich, J.; Fuchs, W.; Rittmann, S.K.-M.R.; Gübitz, G.M.; Ribitsch, D.; Bochmann, G. Comparison of Carbonic Anhydrases for CO2 Sequestration. Int. J. Mol. Sci. 2022, 23, 957. [Google Scholar] [CrossRef]
- Faridi, S.; Satyanarayana, T. Novel Alkalistable α-Carbonic Anhydrase from the Polyextremophilic Bacterium Bacillus Halodurans: Characteristics and Applicability in Flue Gas CO2 Sequestration. Environ. Sci. Pollut. Res. 2016, 23, 15236–15249. [Google Scholar] [CrossRef]
- Capasso, C.; De Luca, V.; Carginale, V.; Cannio, R.; Rossi, M. Biochemical Properties of a Novel and Highly Thermostable Bacterial Alpha-Carbonic Anhydrase from Sulfurihydrogenibium yellowstonense YO3AOP1. J. Enzym. Inhib. Med. Chem. 2012, 27, 892–897. [Google Scholar] [CrossRef] [Green Version]
- Ramanan, R.; Kannan, K.; Vinayagamoorthy, N.; Ramkumar, K.M.; Sivanesan, S.D.; Chakrabarti, T. Purification and Characterization of a Novel Plant-Type Carbonic Anhydrase from Bacillus subtilis. Biotechnol. Bioprocess Eng. 2009, 14, 32–37. [Google Scholar] [CrossRef]
- Kim, S.S.; Kim, N.J.; Hong, S.; Kim, S.S.; Sung, J.; Jin, M.S. The Structural Basis of the Low Catalytic Activities of the Two Minor Beta-Carbonic Anhydrases of the Filamentous Fungus Aspergillus fumigatus. J. Struct. Biol. 2019, 208, 61–68. [Google Scholar] [CrossRef]
- Sridharan, U.; Ragunathan, P.; Kuramitsu, S.; Yokoyama, S.; Kumarevel, T.; Ponnuraj, K. Structural and Functional Characterization of a Putative Carbonic Anhydrase from Geobacillus kaustophilus Reveals Its Cambialistic Function. Biochem. Biophys. Res. Commun. 2021, 547, 96–101. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, Y.; Wang, L.; Jing, Q.; Wang, X.; Xi, X.; Zhao, X.; Wang, H. Molecular Structure of Thermostable and Zinc-Ion-Binding Gamma-Class Carbonic Anhydrases. Biometals 2019, 32, 317–328. [Google Scholar] [CrossRef]
- Alber, B.E.; Ferry, J.G. A Carbonic Anhydrase from the Archaeon Methanosarcina thermophila. Proc. Natl. Acad. Sci. USA 1994, 91, 6909–6913. [Google Scholar] [CrossRef]
- Del Prete, S.; Vullo, D.; Di Fonzo, P.; Osman, S.M.; AlOthman, Z.; Donald, W.A.; Supuran, C.T.; Capasso, C. Sulfonamide Inhibition Profile of the Gamma-Carbonic Anhydrase Identified in the Genome of the Pathogenic Bacterium Burkholderia pseudomallei the Etiological Agent Responsible of Melioidosis. Bioorg. Med. Chem. Lett. 2017, 27, 490–495. [Google Scholar] [CrossRef] [Green Version]
- Del Prete, S.; Vullo, D.; De Luca, V.; Supuran, C.T.; Capasso, C. Biochemical Characterization of the Delta-Carbonic Anhydrase from the Marine Diatom Thalassiosira weissflogii, TweCA. J. Enzym. Inhib. Med. Chem. 2014, 29, 906–911. [Google Scholar] [CrossRef]
- Zhang, H.; Blanco-Ameijeiras, S.; Hopkinson, B.M.; Bernasconi, S.M.; Mejia, L.M.; Liu, C.; Stoll, H. An Isotope Label Method for Empirical Detection of Carbonic Anhydrase in the Calcification Pathway of the Coccolithophore Emiliania huxleyi. Geochim. Cosmochim. Acta 2021, 292, 78–93. [Google Scholar] [CrossRef]
- Xu, Y.; Feng, L.; Jeffrey, P.D.; Shi, Y.; Morel, F.M.M. Structure and Metal Exchange in the Cadmium Carbonic Anhydrase of Marine Diatoms. Nature 2008, 452, 56–61. [Google Scholar] [CrossRef]
- Supuran, C.T.; Capasso, C. The η-Class Carbonic Anhydrases as Drug Targets for Antimalarial Agents. Expert Opin. Ther. Targets 2015, 19, 551–563. [Google Scholar] [CrossRef]
- Kitahara, M.; Fudo, S.; Yoneda, T.; Nukaga, M.; Hoshino, T. Anisotropic Distribution of Ammonium Sulfate Ions in Protein Crystallization. Cryst. Growth Des. 2019, 19, 6004–6010. [Google Scholar] [CrossRef]
- Woods, L.A.; Dolezal, O.; Ren, B.; Ryan, J.H.; Peat, T.S.; Poulsen, S.-A. Native State Mass Spectrometry, Surface Plasmon Resonance, and X-Ray Crystallography Correlate Strongly as a Fragment Screening Combination. J. Med. Chem. 2016, 59, 2192–2204. [Google Scholar] [CrossRef]
- Avvaru, B.S.; Busby, S.A.; Chalmers, M.J.; Griffin, P.R.; Venkatakrishnan, B.; Agbandje-McKenna, M.; Silverman, D.N.; McKenna, R. Apo-Human Carbonic Anhydrase II Revisited: Implications of the Loss of a Metal in Protein Structure, Stability, and Solvent Network. Biochemistry 2009, 48, 7365–7372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergenhem, N.C.; Hallberg, M.; Wisén, S. Molecular Characterization of the Human Carbonic Anhydrase-Related Protein (HCA-RP VIII). Biochim. Biophys. Acta 1998, 1384, 294–298. [Google Scholar] [CrossRef] [PubMed]
- Picaud, S.S.; Muniz, J.R.; Kramm, A.; Pilka, E.S.; Kochan, G.; Oppermann, U.; Yue, W.W. Crystal Structure of Human Carbonic Anhydrase-Related Protein VIII Reveals the Basis for Catalytic Silencing. Proteins 2009, 76, 507–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lakkis, M.M.; Bergenhem, N.C.; O’Shea, K.S.; Tashian, R.E. Expression of the Acatalytic Carbonic Anhydrase VIII Gene, Car8, during Mouse Embryonic Development. Histochem. J. 1997, 29, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Hirota, J.; Ando, H.; Hamada, K.; Mikoshiba, K. Carbonic Anhydrase-Related Protein is a Novel Binding Protein for Inositol 1,4,5-Trisphosphate Receptor Ttype 1. Biochem. J. 2003, 372, 435–441. [Google Scholar] [CrossRef] [Green Version]
- Borén, K.; Andersson, P.; Larsson, M.; Carlsson, U. Characterization of a Molten Globule State of Bovine Carbonic Anhydrase III: Loss of Asymmetrical Environment of the Aromatic Residues Has a Profound Effect on Both the Near- and Far-UV CD Spectrum. Biochim. Biophys. Acta 1999, 1430, 111–118. [Google Scholar] [CrossRef]
- Capasso, C.; Supuran, C.T. Inhibition of Bacterial Carbonic Anhydrases as a Novel Approach to Escape Drug Resistance. Curr. Top. Med. Chem. 2017, 17, 1237–1248. [Google Scholar] [CrossRef] [Green Version]
- Wilbur, K.M.; Anderson, N.G. Electrometric and Colorimetric Determination of Carbonic Anhydrase. J. Biol. Chem. 1948, 176, 147–154. [Google Scholar] [CrossRef]
- Fuchs, W.; Steger, F.; Reich, J.; Ribitsch, D.; Rittmann, S.K.-M.R.; Bochmann, G. A Simple and Straightforward Method for Activity Measurement of Carbonic Anhydrases. Catalysis 2021, 11, 819. [Google Scholar] [CrossRef]
- Verpoorte, J.A.; Mehta, S.; Edsall, J.T. Esterase Activities of Human Carbonic Anhydrases B and C. J. Biol. Chem. 1967, 242, 4221–4229. [Google Scholar] [CrossRef]
- Effendi, S.S.W.; Ng, I.-S. The Prospective and Potential of Carbonic Anhydrase for Carbon Dioxide Sequestration: A Critical Review. Process Biochem. 2019, 87, 55–65. [Google Scholar] [CrossRef]
- Supuran, C.T.; De Simone, G. Carbonic Anhydrases: An Overview. In Carbonic Anhydrases as Biocatalysts: From Theory to Medical and Industrial Applications; Elsevier: Florence, Italy, 2015; pp. 3–13. [Google Scholar] [CrossRef]
- Niks, D.; Hille, R. Reductive Activation of CO2 by Formate Dehydrogenases. In Enzymes of Energy Technology; Methods in Enzymology; Armstrong, F., Ed.; Elsevier Acad. Press. Inc.: San Diego, CA, USA, 2018; Volume 613, pp. 277–295. [Google Scholar] [CrossRef]
- Nielsen, C.F.; Lange, L.; Meyer, A.S. Classification and Enzyme Kinetics of Formate Dehydrogenases for Biomanufacturing via CO2 Utilization. Biotechnol. Adv. 2019, 37, 107408. [Google Scholar] [CrossRef] [PubMed]
- Maia, L.B.; Moura, I.; Moura, J.J.G. Molybdenum and Tungsten-Containing Enzymes: An Overview. In Molibdenum and Tungsten Enzymes: Biochemistry; Hille, R., Schulzke, C., Kirk, M.L., Eds.; RSC Metallobiology Series; Royal Society of Chemistry: Cambridge, UK, 2017; Volume 5, pp. 1–80. [Google Scholar]
- Lemaire, O.N.; Jespersen, M.; Wagner, T. CO2-Fixation Strategies in Energy Extremophiles: What Can We Learn From Acetogens? Front. Microbiol. 2020, 11, 486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moon, M.; Park, G.W.; Lee, J.; Lee, J.-S.; Min, K. Recent Progress in Formate Dehydrogenase (FDH) as a Non-Photosynthetic CO2 Utilizing Enzyme: A Short Review. J. CO2 Util. 2020, 42, 101353. [Google Scholar] [CrossRef]
- Alpdağtaş, S.; Turunen, O.; Valjakka, J.; Binay, B. The Challenges of Using NAD+-Dependent Formate Dehydrogenases for CO2 Conversion. Crit. Rev. Biotechnol. 2022, 42, 953–972. [Google Scholar] [CrossRef]
- Sakai, Y.; Murdanoto, A.P.; Konishi, T.; Iwamatsu, A.; Kato, N. Regulation of the Formate Dehydrogenase Gene, FDH1, in the Methylotrophic Yeast Candida boidinii and Growth Characteristics of an FDH1-Disrupted Strain on Methanol, Methylamine, and Choline. J. Bacteriol. 1997, 179, 4480–4485. [Google Scholar] [CrossRef] [Green Version]
- Tishkov, V.I.; Popov, V.O. Protein Engineering of Formate Dehydrogenase. Biomol. Eng. 2006, 23, 89–110. [Google Scholar] [CrossRef]
- Pagano, P.; Guo, Q.; Ranasinghe, C.; Schroeder, E.; Robben, K.; Hase, F.; Ye, H.; Wickersham, K.; Aspuru-Guzik, A.; Major, D.T.; et al. Oscillatory Active-Site Motions Correlate with Kinetic Isotope Effects in Formate Dehydrogenase. ACS Catal. 2019, 9, 11199–11206. [Google Scholar] [CrossRef]
- Yilmazer, B.; Isupov, M.N.; De Rose, S.A.; Bulut, H.; Benninghoff, J.C.; Binay, B.; Littlechild, J.A. Structural insights into the NAD+-dependent Formate Dehydrogenase Mechanism Revealed from the NADH Complex and the Formate NAD+-Ternary Complex of the Chaetomium thermophilum Enzyme J. Struct. Biol. 2020, 212, 107657. [Google Scholar] [CrossRef]
- Pala, U.; Yelmazer, B.; Corbacioglu, M.; Ruupunen, J.; Valjakka, J.; Turunen, O.; Binay, B. Functional Effects of Active Site Mutations in NAD+-Dependent Formate Dehydrogenases on Transformation of Hydrogen Carbonate to Formate. Protein Eng. Des. Sel. 2018, 31, 327–335. [Google Scholar] [CrossRef]
- Cakar, M.M.; Ruupunen, J.; Mangas-Sanchez, J.; Birmingham, W.R.; Yildirim, D.; Turunen, O.; Turner, N.J.; Valjakka, J.; Binay, B. Engineered Formate Dehydrogenase from Chaetomium Thermophilum, a Promising Enzymatic Solution for Biotechnical CO2 Fixation. Biotechnol. Lett. 2020, 42, 2251–2262. [Google Scholar] [CrossRef]
- Guo, Q.; Gakhar, L.; Wickersham, K.; Francis, K.; Vardi-Kilshtain, A.; Major, D.T.; Cheatum, C.M.; Kohen, A. Structural and Kinetic Studies of Formate Dehydrogenase from Candida boidinii. Biochemistry 2016, 55, 2760–2771. [Google Scholar] [CrossRef] [Green Version]
- Sato, R.; Amao, Y. Studies on the Catalytic Mechanism of Formate Dehydrogenase from Candida boidinii Using Isotope-Labelled Substrate and Co-Enzyme. Catal. Today 2023, 411–412, 113796. [Google Scholar] [CrossRef]
- Jiang, W.; Lin, P.; Yang, R.; Fang, B. Identification of Catalysis, Substrate, and Coenzyme Binding Sites and Improvement Catalytic Efficiency of Formate Dehydrogenase from Candida boidinii. Appl. Microbiol. Biotechnol. 2016, 100, 8425–8437. [Google Scholar] [CrossRef]
- Altas, N.; Aslan, A.S.; Karatas, E.; Chronopoulou, E.; Labrou, N.E.; Binay, B. Heterologous Production of Extreme Alkaline Thermostable NAD+-Dependent Formate Dehydrogenase with Wide-Range pH Activity from Myceliophthora thermophila. Process Biochem. 2017, 61, 110–118. [Google Scholar] [CrossRef]
- Choe, H.; Joo, J.C.; Cho, D.H.; Kim, M.H.; Lee, S.H.; Jung, K.D.; Kim, Y.H. Efficient CO2-Reducing Activity of NAD-Dependent Formate Dehydrogenase from Thiobacillus sp. KNK65MA for Formate Production from CO2 Gas. PLoS ONE 2014, 9, e103111. [Google Scholar] [CrossRef]
- Aslan, A.S.; Valjakka, J.; Ruupunen, J.; Yildirim, D.; Turner, N.J.; Turunen, O.; Binay, B. Chaetomium thermophilum Formate Dehydrogenase Has High Activity in the Reduction of Hydrogen Carbonate (HCO3−) to Formate. Protein Eng. Des. Sel. 2017, 30, 47–55. [Google Scholar] [CrossRef]
- de Bok, F.A.M.; Hagedoorn, P.L.; Silva, P.J.; Hagen, W.R.; Schiltz, E.; Fritsche, K.; Stams, A.J.M. Two W-Containing Formate Dehydrogenases (CO2-Reductases) Involved in Syntrophic Propionate Oxidation by Syntrophobacter fumaroxidans. Eur. J. Biochem. 2003, 270, 2476–2485. [Google Scholar] [CrossRef]
- Maia, L.B.; Fonseca, L.; Moura, I.; Moura, J.J.G. Reduction of Carbon Dioxide by a Molybdenum-Containing Formate Dehydrogenase: A Kinetic and Mechanistic Study. J. Am. Chem. Soc. 2016, 138, 8834–8846. [Google Scholar] [CrossRef]
- Bassegoda, A.; Madden, C.; Wakerley, D.W.; Reisner, E.; Hirst, J. Reversible Interconversion of CO2 and Formate by a Molybdenum-Containing Formate Dehydrogenase. J. Am. Chem. Soc. 2014, 136, 15473–15476. [Google Scholar] [CrossRef] [Green Version]
- Axley, M.J.; Grahame, D.A.; Stadtman, T.C. Escherichia coli Formate-Hydrogen Lyase. Purification and Properties of the Selenium-Dependent Formate Dehydrogenase Component. J. Biol. Chem. 1990, 265, 18213–18218. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, A.R.; Mota, C.; Mourato, C.; Domingos, R.M.; Santos, M.F.A.; Gesto, D.; Guigliarelli, B.; Santos-Silva, T.; Romao, M.J.; Cardoso Pereira, I.A. Toward the Mechanistic Understanding of Enzymatic CO2 Reduction. ACS Catal. 2020, 10, 3844–3856. [Google Scholar] [CrossRef]
- da Silva, S.M.; Pimentel, C.; Valente, F.M.A.; Rodrigues-Pousada, C.; Pereira, I.A.C. Tungsten and Molybdenum Regulation of Formate Dehydrogenase Expression in Desulfovibrio Vulgaris Hildenborough. J. Bacteriol. 2011, 193, 2909–2916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuchmann, K.; Mueller, V. Direct and Reversible Hydrogenation of CO2 to Formate by a Bacterial Carbon Dioxide Reductase. Science 2013, 342, 1382–1385. [Google Scholar] [CrossRef]
- Yu, X.; Niks, D.; Mulchandani, A.; Hille, R. Efficient Reduction of CO2 by the Molybdenum-Containing Formate Dehydrogenase from Cupriavidus necator (Ralstonia eutropha). J. Biol. Chem. 2017, 292, 16872–16879. [Google Scholar] [CrossRef] [Green Version]
- Radon, C.; Mittelstadt, G.; Duffus, B.R.; Burger, J.; Hartmann, T.; Mielke, T.; Teutloff, C.; Leimkuhler, S.; Wendler, P. Cryo-EM Structures Reveal Intricate Fe-S Cluster Arrangement and Charging in Rhodobacter capsulatus Formate Dehydrogenase. Nat. Commun. 2020, 11, 1912. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, T.; Leimkuehler, S. The Oxygen-Tolerant and NAD+-Dependent Formate Dehydrogenase from Rhodobacter capsulatus Is Able to Catalyze the Reduction of CO2 to Formate. FEBS J. 2013, 280, 6083–6096. [Google Scholar] [CrossRef]
- Ruschig, U.; Muller, U.; Willnow, P.; Hopner, T. CO2 Recution to Formate by NADH Catalyzed by Formate Dehydrogenase from Pseudomonas oxalaticus. Eur. J. Biochem. 1976, 70, 325–330. [Google Scholar] [CrossRef]
- Müller, U.; Willnow, P.; Ruschig, U.; Höpner, T. Formate Dehydrogenase from Pseudomonas oxalaticus. Eur. J. Biochem. 1978, 83, 485–498. [Google Scholar] [CrossRef]
- Cakar, M.M.; Mangas-Sanchez, J.; Birmingham, W.R.; Turner, N.J.; Binay, B. Discovery of a New Metal and NAD+-Dependent Formate Dehydrogenase from Clostridium ljungdahlii. Prep. Biochem. Biotechnol. 2018, 48, 327–334. [Google Scholar] [CrossRef]
- Min, K.; Moon, M.; Park, G.W.; Lee, J.-P.; Kim, S.J.; Lee, J.-S. Newly Explored Formate Dehydrogenases from Clostridium Species Catalyze Carbon Dioxide to Formate. Bioresour. Technol. 2022, 348, 126832. [Google Scholar] [CrossRef]
- Almendra, M.J.; Brondino, C.D.; Gavel, O.; Pereira, A.S.; Tavares, P.; Bursakov, S.; Duarte, R.; Caldeira, J.; Moura, J.J.G.; Moura, I. Purification and Characterization of a Tungsten-Containing Formate Dehydrogenase from Desulfovibrio gigas. Biochemistry 1999, 38, 16366–16372. [Google Scholar] [CrossRef]
- Yamamoto, I.; Saiki, T.; Liu, S.M.; Ljundgdahl, L.G. Purification and Properties of NADP-Dependent Formate Dehydrogenase from Clostridium thermoaceticum, a Tungsten Selenium Iron Protein. J. Biol. Chem. 1983, 258, 1826–1832. [Google Scholar] [CrossRef]
- Cordas, C.M.; Moura, J.J.G. Molybdenum and Tungsten Enzymes Redox Properties—A Brief Overview. Coord. Chem. Rev. 2019, 394, 53–64. [Google Scholar] [CrossRef]
- Hartmann, T.; Schwanhold, N.; Leimkuehler, S. Assembly and Catalysis of Molybdenum or Tungsten-Containing Formate Dehydrogenases from Bacteria. Biochim. Biophys. Acta Proteins Proteom. 2015, 1854, 1090–1100. [Google Scholar] [CrossRef]
- Kirk, M.L.; Hille, R. Spectroscopic Studies of Mononuclear Molybdenum Enzyme Centers. Molecules 2022, 27, 4802. [Google Scholar] [CrossRef]
- Jormakka, M.; Tornroth, S.; Byrne, B.; Iwata, S. Molecular Basis of Proton Motive Force Generation: Structure of Formate Dehydrogenase-N. Science 2002, 295, 1863–1868. [Google Scholar] [CrossRef] [Green Version]
- Niks, D.; Hille, R. Molybdenum- and Tungsten-Containing Formate Dehydrogenases and Formylmethanofuran Dehydrogenases: Structure, Mechanism, and Cofactor Insertion. Protein Sci. 2019, 28, 111–122. [Google Scholar] [CrossRef] [Green Version]
- Robinson, W.E.; Bassegoda, A.; Reisner, E.; Hirst, J. Oxidation-State-Dependent Binding Properties of the Active Site in a Mo-Containing Formate Dehydrogenase. J. Am. Chem. Soc. 2017, 139, 9927–9936. [Google Scholar] [CrossRef] [Green Version]
- Meneghello, M.; Oliveira, A.R.; Jacq-Bailly, A.; Pereira, I.A.C.; Leger, C.; Fourmond, V. Formate Dehydrogenases Reduce CO2 Rather than HCO3−: An Electrochemical Demonstration. Angew. Chem. Int. Ed. 2021, 60, 9964–9967. [Google Scholar] [CrossRef]
- Tiberti, M.; Papaleo, E.; Russo, N.; De Gioia, L.; Zampella, G. Evidence for the Formation of a Mo-H Intermediate in the Catalytic Cycle of Formate Dehydrogenase. Inorg. Chem. 2012, 51, 8331–8339. [Google Scholar] [CrossRef]
- Khan, M.R. Immobilized Enzymes: A Comprehensive Review. Bull. Natl. Res. Cent. 2021, 45, 207. [Google Scholar] [CrossRef]
- Bie, J.; Sepodes, B.; Fernandes, P.C.B.; Ribeiro, M.H.L. Enzyme Immobilization and Co-Immobilization: Main Framework, Advances and Some Applications. Processes 2022, 10, 494. [Google Scholar] [CrossRef]
- Porcar, R.; Lavandera, I.; Lozano, P.; Altava, B.; Luis, S.V.; Gotor-Fernandez, V.; Garcia-Verdugo, E. Supported Ionic Liquid-like Phases as Efficient Solid Ionic Solvents for the Immobilisation of Alcohol Dehydrogenases towards the Development of Stereoselective Bioreductions. Green Chem. 2021, 23, 5609–5617. [Google Scholar] [CrossRef]
- Sheldon, R.A.; Basso, A.; Brady, D. New Frontiers in Enzyme Immobilisation: Robust Biocatalysts for a Circular Bio-Based Economy. Chem. Soc. Rev. 2021, 50, 5850–5862. [Google Scholar] [CrossRef] [PubMed]
- Lozano, P.; Garcia-Verdugo, E.; Bernal, J.M.; Izquierdo, D.F.; Isabel Burguete, M.; Sanchez-Gomez, G.; Luis, S.V. Immobilised Lipase on Structured Supports Containing Covalently Attached Ionic Liquids for the Continuous Synthesis of Biodiesel in ScCO2. ChemSusChem 2012, 5, 790–798. [Google Scholar] [CrossRef]
- Ren, S.; Jiang, S.; Yan, X.; Chen, R.; Cui, H. Challenges and Opportunities: Porous Supports in Carbonic Anhydrase Immobilization. J. CO2 Util. 2020, 42, 101305. [Google Scholar] [CrossRef]
- Molina-Fernandez, C.; Luis, P. Immobilization of Carbonic Anhydrase for CO2 Capture and Its Industrial Implementation: A Review. J. CO2 Util. 2021, 47, 101475. [Google Scholar] [CrossRef]
- Rasouli, H.; Nguyen, K.; Iliuta, M.C. Recent Advancements in Carbonic Anhydrase Immobilization and Its Implementation in CO2 Capture Technologies: A Review. Sep. Purif. Technol. 2022, 296, 121299. [Google Scholar] [CrossRef]
- Yuan, Y.; Wang, F.; Li, H.; Su, S.; Gao, H.; Han, X.; Ren, S. Potential Application of the Immobilization of Carbonic Anhydrase Based on Metal Organic Framework Supports. Process Biochem. 2022, 122, 214–223. [Google Scholar] [CrossRef]
- Molina-Fernandez, C.; Peters, A.; Debecker, D.P.; Luis, P. Immobilization of Carbonic Anhydrase in a Hydrophobic Poly(Ionic Liquid): A New Functional Solid for CO2 Capture. Biochem. Eng. J. 2022, 187, 108639. [Google Scholar] [CrossRef]
- Rouf, S.; Greish, Y.E.; Al-Zuhair, S. Immobilization of Formate Dehydrogenase in Metal Organic Frameworks for Enhanced Conversion of Carbon Dioxide to Formate. Chemosphere 2021, 267, 128921. [Google Scholar] [CrossRef]
- Singh, R.K.R.; Tiwari, M.K.; Singh, R.K.R.; Lee, J.-K. From Protein Engineering to Immobilization: Promising Strategies for the Upgrade of Industrial Enzymes. Molecules 2013, 18, 1232–1277. [Google Scholar] [CrossRef] [Green Version]
- Di Spiridione, C.; Aresta, M.; Dibenedetto, A. Improving the Enzymatic Cascade of Reactions for the Reduction of CO2 to CH3OH in Water: From Enzymes Immobilization Strategies to Cofactor Regeneration and Cofactor Suppression. Molecules 2022, 27, 4913. [Google Scholar] [CrossRef]
- Zhu, X.; Du, C.; Gao, B.; He, B. Strategies to Improve the Mass Transfer in the CO2 Capture Process Using Immobilized Carbonic Anhydrase. J. Environ. Manag. 2023, 332, 117370. [Google Scholar] [CrossRef]
- Ren, S.; Chen, R.; Wu, Z.; Su, S.; Hou, J.; Yuan, Y. Enzymatic Characteristics of Immobilized Carbonic Anhydrase and Its Applications in CO2 Conversion. Colloid. Surface B 2021, 204, 111779. [Google Scholar] [CrossRef]
- Shen, J.; Salmon, S. Biocatalytic Membranes for Carbon Capture and Utilization. Membranes 2023, 13, 367. [Google Scholar] [CrossRef] [PubMed]
- Wanjari, S.; Prabhu, C.; Satyanarayana, T.; Vinu, A.; Rayalu, S. Immobilization of Carbonic Anhydrase on Mesoporous Aluminosilicate for Carbonation Reaction. Micropor. Mesopor. Mat. 2012, 160, 151–158. [Google Scholar] [CrossRef]
- Yu, Y.; Chen, B.; Qi, W.; Li, X.; Shin, Y.; Lei, C.; Liu, J. Enzymatic Conversion of CO2 to Bicarbonate in Functionalized Mesoporous Silica. Micropor. Mesopor. Mat. 2012, 153, 166–170. [Google Scholar] [CrossRef] [Green Version]
- Vinoba, M.; Bhagiyalakshmi, M.; Jeong, S.K.; Yoon, Y.I.; Nam, S.C. Carbonic Anhydrase Conjugated to Nanosilver Immobilized onto Mesoporous SBA-15 for Sequestration of CO2. J. Mol. Catal. B Enzym. 2012, 75, 60–67. [Google Scholar] [CrossRef]
- Mao, M.; Zhai, T.; Meng, L.; Meng, Z.; Liu, W. Controllable Preparation of Mesoporous Silica and Its Application in Enzyme-Catalyzed CO2 Reduction. Chem. Eng. J. 2022, 437, 135479. [Google Scholar] [CrossRef]
- Azari, F.N.-G.M. Reversible Denaturation of Carbonic Anhydrase Provides a Method for Its Adsorptive Immobilization. Biotechnol. Bioeng. 1999, 62, 193–199. [Google Scholar] [CrossRef]
- Kim, J.K.; Abdelhamid, M.A.A.; Pack, S.P. Direct Immobilization and Recovery of Recombinant Proteins from Cell Lysates by Using EctP1-Peptide as a Short Fusion Tag for Silica and Titania Supports. Int. J. Biol. Macromol. 2019, 135, 969–977. [Google Scholar] [CrossRef]
- Yong, J.K.J.; Stevens, G.W.; Caruso, F.; Kentish, S.E. In Situ Layer-by-Layer Assembled Carbonic Anhydrase-Coated Hollow Fiber Membrane Contactor for Rapid CO2 Absorption. J. Memb. Sci. 2016, 514, 556–565. [Google Scholar] [CrossRef]
- Ivanovski, V.; Shapovalova, O.E.; Drozdov, A.S. Structural Rearrangements of Carbonic Anhydrase Entrapped in Sol-Gel Magnetite Determined by ATR-FTIR Spectroscopy. Int. J. Mol. Sci. 2022, 23, 5975. [Google Scholar] [CrossRef]
- Moon, H.; Kim, S.; Jo, B.H.; Cha, H.J. Immobilization of Genetically Engineered Whole-Cell Biocatalysts with Periplasmic Carbonic Anhydrase in Polyurethane Foam for Enzymatic CO2 Capture and Utilization. J. CO2 Util. 2020, 39, 101172. [Google Scholar] [CrossRef]
- Jiao, M.; He, J.; Sun, S.; Vriesekoop, F.; Yuan, Q.; Liu, Y.; Liang, H. Fast Immobilization of Human Carbonic Anhydrase II on Ni-Based Metal-Organic Framework Nanorods with High Catalytic Performance. Catalysts 2020, 10, 401. [Google Scholar] [CrossRef] [Green Version]
- Abdelrahim, M.Y.; Martins, C.F.; Neves, L.A.; Capasso, C.; Supuran, C.T.; Coelhoso, I.M.; Crespo, J.G.; Barboiu, M. Supported Ionic Liquid Membranes Immobilized with Carbonic Anhydrases for CO2 Transport at High Temperatures. J. Memb. Sci. 2017, 528, 225–230. [Google Scholar] [CrossRef]
- Martins, C.F.; Neves, L.A.M.; Estevão, L.A.; Rosatella, A.; Alves, V.D.; Afonso, C.A.M.; Crespo, J.G.; Coelhoso, I.M. Effect of Water Activity on Carbon Dioxide Transport in Cholinium-Based Ionic Liquids with Carbonic Anhydrase. Sep. Purif. Technol. 2016, 168, 74–82. [Google Scholar] [CrossRef]
- Vinoba, M.; Bhagiyalakshmi, M.; Jeong, S.K.; Yoon, Y.I.; Nam, S.C. Capture and Sequestration of CO2 by Human Carbonic Anhydrase Covalently Immobilized onto Amine-Functionalized SBA-15. J. Phys. Chem. C 2011, 115, 20209–20216. [Google Scholar] [CrossRef]
- Shen, J.; Yuan, Y.; Salmon, S. Durable and Versatile Immobilized Carbonic Anhydrase on Textile Structured Packing for CO2 Capture. Catalysts 2022, 12, 1108. [Google Scholar] [CrossRef]
- Xv, J.; Zhang, Z.; Pang, S.; Jia, J.; Geng, Z.; Wang, R.; Li, P.; Bilal, M.; Cui, J.; Jia, S. Accelerated CO2 Capture Using Immobilized Carbonic Anhydrase on Polyethyleneimine/Dopamine Co-Deposited MOFs. Biochem. Eng. J. 2022, 189, 108719. [Google Scholar] [CrossRef]
- Shamna, I.; Jeong, S.K.; Margandan, B. Covalent Immobilization of Carbonic Anhydrase on Amine Functionalized Alumino-Siloxane Aerogel Beads for Biomimetic Sequestration of CO2. J. Ind. Eng. Chem. 2021, 100, 288–295. [Google Scholar] [CrossRef]
- Iliuta, I.; Rasouli, H.; Iliuta, M.C. Intensified CO2 Capture in Wall-Coated Microreactors with Immobilized Carbonic Anhydrase: Experimental and Modeling. Sep. Purif. Technol. 2023, 307, 122590. [Google Scholar] [CrossRef]
- Kimmel, J.D.; Arazawa, D.T.; Ye, S.-H.; Shankarraman, V.; Wagner, W.R.; Federspiel, W.J. Carbonic Anhydrase Immobilized on Hollow Fiber Membranes Using Glutaraldehyde Activated Chitosan for Artificial Lung Applications. J. Mater. Sci. Mater. Med. 2013, 24, 2611–2621. [Google Scholar] [CrossRef]
- Peirce, S.; Russo, M.E.; Isticato, R.; Lafuente, R.F.; Salatino, P.; Marzocchella, A. Structure and Activity of Magnetic Cross-Linked Enzyme Aggregates of Bovine Carbonic Anhydrase as Promoters of Enzymatic CO2 Capture. Biochem. Eng. J. 2017, 127, 188–195. [Google Scholar] [CrossRef]
- Chang, S.; He, Y.; Li, Y.; Cui, X. Study on the Immobilization of Carbonic Anhydrases on Geopolymer Microspheres for CO2 Capture. J. Clean. Prod. 2021, 316, 128163. [Google Scholar] [CrossRef]
- Zhang, X.; Shao, W.; Chen, B.; Wang, M. Cross-Linking of Carbonic Anhydrase and Formate Dehydrogenase Based on Amino Acid Specific Recognition: Conversion of Carbon Dioxide to Formic Acid. Enzym. Microb. Technol. 2021, 146, 109763. [Google Scholar] [CrossRef]
- Nelson, J.M.; Griffin, E.G. Adsorption of Invertase. J. Am. Chem. Soc. 1916, 38, 1109–1115. [Google Scholar] [CrossRef] [Green Version]
- Jesionowski, T.; Zdarta, J.; Krajewska, B. Enzyme immobilization by adsorption: A review. Adsorption 2014, 20, 801–821. [Google Scholar] [CrossRef] [Green Version]
- Nabavi Zadeh, P.S.; Åkerman, B. Immobilization of Enzymes in Mesoporous Silica Particles: Protein Concentration and Rotational Mobility in the Pores. J. Phys. Chem. B 2017, 121, 2575–2583. [Google Scholar] [CrossRef] [PubMed]
- Crumbliss, A.L.; Perine, S.C.; Stonehuerner, J.; Tubergen, K.R.; Zhao, J.; Henkens, R.W.; O’Daly, J.P. Colloidal Gold as a Biocompatible Immobilization Matrix Suitable for the Fabrication of Enzyme Electrodes by Electrodeposition. Biotechnol. Bioeng. 1992, 40, 483–490. [Google Scholar] [CrossRef]
- Suvannasara, P.; Juntapram, K.; Praphairaksit, N.; Siralertmukul, K.; Muangsin, N. Mucoadhesive 4-Carboxybenzenesulfonamide-Chitosan with Antibacterial Properties. Carbohydr. Polym. 2013, 94, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-J.; Yin, L.; Wang, Z.-F.; Jing, Y.-C.; Jiang, Z.-L.; Ding, Y.; Wang, H.-S. Enzyme-Immobilized Metal-Organic Frameworks: From Preparation to Application. Chem. Asian J. 2022, 17, e202200751. [Google Scholar] [CrossRef]
- Zhou, H.-C.; Long, J.R.; Yaghi, O.M. Introduction to Metal–Organic Frameworks. Chem. Rev. 2012, 112, 673–674. [Google Scholar] [CrossRef]
- Zhang, W.; Taheri-Ledari, R.; Saeidirad, M.; Qazi, F.S.; Kashtiaray, A.; Ganjali, F.; Tian, Y.; Maleki, A. Regulation of Porosity in MOFs: A Review on Tunable Scaffolds and Related Effects and Advances in Different Applications. J. Environ. Chem. Eng. 2022, 10, 108836. [Google Scholar] [CrossRef]
- Silva, A.R.M.; Alexandre, J.Y.N.H.; Souza, J.E.S.; Lima Neto, J.G.; de Sousa Junior, P.G.; Rocha, M.V.P.; dos Santos, J.C.S. The Chemistry and Applications of Metal-Organic Frameworks (MOFs) as Industrial Enzyme Immobilization Systems. Molecules 2022, 27, 4529. [Google Scholar] [CrossRef]
- Liu, Q.; Bai, X.; Pham, H.; Hu, J.; Dinu, C.Z. Active Nanointerfaces Based on Enzyme Carbonic Anhydrase and Metal-Organic Framework for Carbon Dioxide Reduction. Nanomaterials 2021, 11, 1008. [Google Scholar] [CrossRef]
- Huang, Y.; Zhang, S.; Chen, H.; Zhao, L.; Zhang, Z.; Cheng, P.; Chen, Y. A Zinc Coordination Complex Mimicking Carbonic Anhydrase for CO2 Hydrolysis and Sequestration. Inorg. Chem. 2019, 58, 9916–9921. [Google Scholar] [CrossRef]
- Bien, C.E.; Chen, K.K.; Chien, S.-C.; Reiner, B.R.; Lin, L.-C.; Wade, C.R.; Ho, W.S.W. Bioinspired Metal-Organic Framework for Trace CO2 Capture. J. Am. Chem. Soc. 2018, 140, 12662–12666. [Google Scholar] [CrossRef]
- Han, L.; Pham, T.; Zhuo, M.; Forrest, K.A.; Suepaul, S.; Space, B.; Zaworotko, M.J.; Shi, W.; Chen, Y.; Cheng, P.; et al. Molecular Sieving and Direct Visualization of CO2 in Binding Pockets of an Ultramicroporous Lanthanide Metal-Organic Framework Platform. ACS Appl. Mater. Interfaces 2019, 11, 23192–23197. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Nan, Y.; Zhao, Y.; Wang, X.; Huang, S.; Shi, J. Immobilization of Carbonic Anhydrase for Facilitated CO2 Capture and Separation. Chin. J. Chem. Eng. 2020, 28, 2817–2831. [Google Scholar] [CrossRef]
- Neves, L.A.; Afonso, C.; Coelhoso, I.M.; Crespo, J.G. Integrated CO2 Capture and Enzymatic Bioconversion in Supported Ionic Liquid Membranes. Sep. Purif. Technol. 2012, 97, 34–41. [Google Scholar] [CrossRef]
- Bednár, A.; Nemestóthy, N.; Bakonyi, P.; Fülöp, L.; Zhen, G.; Lu, X.; Kobayashi, T.; Kumar, G.; Xu, K.; Bélafi-Bakó, K. Enzymatically-Boosted Ionic Liquid Gas Separation Membranes Using Carbonic Anhydrase of Biomass Origin. Chem. Eng. J. 2016, 303, 621–626. [Google Scholar] [CrossRef]
- de Castro, A.M.; Neves, L.A.; Corvo, M.C.; Cabrita, E.J.; Crespo, J.G. Effect of Carbonic Anhydrase on CO2 Absorption Promoted by Choline Hydroxide Using Supported Liquid Membranes. Sep. Purif. Technol. 2022, 280, 119921. [Google Scholar] [CrossRef]
- Migliardini, F.; De Luca, V.; Carginale, V.; Rossi, M.; Corbo, P.; Supuran, C.T.; Capasso, C. Biomimetic CO2 Capture Using a Highly Thermostable Bacterial α-Carbonic Anhydrase Immobilized on a Polyurethane Foam. J. Enzym. Inhib. Med. Chem. 2014, 29, 146–150. [Google Scholar] [CrossRef]
- Zaidi, S.; Srivastava, N.; Khare, S.K. Microbial Carbonic Anhydrase Mediated Carbon Capture, Sequestration & Utilization: A Sustainable Approach to Delivering Bio-Renewables. Bioresour. Technol. 2022, 365, 128174. [Google Scholar] [CrossRef]
- Rasouli, H.; Iliuta, I.; Bougie, F.; Garnier, A.; Iliuta, M.C. Enhanced CO2 Capture in Packed-Bed Column Bioreactors with Immobilized Carbonic Anhydrase. Chem. Eng. J. 2022, 432, 134029. [Google Scholar] [CrossRef]
- Vinoba, M.; Bhagiyalakshmi, M.; Jeong, S.K.; Yoon, Y.I.I.; Nam, S.C. Immobilization of Carbonic Anhydrase on Spherical SBA-15 for Hydration and Sequestration of CO2. Colloids Surf. B Biointerfaces 2012, 90, 91–96. [Google Scholar] [CrossRef]
- Uygun, M.; Singh, V.V.; Kaufmann, K.; Uygun, D.A.; De Oliveira, S.D.S.; Wang, J. Micromotor-Based Biomimetic Carbon Dioxide Sequestration: Towards Mobile Microscrubbers. Angew. Chem.-Int. Ed. 2015, 54, 12900–12904. [Google Scholar] [CrossRef]
- Xu, X.; Kentish, S.E.; Martin, G.J.O. Direct Air Capture of CO2 by Microalgae with Buoyant Beads Encapsulating Carbonic Anhydrase. ACS Sustain. Chem. Eng. 2021, 9, 9698–9706. [Google Scholar] [CrossRef]
- Sifat, N.S.; Haseli, Y. A Critical Review of CO2 Capture Technologies and Prospects for Clean Power Generation. Energies 2019, 12, 4143. [Google Scholar] [CrossRef] [Green Version]
- Fu, L.; Ren, Z.; Si, W.; Ma, Q.; Huang, W.; Liao, K.; Huang, Z.; Wang, Y.; Li, J.; Xu, P. Research Progress on CO2 Capture and Utilization Technology. J. CO2 Util. 2022, 66, 102260. [Google Scholar] [CrossRef]
- Fagorite, I.V.; Chijioke, C.F.; Opara, I.A.; Onyekuru, S.O.; Oguzie, E.E. Environmental and Safety Issues Associated with Geological Carbon Storage: A Review. Euro-Mediterr. J. Environ. Integr. 2022, 7, 445–461. [Google Scholar] [CrossRef]
- Farrukh, S.; Wu, D.; Al-Dadah, R.; Gao, W.; Wang, Z. A Review of Integrated Cryogenic Energy Assisted Power Generation Systems and Desalination Technologies. Appl. Therm. Eng. 2023, 221, 119836. [Google Scholar] [CrossRef]
- International Energy Agency. CO2 Emissions in 2022. Available online: https://iea.blob.core.windows.net/assets/3c8fa115-35c4-4474-b237-1b00424c8844/CO2Emissionsin2022.pdf (accessed on 30 June 2023).
- International Energy Agency. Global Energy-Related CO2 Emissions by Sector. Available online: https://www.iea.org/data-and-statistics/charts/global-energy-related-co2-emissions-by-sector (accessed on 30 June 2023).
- Hepburn, C.; Adlen, E.; Beddington, J.; Carter, E.A.; Fuss, S.; Mac Dowell, N.; Minx, J.C.; Smith, P.; Williams, C.K. The Technological and Economic Prospects for CO2 Utilization and Removal. Nature 2019, 575, 87–97. [Google Scholar] [CrossRef] [Green Version]
- Gundersen, M.T.; Von Solms, N.; Woodley, J.M. Enzymatically Assisted CO2 Removal from Flue-Gas. In Energy Procedia; Department of Chemical and Biochemical Engineering, Technical University of Denmark: Lyngby, Denmark, 2014; Volume 63, pp. 624–632. [Google Scholar] [CrossRef]
- Zhang, S.; Du, M.; Shao, P.; Wang, L.; Ye, J.; Chen, J.; Chen, J. Carbonic Anhydrase Enzyme-MOFs Composite with a Superior Catalytic Performance to Promote CO2 Absorption into Tertiary Amine Solution. Environ. Sci. Technol. 2018, 52, 12708–12716. [Google Scholar] [CrossRef]
- Gladis, A.; Lomholdt, N.F.; Fosbøl, P.L.; Woodley, J.M.; von Solms, N. Pilot Scale Absorption Experiments with Carbonic Anhydrase-Enhanced MDEA-Benchmarking with 30 wt% MEA. Int. J. Greenh. Gas Control. 2019, 82, 69–85. [Google Scholar] [CrossRef]
- Kim, T.-J.; Lang, A.; Chikukwa, A.; Sheridan, E.; Dahl, P.I.; Leimbrink, M.; Skiborowski, M.; Roubroeks, J. Enzyme Carbonic Anhydrase Accelerated CO2 Absorption in Membrane Contactor. In Energy Procedia; SINTEF Materials and Chemistry: Trondheim, Norway, 2017; Volume 114, pp. 17–24. [Google Scholar] [CrossRef]
- Thee, H.; Smith, K.H.; Da Silva, G.; Kentish, S.E.; Stevens, G.W. Carbonic Anhydrase Promoted Absorption of CO2 into Potassium Carbonate Solutions. Greenh. Gases Sci. Technol. 2015, 5, 108–114. [Google Scholar] [CrossRef]
- Hu, G.; Nicholas, N.J.; Smith, K.H.; Mumford, K.A.; Kentish, S.E.; Stevens, G.W. Carbon Dioxide Absorption into Promoted Potassium Carbonate Solutions: A Review. Int. J. Greenh. Gas Control 2016, 53, 28–40. [Google Scholar] [CrossRef]
- Jin, P.; Zhang, S.; Liu, Y.; Zhang, W.; Wang, R. Application of Bacillus mucilaginosus in the Carbonation of Steel Slag. Appl. Microbiol. Biotechnol. 2021, 105, 8663–8674. [Google Scholar] [CrossRef] [PubMed]
- Jo, B.H.; Seo, J.H.; Yang, Y.J.; Baek, K.; Choi, Y.S.; Pack, S.P.; Oh, S.H.; Cha, H.J. Bioinspired Silica Nanocomposite with Autoencapsulated Carbonic Anhydrase as a Robust Biocatalyst for CO2 Sequestration. ACS Catal. 2014, 4, 4332–4340. [Google Scholar] [CrossRef]
- Yang, G.; Li, L.; Li, F.; Zhang, C.; Lyu, J. Mechanism of Carbonate Mineralization Induced by Microbes: Taking Curvibacter lanceolatus Strain HJ-1 as an Example. Micron 2021, 140, 102980. [Google Scholar] [CrossRef]
- Bose, H.; Satyanarayana, T. Suitability of the Alkalistable Carbonic Anhydrase from a Polyextremophilic Bacterium Aeribacillus pallidus TSHB1 in Biomimetic Carbon Sequestration. Bioprocess Biosyst. Eng. 2016, 39, 1515–1525. [Google Scholar] [CrossRef]
- De Luca, V.; Vullo, D.; Scozzafava, A.; Carginale, V.; Rossi, M.; Supuran, C.T.; Capasso, C. An Alpha-Carbonic Anhydrase from the Thermophilic Bacterium Sulphurihydrogenibium azorense Is the Fastest Enzyme Known for the CO2 Hydration Reaction. Bioorg. Med. Chem. 2013, 21, 1465–1469. [Google Scholar] [CrossRef]
- Di Lorenzo, F.; Ruiz-Agudo, C.; Ibañez-Velasco, A.; Gil-San Millán, R.; Navarro, J.A.R.; Ruiz-Agudo, E.; Rodriguez-Navarro, C. The Carbonation of Wollastonite: A Model Reaction to Test Natural and Biomimetic Catalysts for Enhanced CO2 Sequestration. Minerals 2018, 8, 209. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Lawal, A.; Stephenson, P.; Sidders, J.; Ramshaw, C. Post-Combustion CO2 Capture with Chemical Absorption: A State-of-the-Art Review. Chem. Eng. Res. Des. 2011, 89, 1609–1624. [Google Scholar] [CrossRef] [Green Version]
- Heldebrant, D.J.; Koech, P.K.; Glezakou, V.-A.; Rousseau, R.; Malhotra, D.; Cantu, D.C. Water-Lean Solvents for Post-Combustion CO2 Capture: Fundamentals, Uncertainties, Opportunities, and Outlook. Chem. Rev. 2017, 117, 9594–9624. [Google Scholar] [CrossRef]
- Sharif, M.; Zhang, T.; Wu, X.; Yu, Y.; Zhang, Z. Evaluation of CO2 Absorption Performance by Molecular Dynamic Simulation for Mixed Secondary and Tertiary Amines. Int. J. Greenh. Gas. Control 2020, 97, 103059. [Google Scholar] [CrossRef]
- Bernhardsen, I.M.; Krokvik, I.R.T.; Jens, K.-J.; Knuutila, H.K. Performance of MAPA Promoted Tertiary Amine Systems for CO2 Absorption: Influence of Alkyl Chain Length and Hydroxyl Groups. Energy Procedia 2017, 114, 1682–1688. [Google Scholar] [CrossRef]
- Liu, B.; Cui, Z.; Tian, W. The Kinetics Investigation of CO2 Absorption into TEA and DEEA Amine Solutions Containing Carbonic Anhydrase. Processes 2021, 9, 2140. [Google Scholar] [CrossRef]
- Bui, M.; Adjiman, C.S.; Bardow, A.; Anthony, E.J.; Boston, A.; Brown, S.; Fennell, P.S.; Fuss, S.; Galindo, A.; Hackett, L.A.; et al. Carbon Capture and Storage (CCS): The Way Forward. Energy Environ. Sci. 2018, 11, 1062–1176. [Google Scholar] [CrossRef] [Green Version]
- Power, I.M.; Harrison, A.L.; Dipple, G.M. Accelerating Mineral Carbonation Using Carbonic Anhydrase. Environ. Sci. Technol. 2016, 50, 2610–2618. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, H.; Bailey, E.; Wysokowski, M.; Jesionowski, T. Forced Biomineralization: A Review. Biomimetics 2021, 6, 46. [Google Scholar] [CrossRef]
- Bose, H.; Satyanarayana, T. Microbial Carbonic Anhydrases in Biomimetic Carbon Sequestration for Mitigating Global Warming: Prospects and Perspectives. Front. Microbiol. 2017, 8, 1615. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Dreisinger, D.B.; Jarvis, M.; Hitchins, T. The Technology of CO2 Sequestration by Mineral Carbonation: Current Status and Future Prospects. Can. Metall. Q. 2018, 57, 46–58. [Google Scholar] [CrossRef] [Green Version]
- Hills, C.D.; Tripathi, N.; Carey, P.J. Mineralization Technology for Carbon Capture, Utilization, and Storage. Front. Energy Res. 2020, 8, 142. [Google Scholar] [CrossRef]
- Bhatia, S.K.; Bhatia, R.K.; Jeon, J.-M.; Kumar, G.; Yang, Y.-H. Carbon Dioxide Capture and Bioenergy Production Using Biological System–A Review. Renew. Sust. Energy Rev. 2019, 110, 143–158. [Google Scholar] [CrossRef]
- Zajac, M.; Krol, M.; Bullerjahn, F.; Deja, J. Effect of Temperature on Carbon Dioxide Mineralisation in Recycled Cement Paste. Adv. Cem. Res. 2023, 35, 1–12. [Google Scholar] [CrossRef]
- Yin, B.; Xu, H.; Fan, F.; Qi, D.; Hua, X.; Xu, T.; Liu, C.; Hou, D. Superhydrophobic Coatings Based on Bionic Mineralization for Improving the Durability of Marine Concrete. Constr. Build. Mater. 2023, 362, 129705. [Google Scholar] [CrossRef]
- Heldebrant, D.J.; Kothandaraman, J.; Mac Dowell, N.; Brickett, L. Next Steps for Solvent-Based CO2 Capture; Integration of Capture, Conversion, and Mineralisation. Chem. Sci. 2022, 13, 6445–6456. [Google Scholar] [CrossRef]
- Rodriguez-Navarro, C.; Cizer, O.; Kudlacz, K.; Ibanez-Velasco, A.; Ruiz-Agudo, C.; Elert, K.; Burgos-Cara, A.; Ruiz-Agudo, E. The Multiple Roles of Carbonic Anhydrase in Calcium Carbonate Mineralization. CrystEngComm 2019, 21, 7407–7423. [Google Scholar] [CrossRef]
- Fernández, M.S.; Montt, B.; Ortiz, L.; Neira-Carrillo, A.; Arias, J.L. Effect of Carbonic Anhydrase Immobilized on Eggshell Membranes on Calcium Carbonate Crystallization In Vitro. In Biomineralization; Endo, K., Kogure, T., Nagasawa, H., Eds.; Springer: Singapore, 2018; pp. 31–37. [Google Scholar]
- Jin, P.; Wang, R.; Zhang, S.; Chen, Y. Effect of Carbonic Anhydrase Bacteria on the Carbonation Process of γ-C2S. Adv. Cem. Res. 2021, 34, 15–27. [Google Scholar] [CrossRef]
- Sharma, V.K.; Hutchison, J.M.; Allgeier, A.M. Redox Biocatalysis: Quantitative Comparisons of Nicotinamide Cofactor Regeneration Methods. ChemSusChem 2022, 15, e2022008. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Jaen, S.; Virginie, M.; Bonin, J.; Robert, M.; Wojcieszak, R.; Khodakov, A.Y. Highlights and Challenges in the Selective Reduction of Carbon Dioxide to Methanol. Nat. Rev. Chem. 2021, 5, 564–579. [Google Scholar] [CrossRef]
- Corrado, M.L.; Knaus, T.; Schwaneberg, U.; Mutti, F.G. High-Yield Synthesis of Enantiopure 1,2-Amino Alcohols from L-Phenylalanine via Linear and Divergent Enzymatic Cascades. Org. Process Res. Dev. 2022, 26, 2085–2095. [Google Scholar] [CrossRef]
- Singh, P.; Srivastava, R. Utilization of Bio-Inspired Catalyst for CO2 Reduction into Green Fuels: Recent Advancement and Future Perspectives. J. CO2 Util. 2021, 53, 101748. [Google Scholar] [CrossRef]
- Armstrong, F.A.; Cheng, B.; Herold, R.A.; Megarity, C.F.; Siritanaratkul, B. From Protein Film Electrochemistry to Nanoconfined Enzyme Cascades and the Electrochemical Leaf. Chem. Rev. 2023, 123, 5421–5458. [Google Scholar] [CrossRef]
- Meneghello, M.; Leger, C.; Fourmond, V. Electrochemical Studies of CO2-Reducing Metalloenzymes. Chem. Eur. J. 2021, 27, 17542–17553. [Google Scholar] [CrossRef]
- Rasheed, T.; Shafi, S.; Anwar, M.T.; Rizwan, K.; Ahmad, T.; Bilal, M. Revisiting Photo and Electro-Catalytic Modalities for Sustainable Conversion of CO2. Appl. Catal. A-Gen. 2021, 623, 118248. [Google Scholar] [CrossRef]
- Cadoux, C.; Milton, R.D. Recent Enzymatic Electrochemistry for Reductive Reactions. ChemElectroChem 2020, 7, 1974–1986. [Google Scholar] [CrossRef]
- Xing, X.; Liu, Y.; Lin, R.-D.; Zhang, Y.; Wu, Z.-L.; Yu, X.-Q.; Li, K.; Wang, N. Development of an Integrated System for Highly Selective Photoenzymatic Synthesis of Formic Acid from CO2. ChemSusChem 2023, 16, e2022019. [Google Scholar] [CrossRef]
- Wang, Q.; Pan, Z. Advances and Challenges in Developing Cocatalysts for Photocatalytic Conversion of Carbon Dioxide to Fuels. Nano Res. 2022, 15, 10090–10109. [Google Scholar] [CrossRef]
- Fang, X.; Kalathil, S.; Reisner, E. Semi-Biological Approaches to Solar-to-Chemical Conversion. Chem. Soc. Rev. 2020, 49, 4926–4952. [Google Scholar] [CrossRef]
- Ozgen, F.F.; Runda, M.E.; Schmidt, S. Photo-Biocatalytic Cascades: Combining Chemical and Enzymatic Transformations Fueled by Light. ChemBioChem 2021, 22, 790–806. [Google Scholar] [CrossRef]
- Chen, H.; Huang, Y.; Sha, C.; Moradian, J.M.; Yong, Y.-C.; Fang, Z. Enzymatic Carbon Dioxide to Formate: Mechanisms, Challenges and Opportunities. Renew. Sust. Energy Rev. 2023, 178, 113271. [Google Scholar] [CrossRef]
- Immanuel, S.; Sivasubramanian, R.; Gul, R.; Dar, M.A. Recent Progress and Perspectives on Electrochemical Regeneration of Reduced Nicotinamide Adenine Dinucleotide (NADH). Chem. Asian J. 2020, 15, 4256–4270. [Google Scholar] [CrossRef]
- Lee, Y.S.; Gerulskis, R.; Minteer, S.D. Advances in Electrochemical Cofactor Regeneration: Enzymatic and Non-Enzymatic Approaches. Curr. Opin. Biotechnol. 2022, 73, 14–21. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, X.; Ji, X. Developing and Regenerating Cofactors for Sustainable Enzymatic CO2 Conversion. Processes 2022, 10, 230. [Google Scholar] [CrossRef]
- Bachosz, K.; Zdarta, J.; Bilal, M.; Meyer, A.S.; Jesionowski, T. Enzymatic Cofactor Regeneration Systems: A New Perspective on Efficiency Assessment. Sci. Total Environ. 2023, 868, 161630. [Google Scholar] [CrossRef]
- Findrik, Z.; Vasic-Racki, D. Overview on Reactions with Multi-Enzyme Systems. Chem. Biochem. Eng. Q. 2009, 23, 545–553. [Google Scholar]
- Neves, L.A.; Afonso, C.A.M.; Coelhoso, L.M.; Crespo, J.G. CO2 Capture by Enzymatic Bioconversion in a Membrane Contactor with Task Specific Ionic Liquids. In Procedia Engineering; Universidade Nova de Lisboa: Lisbon, Portugal, 2012; Volume 44, pp. 557–558. [Google Scholar] [CrossRef]
- Qadir, M.I.; Dupont, J. Thermo- and Photocatalytic Activation of CO2 in Ionic Liquids. Angew. Chem. Int. Ed. 2023, e202301497. [Google Scholar] [CrossRef]
- Chenault, H.K.; Whitesides, G.M. Regeneration of Nicotinamide Cofactors for Use in Organic Synthesis. Appl. Biochem. Biotechnol. 1987, 14, 147–197. [Google Scholar] [CrossRef] [PubMed]
- Chenault, H.K.; Simon, E.S.; Whitesides, G.M. Cofactor Regeneration for Enzyme-Catalysed Synthesis. Biotechnol. Genet. Eng. Rev. 1988, 6, 221–270. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Niks, D.; Ge, X.; Liu, H.; Hille, R.; Mulchandani, A. Synthesis of Formate from CO2 Gas Catalyzed by an O2-Tolerant NAD-Dependent Formate Dehydrogenase and Glucose Dehydrogenase. Biochemistry 2019, 58, 1861–1868. [Google Scholar] [CrossRef]
- Liao, Q.; Liu, W.; Meng, Z. Strategies for Overcoming the Limitations of Enzymatic Carbon Dioxide Reduction. Biotechnol. Adv. 2022, 60, 108024. [Google Scholar] [CrossRef]
- El-Zahab, B.; Donnelly, D.; Wang, P. Particle-Tethered NADH for Production of Methanol from CO2 Catalyzed by Coimmobilized Enzymes. Biotechnol. Bioeng. 2008, 99, 508–514. [Google Scholar] [CrossRef]
- Ji, X.; Su, Z.; Wang, P.; Ma, G.; Zhang, S. Tethering of Nicotinamide Adenine Dinucleotide inside Hollow Nanofibers for High-Yield Synthesis of Methanol from Carbon Dioxide Catalyzed by Coencapsulated Multienzymes. ACS Nano 2015, 9, 4600–4610. [Google Scholar] [CrossRef]
- Ren, S.; Wang, Z.; Bilal, M.; Feng, Y.; Jiang, Y.; Jia, S.; Cui, J. Co-Immobilization Multienzyme Nanoreactor with Co-Factor Regeneration for Conversion of CO2. Int. J. Biol. Macromol. 2020, 155, 110–118. [Google Scholar] [CrossRef]
- Shukia, S.K.; Khokarale, S.G.; Bui, T.Q.; Mikkola, J.-P.T. Ionic Liquids: Potential Materials for Carbon Dioxide Capture and Utilization. Front. Mater. 2019, 6, 42. [Google Scholar] [CrossRef] [Green Version]
- Zeng, S.; Zhang, X.X.; Bai, L.; Zhang, X.X.; Wang, H.; Wang, J.; Bao, D.; Li, M.; Liu, X.; Zhang, S. Ionic-Liquid-Based CO2 Capture Systems: Structure, Interaction and Process. Chem. Rev. 2017, 117, 9625–9673. [Google Scholar] [CrossRef]
- Zhang, Z.; Muschiol, J.; Huang, Y.; Sigurdardottir, S.B.; von Solms, N.; Daugaard, A.E.; Wei, J.; Luo, J.; Xu, B.-H.; Zhang, S.; et al. Efficient Ionic Liquid-Based Platform for Multi-Enzymatic Conversion of Carbon Dioxide to Methanol. Green Chem. 2018, 20, 4339–4348. [Google Scholar] [CrossRef] [Green Version]
- The Methanol Institute. Available online: https://www.methanol.org/applications/ (accessed on 7 July 2023).
- Cazelles, R.; Drone, J.; Fajula, F.; Ersen, O.; Moldovan, S.; Galarneau, A. Reduction of CO2 to Methanol by a Polyenzymatic System Encapsulated in Phospholipids-Silica Nanocapsules. New J. Chem. 2013, 37, 3721–3730. [Google Scholar] [CrossRef]
- Singh, R.K.R.; Singh, R.K.R.; Sivakumar, D.; Kondaveeti, S.; Kim, T.; Li, J.; Sung, B.H.; Cho, B.-K.; Kim, D.R.; Kim, S.C.; et al. Insights into Cell-Free Conversion of CO2 to Chemicals by a Multienzyme Cascade Reaction. ACS Catal. 2018, 8, 11085–11093. [Google Scholar] [CrossRef]
- Dave, B.C.; Rao, M.S.; Burt, M.C. Converting Carbon Dioxide to Methanol Comprises Serial Reduction of the Carbon Dioxide to Methanol By Dehydrogenase Enzymes in the Presence of Reduced Nicotinamide Adenine Dinucleotide. US20070042479(A1), 18 August 2005. [Google Scholar]
- Alvarez-Malmagro, J.; Oliveira, A.R.; Gutierrez-Sanchez, C.; Villajos, B.; Pereira, I.A.C.; Velez, M.; Pita, M.; De Lacey, A.L. Bioelectrocatalytic Activity of W-Formate Dehydrogenase Covalently Immobilized on Functionalized Gold and Graphite Electrodes. ACS Appl. Mater. Interfaces 2021, 13, 11891–11900. [Google Scholar] [CrossRef]
- Barin, R.; Rashid-Nadimi, S.; Biria, D.; Asadollahi, M.A. Direct Electrochemical Regeneration of 1,4-NADH at the Copper Foam and Bimetallic Copper Foam. Electrochim. Acta 2017, 247, 1095–1102. [Google Scholar] [CrossRef]
- Song, H.; Ma, C.; Liu, P.; You, C.; Lin, J.; Zhu, Z. A Hybrid CO2 Electroreduction System Mediated by Enzyme-Cofactor Conjugates Coupled with Cu Nanoparticle-Catalyzed Cofactor Regeneration. J. CO2 Util. 2019, 34, 568–575. [Google Scholar] [CrossRef]
- Addo, P.K.; Arechederra, R.L.; Waheed, A.; Shoemaker, J.D.; Sly, W.S.; Minteer, S.D. Methanol Production via Bioelectrocatalytic Reduction of Carbon Dioxide: Role of Carbonic Anhydrase in Improving Electrode Performance. Electrochem. Solid-State Lett. 2011, 14, E9–E13. [Google Scholar] [CrossRef]
- Yuan, M.; Sahin, S.; Cai, R.; Abdellaoui, S.; Hickey, D.P.; Minteer, S.D.; Milton, R.D. Creating a Low-Potential Redox Polymer for Efficient Electroenzymatic CO2 Reduction. Angew. Chem. Int. Ed. 2018, 57, 6582–6586. [Google Scholar] [CrossRef]
- Kim, S.-H.; Chung, G.-Y.; Kim, S.-H.; Vinothkumar, G.; Yoon, S.-H.; Jung, K.-D. Electrochemical NADH Regeneration and Electroenzymatic CO2 Reduction on Cu Nanorods/Glassy Carbon Electrode Prepared by Cyclic Deposition. Electrochim. Acta 2016, 210, 837–845. [Google Scholar] [CrossRef]
- Chen, Y.; Li, P.; Zhou, J.; Buru, C.T.; Dordevic, L.; Li, P.; Zhang, X.; Cetin, M.M.; Stoddart, J.F.; Stupp, S.I.; et al. Integration of Enzymes and Photosensitizers in a Hierarchical Mesoporous Metal–Organic Framework for Light-Driven CO2 Reduction. J. Am. Chem. Soc. 2020, 142, 1768–1773. [Google Scholar] [CrossRef]
- Barin, R.; Biria, D.; Rashid-Nadimi, S.; Asadollahi, M.A. Enzymatic CO2 Reduction to Formate by Formate Dehydrogenase from Candida Boidinii Coupling with Direct Electrochemical Regeneration of NADH. J. CO2 Util. 2018, 28, 117–125. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, J.; Ji, M.; Liu, Y.; Wang, N.; Zhang, X.; Zhang, S.; Ji, X. Encapsulation of Multiple Enzymes in a Metal-Organic Framework with Enhanced Electro-Enzymatic Reduction of CO2 to Methanol. Green Chem. 2021, 23, 2362–2371. [Google Scholar] [CrossRef]
- Zhang, T.R.; Lu, S.Y. Sacrificial Agents for Photocatalytic Hydrogen Production: Effects, Cost, and Development. Chem. Catalysis 2022, 2, 1502–1505. [Google Scholar] [CrossRef]
- Hernandez-Ibanez, N.; Gomis-Berenguer, A.; Montiel, V.; Ania, C.O.; Iniesta, J. Fabrication of a Biocathode for Formic Acid Production upon the Immobilization of Formate Dehydrogenase from Candida boidinii on a Nanoporous Carbon. Chemosphere 2022, 291, 133117. [Google Scholar] [CrossRef]
- Cheng, Y.; Shi, J.; Wu, Y.; Wang, X.; Sun, Y.; Cai, Z.; Chen, Y.; Jiang, Z. Intensifying Electron Utilization by Surface-Anchored Rh Complex for Enhanced Nicotinamide Cofactor Regeneration and Photoenzymatic CO2 Reduction. Research 2021, 2021, 8175709. [Google Scholar] [CrossRef]
- Ottone, C.; Pugliese, D.; Laurenti, M.; Hernandez, S.; Cauda, V.; Grez, P.; Wilson, L. ZnO Materials as Effective Anodes for the Photoelectrochemical Regeneration of Enzymatically Active NAD+. ACS Appl. Mater. Interfaces 2021, 13, 10719–10727. [Google Scholar] [CrossRef]
- Tensi, L.; Macchioni, A. Extremely Fast NADH-Regeneration Using Phosphonic Acid as Hydride Source and Iridium-Pyridine-2-Sulfonamidate Catalysts. ACS Catal. 2020, 10, 7945–7949. [Google Scholar] [CrossRef]
- Liao, Q.; Guo, M.; Mao, M.; Gao, R.; Meng, Z.; Fan, X.; Liu, W. Construction and Optimization of a Photo-Enzyme Coupled System for Sustainable CO2 Conversion to Methanol. Process Biochem. 2023, 129, 44–55. [Google Scholar] [CrossRef]
- Guo, M.; Gu, F.; Meng, L.; Liao, Q.; Meng, Z.; Liu, W. Synthesis of Formaldehyde from CO2 Catalyzed by the Coupled Photo-Enzyme System. Sep. Purif. Technol. 2022, 286, 120480. [Google Scholar] [CrossRef]
- Kim, J.A.; Kim, S.; Lee, J.; Baeg, J.-O.; Kim, J.A. Photochemical Production of NADH Using Cobaloxime Catalysts and Visible-Light Energy. Inorg. Chem. 2012, 51, 8057–8063. [Google Scholar] [CrossRef] [PubMed]
- Laun, K.; Duffus, B.R.; Kumar, H.; Oudsen, J.-P.H.; Karafoulidi-Retsou, C.; Waffo, A.T.; Hildebrandt, P.; Ly, K.H.; Leimkuehler, S.; Katz, S.; et al. A Minimal Light-Driven System to Study the Enzymatic CO2 Reduction of Formate Dehydrogenase. ChemCatChem 2022, 14, e202201067. [Google Scholar] [CrossRef]
- Ji, X.; Wang, J.; Kang, Y.; Mei, L.; Su, Z.; Wang, S.; Ma, G.; Shi, J.; Zhang, S. Enhanced Solar Energy Harvest and Electron Transfer through Intra- and Intermolecular Dual Channels in Chlorosome-Mimicking Supramolecular Self-Assemblies. ACS Catal. 2018, 8, 10732–10745. [Google Scholar] [CrossRef]
- Lin, G.; Zhang, Y.; Hua, Y.; Zhang, C.; Jia, C.; Ju, D.; Yu, C.; Li, P.; Liu, J. Bioinspired Metalation of the Metal-Organic Framework MIL-125-NH2 for Photocatalytic NADH Regeneration and Gas-Liquid-Solid Three-Phase Enzymatic CO2 Reduction. Angew. Chem. Int. Ed. 2022, 61, e202206283. [Google Scholar] [CrossRef] [PubMed]
- Ong, W.-J.; Tan, L.-L.; Ng, Y.H.; Yong, S.-T.; Chai, S.-P. Graphitic Carbon Nitride (g-C3N4)-Based Photocatalysts for Artificial Photosynthesis and Environmental Remediation: Are We a Step Closer To Achieving Sustainability? Chem. Rev. 2016, 116, 7159–7329. [Google Scholar] [CrossRef]
- Zeng, P.; Ji, X.; Su, Z.; Zhang, S. WS2/g-C3N4 Composite as an Efficient Heterojunction Photocatalyst for Biocatalyzed Artificial Photosynthesis. RSC Adv. 2018, 8, 20557–20567. [Google Scholar] [CrossRef]
- Meng, J.; Tian, Y.; Li, C.; Lin, X.; Wang, Z.; Sun, L.; Zhou, Y.; Li, J.; Yang, N.; Zong, Y.; et al. A Thiophene-Modified Doubleshell Hollow g- C3N4 Nanosphere Boosts NADH Regeneration via Synergistic Enhancement of Charge Excitation and Separation. Catal. Sci. Technol. 2019, 9, 1911–1921. [Google Scholar] [CrossRef]
- Gao, Y.; Li, W.; Sun, X.; Zhao, Y.; Ji, H.; Luo, H.; Wu, G.; Wan, L.; Zhang, L. Boosting the Performance of Formate Dehydrogenase by Silver Nanoclusters for Photoreduction of CO2 to Formate. ACS Sustain. Chem. Eng. 2022, 10, 14888–14896. [Google Scholar] [CrossRef]
- Gupta, P.; Verma, N. Conversion of CO2 to Formate Using Activated Carbon Fiber-Supported g-C3N4-NiCoWO4 Photoanode in a Microbial Electrosynthesis System. Chem. Eng. J. 2022, 446, 137029. [Google Scholar] [CrossRef]
- Tian, Y.; Zhou, Y.; Zong, Y.; Li, J.; Yang, N.; Zhang, M.; Guo, Z.; Song, H. Construction of Functionally Compartmental Inorganic Photocatalyst-Enzyme System via Imitating Chloroplast for Efficient Photoreduction of CO2 to Formic Acid. ACS Appl. Mater. Interfaces 2020, 12, 34795–34805. [Google Scholar] [CrossRef]
- Yu, S.; Lv, P.; Xue, P.; Wang, K.; Yang, Q.; Zhou, J.; Wang, M.; Wang, L.; Chen, B.; Tan, T. Light-Driven Enzymatic Nanosystem for Highly Selective Production of Formic Acid from CO2. Chem. Eng. J. 2021, 420, 127649. [Google Scholar] [CrossRef]
- Ji, X.; Kang, Y.; Su, Z.; Wang, P.; Ma, G.; Zhang, S. Graphene Oxide and Polyelectrolyte Composed One-Way Expressway for Guiding Electron Transfer of Integrated Artificial Photosynthesis. ACS Sustain. Chem. Eng. 2018, 6, 3060–3069. [Google Scholar] [CrossRef]
- Ji, X.; Su, Z.; Wang, P.; Ma, G.; Zhang, S. Integration of Artificial Photosynthesis System for Enhanced Electronic Energy-Transfer Efficacy: A Case Study for Solar-Energy Driven Bioconversion of Carbon Dioxide to Methanol. Small 2016, 12, 4753–4762. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Lee, S.; Anjong, T.F.; Jang, H.Y.; Kim, J.-Y.; Lee, C.; Park, S.; Lee, H.J.; Yoon, J.; Kim, J. Artificial Photocatalytic System Using Polydiacetylene-(-NH-Phen)Ru(Bpy)2 for Cofactor Regeneration and CO2 Reduction. J. Phys. Chem. C 2016, 120, 28407–28414. [Google Scholar] [CrossRef]
- Sokol, K.P.; Robinson, W.E.; Oliveira, A.R.; Warnan, J.; Nowaczyk, M.M.; Ruff, A.; Pereira, I.A.C.; Reisner, E. Photoreduction of CO2 with a Formate Dehydrogenase Driven by Photosystem II Using a Semi-Artificial Z-Scheme Architecture. J. Am. Chem. Soc. 2018, 140, 16418–16422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuler, E.; Ermolich, P.A.; Shiju, N.R.; Gruter, G.-J. Monomers from CO2: Superbases as Catalysts for Formate-to-Oxalate Coupling. ChemSusChem 2021, 14, 1517–1523. [Google Scholar] [CrossRef]
- Murcia-Valderrama, M.A.; van Putten, R.-J.; Gruter, G.-J.M. The Potential of Oxalic—And Glycolic Acid Based Polyesters (Review). Towards CO2 as a Feedstock (Carbon Capture and Utilization–CCU). Eur. Polym. J. 2019, 119, 445–468. [Google Scholar] [CrossRef]
- Schwarz, F.M.; Mueller, V. Whole-Cell Biocatalysis for Hydrogen Storage and Syngas Conversion to Formate Using a Thermophilic Acetogen. Biotechnol. Biofuels 2020, 13, 32. [Google Scholar] [CrossRef] [Green Version]
- Luxuan, L.; Zhijun, X.; Huang, X. Whole-Cell-Based Photosynthetic Biohybrid Systems for Energy and Environmental Applications. ChemPlusChem 2021, 86, 1021–1036. [Google Scholar]
- Hwang, H.; Yeon, Y.; Lee, S.; Choe, H.; Cho, D.; Park, S.; Kim, Y. Electro-Biocatalytic Production of Formate from Carbon Dioxide using an Oxygen-Stable Whole-Cell Biocatalyst. Bioresour. Technol. 2015, 185, 35–39. [Google Scholar] [CrossRef]
- Le, T.Q.A. Recent Applications and Strategies to Enhance Performance of Electrochemical Reduction of CO2 Gas into Value-Added Chemicals Catalyzed by Whole-Cell Biocatalysts. Processes 2023, 11, 766. [Google Scholar] [CrossRef]
- Alissandratos, A.; Kim, H.-K.; Easton, C.J. Formate Production through Carbon Dioxide Hydrogenation with Recombinant Whole Cell Biocatalysts. Bioresour. Technol. 2014, 164, 7–11. [Google Scholar] [CrossRef]
- Roger, M.; Brown, F.; Gabrielli, W.; Sargent, F. Efficient Hydrogen-Dependent Carbon Dioxide Rreduction by Escherichia coli. Curr. Biol. 2018, 28, 140–145.e2. [Google Scholar] [CrossRef] [Green Version]
- Leo, F.; Schwarz, F.M.; Schuchmann, K.; Mueller, V. Capture of Carbon Dioxide and Hydrogen by Engineered Escherichia coli: Hydrogen-Dependent CO2 Reduction to Formate. Appl. Microbiol. Biotechnol. 2021, 105, 5861–5872. [Google Scholar] [CrossRef]
- Schwarz, F.M.; Oswald, F.; Müller, V. Acetogenic Conversion of H2 and CO2 into Formic Acid and Vice Versa in a Fed-Batch-Operated Stirred-Tank Bioreactor. ACS Sustain. Chem. Eng. 2021, 9, 6810–6820. [Google Scholar] [CrossRef]
- Le, Q.A.T.; Kim, H.G.; Kim, Y.H. Electrochemical Synthesis of Formic Acid from CO2 Catalyzed by Shewanella oneidensis MR-1 Whole-Cell Biocatalyst. Enzym. Microb. Technol. 2018, 116, 1–5. [Google Scholar] [CrossRef]
- Singh, S.; Noori, M.T.; Verma, N. Efficient Bio-Electroreduction of CO2 to Formate on a Iron Phthalocyanine-Dispersed CDC in Microbial Electrolysis System. Electrochim. Acta 2020, 338, 135887. [Google Scholar] [CrossRef]
- Phan, U.T.; Jeon, B.W.; Kim, Y.H. Microbial Engineering of Methylorubrum extorquens AM1 to Enhance CO2 Conversion into Formate. Enzym. Microb. Technol. 2023, 168, 110264. [Google Scholar] [CrossRef]
- Chen, H.; Li, J.; Fan, Q.; Zheng, T.; Zhang, Y.; Yong, Y.-C.; Fang, Z. A feasible strategy for microbial electrocatalytic CO2 reduction via whole-cell-packed and exogenous-mediator-free rGO/Shewanella biohydrogel. Chem. Eng. J. 2023, 460, 141863. [Google Scholar] [CrossRef]
- Guntermann, N.; Mengers, H.G.; Franciò, G.; Blank, L.M.; Leitner, W. Bio-energy Conversion with Carbon Capture and Utilization (BECCU): Integrated Biomass Fermentation and Chemo-Catalytic CO2 Hydrogenation for Bioethanol and Formic Acid Coproduction. Green Chem. 2021, 23, 9860–9864. [Google Scholar] [CrossRef]
- Rowe, S.F.; Le Gall, G.; Ainsworth, E.V.; Davies, J.A.; Lockwood, C.W.J.; Shi, L.; Elliston, A.; Roberts, I.N.; Waldron, K.W.; Richardson, D.J.; et al. Light-Driven H2 Evolution and C=C or C=O Bond Hydrogenation by Shewanella oneidensis: A Versatile Strategy for Photocatalysis by Nonphotosynthetic Microorganisms. ACS Catal. 2017, 7, 7558–7566. [Google Scholar] [CrossRef] [Green Version]
- Tuyishime, P.; Sinumvayo, J.P. Novel Outlook in Engineering Synthetic Methylotrophs and Formatotrophs: A Course for Advancing C1-Based Chemicals Production. World J. Microbiol. Biotechnol. 2020, 36, 118. [Google Scholar] [CrossRef] [PubMed]
- Yishai, O.; Lindner, S.N.; Gonzalez de la Cruz, J.; Tenenboim, H.; Bar-Even, A. The formate bio-economy. Curr. Opin. Chem. Biol. 2016, 35, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Shengyuan Guo, S.; Asset, T.; Atanassov, P. Catalytic Hybrid Electrocatalytic/Biocatalytic Cascades for Carbon Dioxide Reduction and Valorization. ACS Catalysis 2021, 11, 5172–5188. [Google Scholar] [CrossRef]
- Liang, B.; Zhao, Y.; Yang, J. Recent Advances in Developing Artificial Autotrophic Microorganism for Reinforcing CO2 Fixation. Front. Microbiol. 2020, 11, 592631. [Google Scholar] [CrossRef]
- Collas, F.; Dronsella, B.B.; Kubis, A.; Schann, K.; Binder, S.; Arto, N.; Claassens, N.J.; Kensy, F.; Orsi, E. Engineering the Biological Conversion of Formate into Crotonate in Cupriavidus necator. Metab. Eng. 2023, in press. [Google Scholar] [CrossRef]
- Cotton, C.A.; Claassens, N.J.; Benito-Vaquerizo, S.; Bar-Even, A. Renewable Methanol and Formate as Microbial Feedstocks. Curr. Opin. Biotechnol. 2020, 62, 168–180. [Google Scholar] [CrossRef]
Figure 1.
Three pathways to incorporating CO2 into soluble carbon forms by using enzymes. (A) Formation of hydrogen carbonate using carbonic anhydrase; (B) photosynthesis performed using RuBisCO protein; (C) reduction to achieve C1 in reduced form, formic acid, catalyzed using FDHs. Approaches A and C are the aims of the present work (see text for details).
Figure 1.
Three pathways to incorporating CO2 into soluble carbon forms by using enzymes. (A) Formation of hydrogen carbonate using carbonic anhydrase; (B) photosynthesis performed using RuBisCO protein; (C) reduction to achieve C1 in reduced form, formic acid, catalyzed using FDHs. Approaches A and C are the aims of the present work (see text for details).

Figure 2.
(A) Number of articles published per year related to carbon capture storage (red) and carbon capture utilization (blue); the insert is a vertical expansion, scale 0–15, by year, published in the last century (the year 2023 contains the number of articles at halfway through the year). (B,C) Percentage of articles related to CCS (B) and CCU (C) classified by research area. Source: compiled by the authors, based on the Web of Science database (https://clarivate.com), accessed on 2 July 2023.
Figure 2.
(A) Number of articles published per year related to carbon capture storage (red) and carbon capture utilization (blue); the insert is a vertical expansion, scale 0–15, by year, published in the last century (the year 2023 contains the number of articles at halfway through the year). (B,C) Percentage of articles related to CCS (B) and CCU (C) classified by research area. Source: compiled by the authors, based on the Web of Science database (https://clarivate.com), accessed on 2 July 2023.

Scheme 1.
Acid–base equilibria for CO2 water-soluble species as a function of the pH (Equations (1)–(4)) and reduction CO2 equation to generate formic acid using the cofactor NADH (Equation (5)).
Figure 3.
(A) Free formation energies of some C1 compounds under standard conditions. (B) Redox potentials vs. the normal hydrogen electrode (NHE) at pH 7 of some relevant compounds, photosynthesizers, and sacrificial agents (see Section 7.4) commented in the text [32,33]. The redox potential values are shown between parentheses. The HOMO and LUMO energy orbitals for some dyes are indicated in green and purple, respectively.
Figure 3.
(A) Free formation energies of some C1 compounds under standard conditions. (B) Redox potentials vs. the normal hydrogen electrode (NHE) at pH 7 of some relevant compounds, photosynthesizers, and sacrificial agents (see Section 7.4) commented in the text [32,33]. The redox potential values are shown between parentheses. The HOMO and LUMO energy orbitals for some dyes are indicated in green and purple, respectively.

Figure 4.
(A) Three-dimensional structure of human CA (5ZXW pdb code [68]). (B) Active center of the enzyme showing the most relevant amino acids (see text for details). (C) CA mechanism of action.
Figure 4.
(A) Three-dimensional structure of human CA (5ZXW pdb code [68]). (B) Active center of the enzyme showing the most relevant amino acids (see text for details). (C) CA mechanism of action.

Figure 5.
(A) Three-dimensional structure of CbFDH with NAD+ and azide anion (6D4C pdb code [90]). The two dimers are colored in red and green, while the NAD+ cofactor is colored in orange. (B) Active center of CbFDH showing the arrangement of the cofactor (pink) as well as the most relevant amino acids closest the NAD+, as discussed in the text. (C) Mechanism of action of NAD+-dependent FDHs for formate oxidation (blue arrows) or formate reduction (inverse sense, red arrows); the NAD nicotinamide group in its oxidized and reduced state is displayed. (D) Active center of CtFDH (6T8Z pdb code [91]) showing the four residues mutated in both works cited in the text [92,93], the numbering corresponding to CtFDH.
Figure 5.
(A) Three-dimensional structure of CbFDH with NAD+ and azide anion (6D4C pdb code [90]). The two dimers are colored in red and green, while the NAD+ cofactor is colored in orange. (B) Active center of CbFDH showing the arrangement of the cofactor (pink) as well as the most relevant amino acids closest the NAD+, as discussed in the text. (C) Mechanism of action of NAD+-dependent FDHs for formate oxidation (blue arrows) or formate reduction (inverse sense, red arrows); the NAD nicotinamide group in its oxidized and reduced state is displayed. (D) Active center of CtFDH (6T8Z pdb code [91]) showing the four residues mutated in both works cited in the text [92,93], the numbering corresponding to CtFDH.

Figure 6.
(A) Up: three-dimensional structure of FDH N from E. coli (1KQF pdb code [119]). The three polypeptide chains are shown in blue (chain A), pink (chain B), and green (chain C); the molybdopterin (in chain A), the five F4S4 clusters (in chains A and B), and the two heme (chain C) metal cofactors are displayed in pink, green, and red, respectively. (A) Down: a detailed picture of the cofactors at the same scale. (B) Up: graphic of the molybdenum cofactor in FDH N from E. coli. (B) Down: chemical formula of the Mo cofactor. (C) FDH mechanism of action for CO2 reduction (red arrows) or formate oxidation (inverse sense, blue arrows) [27]; the direction of the reaction depends on the experimental conditions (see text for details).
Figure 6.
(A) Up: three-dimensional structure of FDH N from E. coli (1KQF pdb code [119]). The three polypeptide chains are shown in blue (chain A), pink (chain B), and green (chain C); the molybdopterin (in chain A), the five F4S4 clusters (in chains A and B), and the two heme (chain C) metal cofactors are displayed in pink, green, and red, respectively. (A) Down: a detailed picture of the cofactors at the same scale. (B) Up: graphic of the molybdenum cofactor in FDH N from E. coli. (B) Down: chemical formula of the Mo cofactor. (C) FDH mechanism of action for CO2 reduction (red arrows) or formate oxidation (inverse sense, blue arrows) [27]; the direction of the reaction depends on the experimental conditions (see text for details).

Figure 7.
Different methods of enzyme immobilization.

Scheme 2.
Reactions of CO2 with primary, secondary (Equation (6)), and tertiary (Equation (7)) amines in water solutions.
Figure 8.
The three different (enzymatic, electrochemical, and photochemical) approaches to regenerating cofactor NADH and producing formate with FDH.
Figure 8.
The three different (enzymatic, electrochemical, and photochemical) approaches to regenerating cofactor NADH and producing formate with FDH.

Figure 10.
(A) Schematic view of an electrochemical device for obtaining formic acid from CO2. (B) Scheme of three possible mechanisms for transferring electrons from the cathode to the cofactor and finally to FDH: without any mediator species, with a mediator (typically a photosynthesizer molecule), and also with an enzyme intercalated between the mediator and the cofactor.
Figure 10.
(A) Schematic view of an electrochemical device for obtaining formic acid from CO2. (B) Scheme of three possible mechanisms for transferring electrons from the cathode to the cofactor and finally to FDH: without any mediator species, with a mediator (typically a photosynthesizer molecule), and also with an enzyme intercalated between the mediator and the cofactor.

Figure 11.
The two reduction steps of NAD+. The addition of one electron produces the radical NAD*. This radical can either be reduced by the addition of an additional electron and a proton (up), to produce 1,4-NADH (up) or combine with another radical to generate the 4,4′-dimer (NAD)2.
Figure 11.
The two reduction steps of NAD+. The addition of one electron produces the radical NAD*. This radical can either be reduced by the addition of an additional electron and a proton (up), to produce 1,4-NADH (up) or combine with another radical to generate the 4,4′-dimer (NAD)2.

Figure 12.
Scheme of the photochemical reduction of CO2 using FDH.


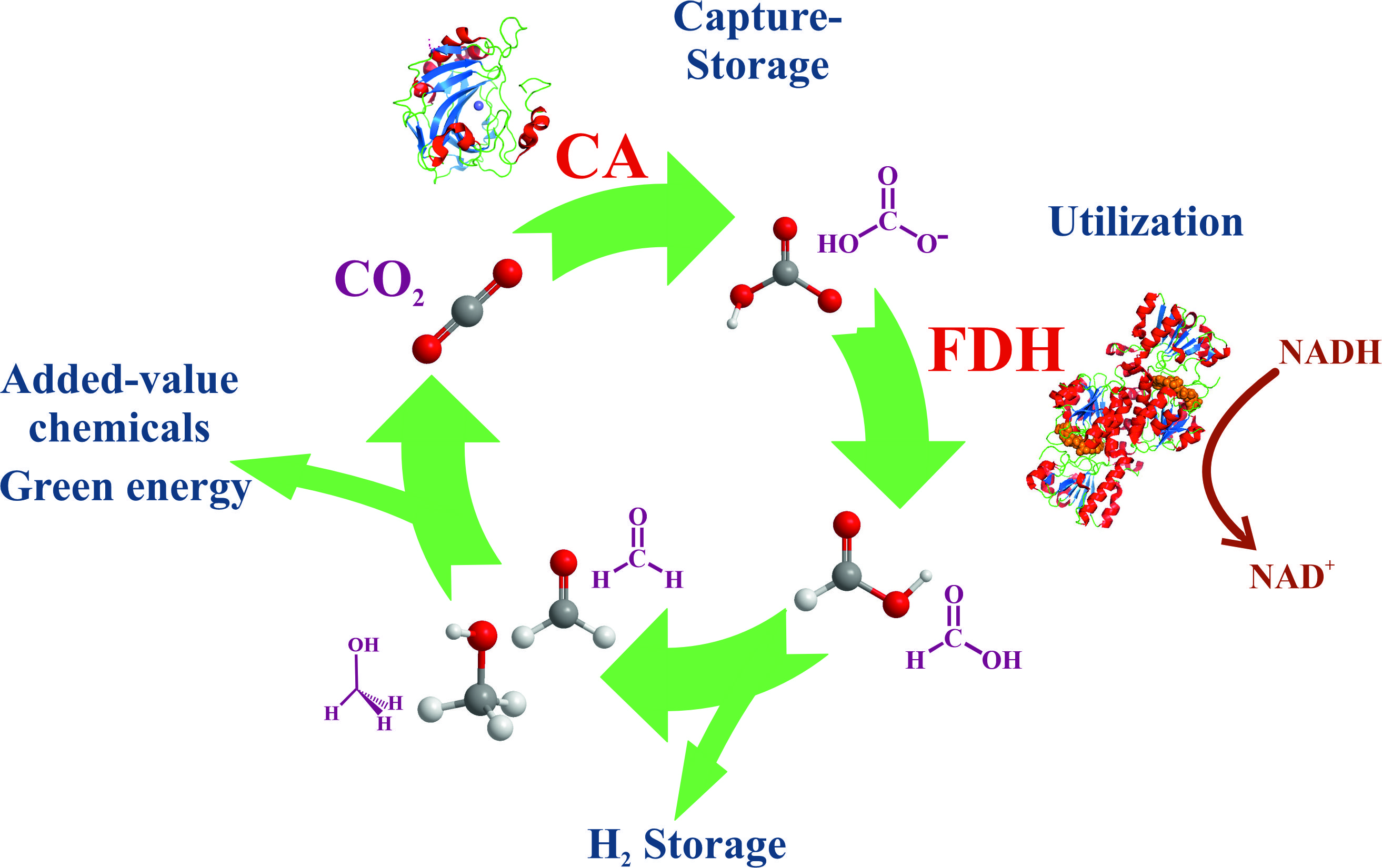{kind=link}
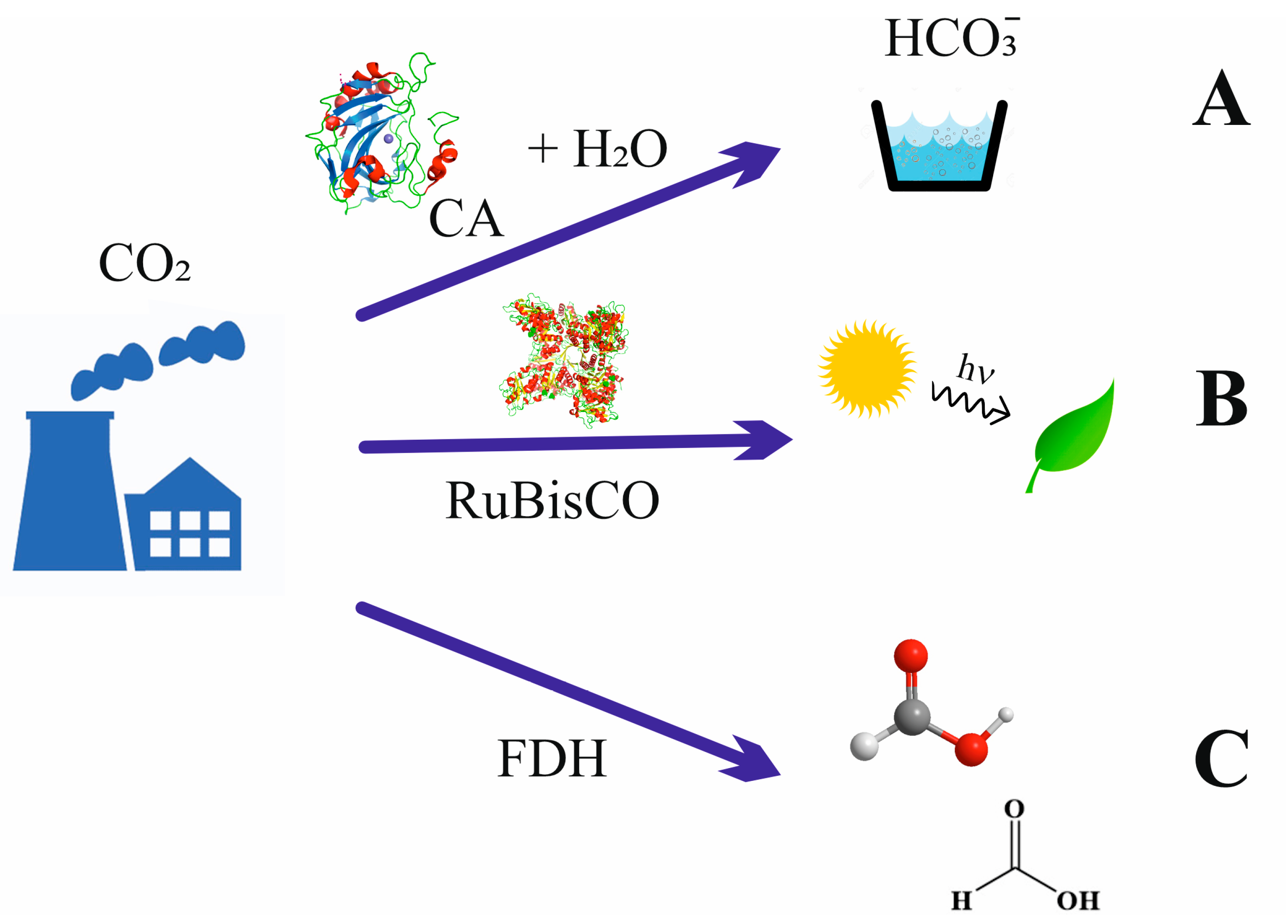{kind=link}
{kind=link}
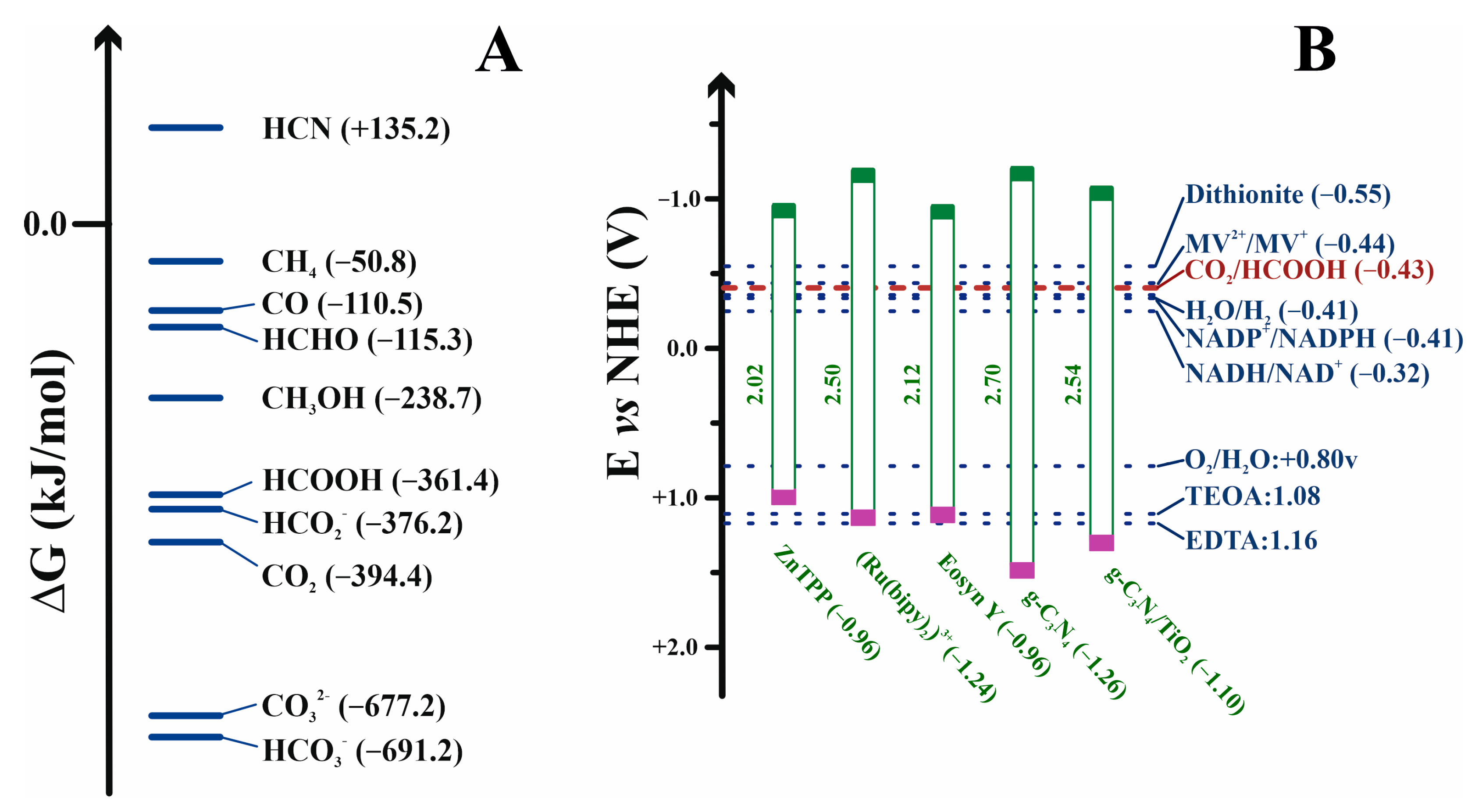{kind=link}
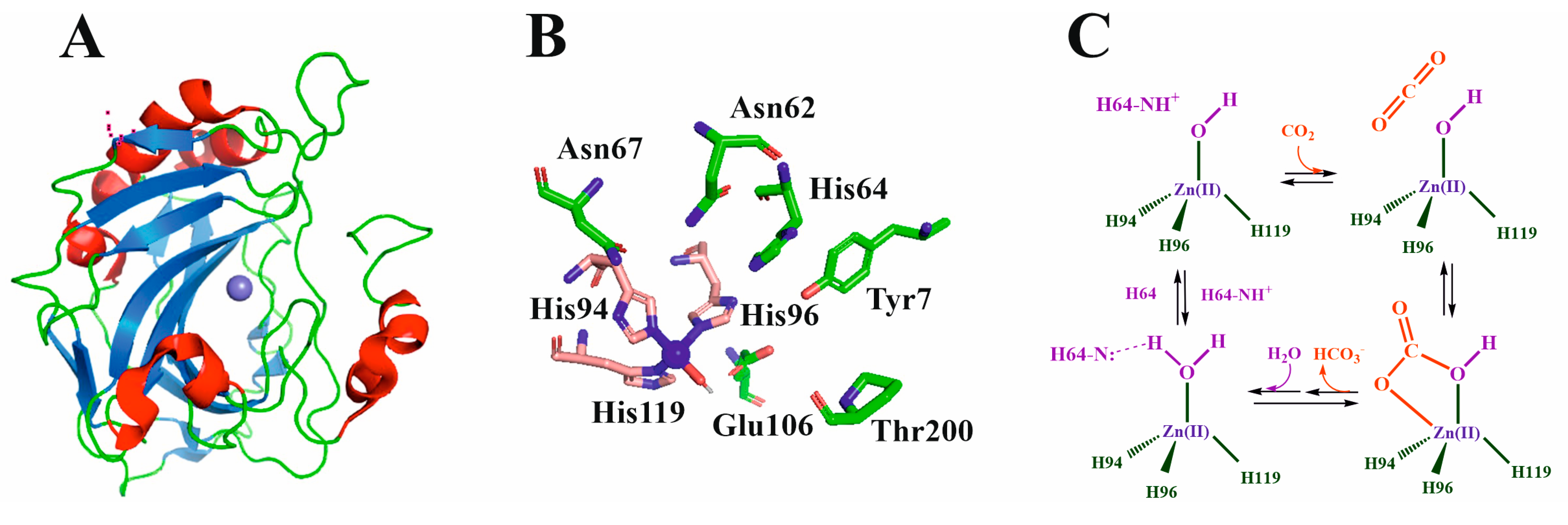{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
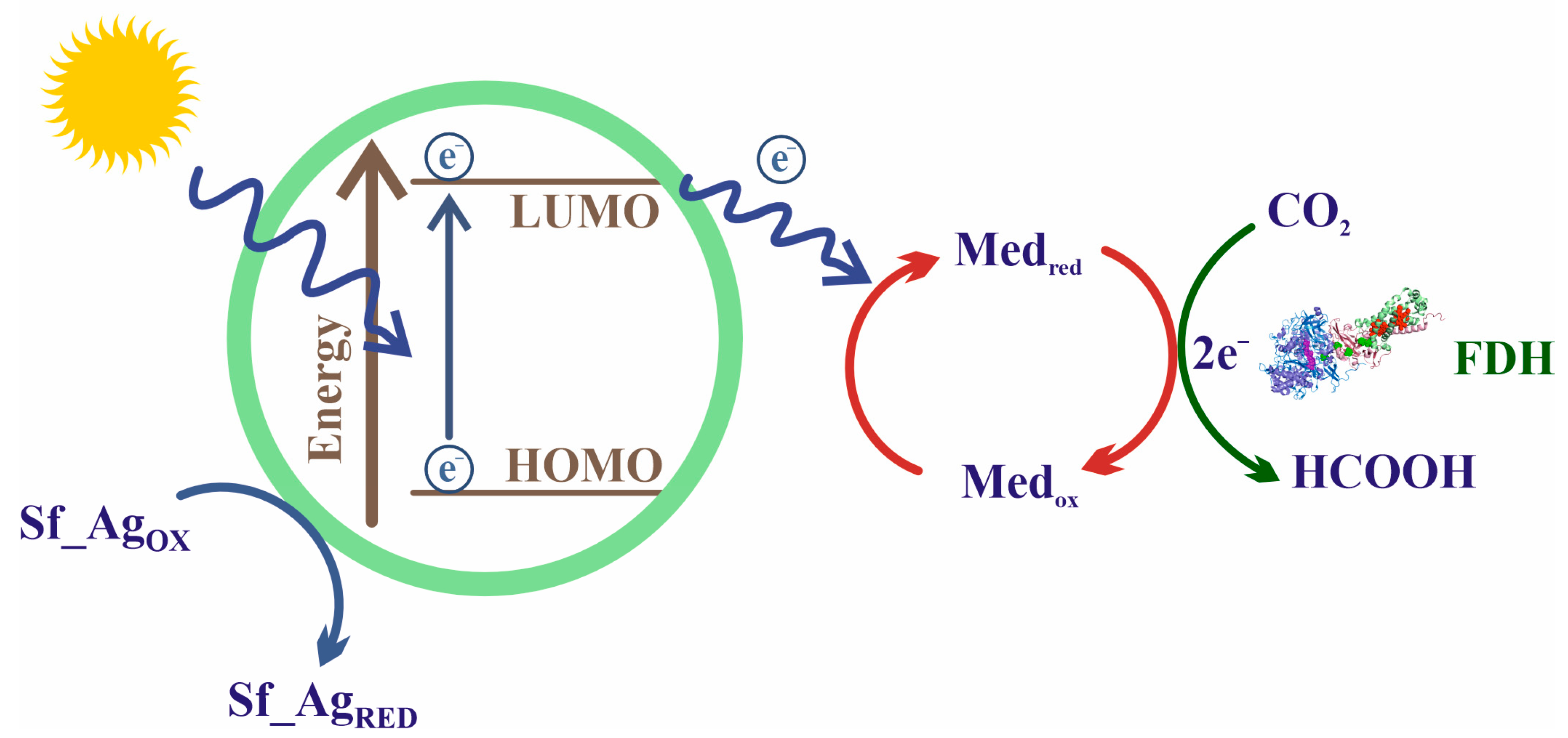{kind=link}
Table 1.
Features of representative carbonic anhydrases.
| Type | Organism | MW (kDa) | Activity (WAU/mg) | Activity (k−1) | Metal Coordination | Ref. |
|---|---|---|---|---|---|---|
| α | Bovine | 29.8 | 2540 | Zn(II), 3 His, H2O | [53] | |
| Homo sapiens (HCAI) | 28.7 | 920 | 2.0 × 105 | [54] | ||
| Homo sapiens (HCAII) | 29.1 | 8000 | 1.4 × 106 | [54] | ||
| Persephonella marina | 26.9 | 1748 | [55] | |||
| Thermosulfurimonas dismutans | 27.9 | 2032 | [53] | |||
| Thermovibrio ammonificans | 25.3 | 1016 | [53] | |||
| Bacillus halodurans | 37.0 | 3425 | [56] | |||
| Sulphurihydrogenibium yellowstonense | 26.0 | 7254 | [57] | |||
| β | Bacillus subtilis | 37.0 | 714 | Zn(II), His, 2 Cys | [58] | |
| Acetobacterium woodii | 22.0 | 1814 | [55] | |||
| Methanobacterium thermoautotrophicum | 19.9 | 580 | [55] | |||
| Aspergillus fumigatus | 23.0 | 20 | [59] | |||
| γ | Geobacillus kaustophilus | 22.0 | 179 | Zn(II) or Fe(II), 3 His, H2O | [60] | |
| Thermus thermophilus HB8 | 24.3 a | 0.9 | [61] | |||
| Methanosarcina thermophila | 40.0 | 4872 | [62] | |||
| Burkholderia pseudomallei | 28.2 | 5.3 × 105 | [63] | |||
| Vibrio cholerae | 26.3 | 7.39 × 105 | [63] | |||
| Porphyromonasgingivalis | 26.2 | 4.1 × 105 | [63] | |||
| δ | Thalassiosira weissflogii, TWCA1 | 27.0 | 1.3 × 105 | Zn(II), 3 His, H2O | [64] | |
| δ | Emiliania huxleyi | 18.3 | 1.3 × 106 (b) | Zn(II), 3 His, H2O | [65] | |
| ζ | Thalassiosira weissflogii, CDCA1 | 69 a | 1.5 × 106 | Cd(II) or Zn(II), His, 2 Cys | [42,66] | |
| η | Plasmodium falciparum | 26.2 | 1.4 × 105 | Zn(II), 2 His, Gln, H2O | [43,67] | |
| θ | Thalassiosirapseudonana | 26.0 | 122 | Cd(II) or Zn(II), His, 2 Cys | [38] | |
| Phaeodactylum tricornutum | 31.1 | 30.9 | [47] | |||
| ι | Burkholderia territorii | 28.2 | 3.0 × 105 | Cd(II) or Zn(II), His, 2 Cys | [46] | |
| Anabaena sp. PCC 7120 c | 19.3 | 16.7 | [52] | |||
| Bigelowiella natansd | 55.3 | 85.8 | [52] |
Table 2.
Kinetic and thermodynamic parameters of most relevant FDHs, classified into their two groups. Molecular weights and pH conditions in which these conditions were determined are also indicated. For metal-dependent FDHs, the types and number of their subunits as well as the cofactors of their active centers are also indicated.
Table 2.
Kinetic and thermodynamic parameters of most relevant FDHs, classified into their two groups. Molecular weights and pH conditions in which these conditions were determined are also indicated. For metal-dependent FDHs, the types and number of their subunits as well as the cofactors of their active centers are also indicated.
| A. NAD-Dependent FDHs. | ||||||||||||
| Formate Oxidation | CO2 reduction | |||||||||||
| Organism | MW (kDa) a | pH | kcat (s−1) (U mg−1) b | KM (mM) | kcat/KM (mM−1 s−1) | pH | kcat (s−1) (U mg−1) | KM (mM) | kcat/KM (mM−1 s−1) | ref | ||
| Myceliophthora thermophila | 42 | 10.5 | 0.32 | 7.2 | 0.04 | 7.0 | 0.10 | 0.43 | 0.23 | [97] | ||
| Ancylobacter aquaticus | 45 | 6.0 | (21.6) | 6.0 | (23) | 4.5 | [98] | |||||
| Candida boidinii | 41 | 7.0 | 1.081 (6.1) | 8.55 | 0.13 | 5.5 | 0.015 | 2.6 | 0.006 | [98] | ||
| Thiobacillus sp. KNK65MA | 45 | 6.5 | 1.769 (10.9) | 16.24 | 0.11 | 5.5 | 0.32 | 0.95 | 0.34 | [98] | ||
| Candida methylica | 42 | 8.0 | 1.31 (13.2) | 7.01 | 0.19 | 8.0 | 0.008 | 0.078 | 0.01 | [97] | ||
| Chaetomium thermophilum | 45 | 5.0 | 2.04(3.1) | 3.30 | 0.62 | 5.0 | 0.023 | 3.29 | 0.069 | [93,99] | ||
| Ceriporiopsis subvermispora | 40 | 6.5 | (1.3) | 6.0 | (0.8) | [98] | ||||||
| Moraxella sp. C-1 | 45 | 5.5 | (14.3) | 5.5 | (2.8) | [98] | ||||||
| Paracoccus sp. 12-A | 45 | 5.5 | (12.2) | 5.5 | (6.5) | [98] | ||||||
| B. Metal-Dependent FDHs | ||||||||||||
| Formate Oxidation | CO2 reduction | |||||||||||
| Organism | MW a (kDa) | Subunits b | Cofactors | pH | kcat(s−1) (U mg−1)a | KM(mM) | kcat/KM (mM−1 s−1) | pH | kcat(s−1) (U mg−1) a | KM(mM) | kcat/KM (mM−1 s−1) | ref |
| Syntrophobacter fumaroxidans FDH-1 | 175 | (αβγ)2 | W, SeCys 4 [Fe2S2] [Fe4S4] | 7.0 | (700) | 0.04 | - | 282 (900) | - | - | [100] | |
| Syntrophobacter fumaroxidans FDH-2 | 125 | (αβ)2 | W, SeCys 2 [Fe2S2] | 7.0 | (2700) | 0.01 | - | 282 (89) | - | - | [100] | |
| Desulfovibrio desulfuricans | 135 | (αβγ)3 | Mo, SeCys 2 MGDs 4 c-heme 2 [Fe4S4] | 8.0 | 543 | 0.0571 | 9526 | 7.0 | 46.6 | 0.0157 | 2968 | [101] |
| Escherichia coli FDH-H | 79 | αβ | Mo, SeCys 2 MGDs 1 [Fe4S4] | 7.5 | 2800 | 26 | 107.7 | 7.5 | 1.0 | 8.3 | 0.12 | [102,103] |
| Desulfovibrio vulgaris Hildenborough | 97.4 | (αβχ)3 | Mo, SeCys 4 [Fe4S4] 4 c-heme | 7.6 | 1310 (77) | 0.017 | 77.06 | 315(1.0) | 0.42 | 750 | [104,105] | |
| Acetobacterium woodii | 169 | (αβ)3 | Mo SeCys [4Fe-4S]) | 7.0 | (600) | 1.0 | - | 7.0 | 372 (132) | 3.8 | 97.9 | [106] |
| Cupriavidus necator | 178 | (αβγ)3 | Mo, 4 [Fe4S4] FMN 3 [Fe2S2] | 7.0 | 140 | 0.082 | 1707 | 7.0 | 11 | 2.7 | 4.07 | [107] |
| Rhodobacter capsulatus | 180 | (αβγ)2 | Mo, 4 Fe4S4, 1Fe2S2, 2 MGD, FMN | 5.0 | 36.5 | 281 | 0.13 | 7.7 | 1.48 | - | - | [108,109] |
| Pseudomonas oxalaticus | 315 | - | Mo, 2 FMN | - | 0.135 | - | 6.2 | 3.0 | 40 | 0.075 | [110,111] | |
| Clostridium Ijungdahlii | 80 | - | W Cys 2 MGD [Fe4S4] | 9.0 | 14.77 | 1.40 | 10.55 | 7.0 | 0.73 | 7.27 | 0.17 | [112,113] |
| Clostridium autoethanogenum | 74 | W 2 MGD [Fe4S4] | 9.0 | 1.04 | 4.51 | 0.231 | 7.0 | 4.00 | 23.15 | 0.17 | [113] | |
| Clostridium coskatii | 62 | W 2 MGD [Fe4S4] | 9.0 | 0.62 | 5.57 | 0.111 | 7.0 | 5.62 | 59.65 | 0.094 | [113] | |
| Clostridium ragsdalei | 74 | W | 9.0 | 11.88 | 44.83 | 0.265 | 7.0 | 3.28 | 31.20 | 0.11 | [113] | |
| Desulfovibrio gigas | 121 | (αβ)2 | W, SCys 4 [Fe4S4] | 8.0 | (34.1) | - | - | - | - | - | - | [114] |
| Moorella thermoacetica | (αβ)2 | W, SeCys 4 [Fe4S4] | 7.5 | (1100) | - | - | - | - | - | - | [115] | |
Sub-Table (A): a Molecular weight of the monomer (most FDHs are associated with dimers). b Data between parenthesis refer to the activity in units (μmol min−1) of formate/CO2 oxidized/reduced per mg of FDH; Sub-Table (B): a Molecular weight of the monomer. b The number refers to the number of subunits, that is, the composition of dimers or trimers of each FDH. MGD: molybdopterin guanidine dinucleotide. FMN: flavin mononucleotide.
Table 3.
Immobilization of Carbonic Anhydrase using different methods.
| Method | Support | Immobilization Conditions | Main Results | Ref |
|---|---|---|---|---|
| Physical adsorption | Mesoporous aluminosilicate | Phosphate buffer 100 mM, pH 7, 0.2 mL of enzyme (1 mg/mL) in 10 mg of support for 6 hr at 120 rpm. | CA loading up to 3 mg/mL optimal for CA activity. Carbonation activity was 55% (8 cycles of reuse | [140] |
| Carboxylic acid group-functionalized mesoporous silica (FMS) | 0.6–2.0 mg FMS with functional groups incubated with 70–1000 IL of 2.0 mg/mL BCA II in water (pH 6.5) for 2 h at 21 °C under 1200 rpm shaking. | High protein loading density 0.5 mg of protein/mg. High stability and activity (95.6%) of the immobilized enzyme | [141] | |
| Silver nanoparticles amine-functionalized mesoporous SBA-15 | 10 mg dispersed separately in 2 mL free HCA in buffer (3 mg/mL HCA in 100 mM sodium phosphate, pH 6.4) followed by incubation at 25 °C with shaking for 4 h. | The activity of the materials was ∼25-fold higher than the activity of the free HCA for converting CO2 to CaCO3 after 30 cycles. | [142] | |
| Mesoporous silica nanoparticles with polydopamine (PDA) and polyethyleneimine (PEI) | 5 mg FDH/1.6 mg CA added to 2 mL of phosphate buffer (0.05 M, pH 6). PDA/PEI-mSiO2 (0.05 g) and 5 μL GA aqueous solution (20 wt%) were added (30 °C for 24 h). | Optimal FDH and CA concentration was 2.5 and 0.8 mg/mL respectively, (specific activity of 0.045 mM/h/mg). 10 reuse cycles with 86.7% of activity. | [143] | |
| Palmityl-substituted sepharose 4B | Denatured Bovine CA solution by incubation for 15–120 min (58–65 °C), Tris-sulfate buffer, pH 7.5 (0.0125–0.5 mg/mL). | Denaturation and renaturation processes of the enzyme in the matrix. After renaturation at 60 °C, 85% of immobilization was achieved (70% of the original activity after 1 h). | [144] | |
| Silica or titania particles | The clarified lysates expressed BCA-peptide (0.088 mg protein) were incubated with 30 mg silica or 20 mg titania for 10 min with shaking at room temperature in 25 mM sodium phosphate buffer (pH 6.5). | After 10 days, 90 ± 4% and 95 ± 3% of silica and titania particles’ original activity remained. Immobilized BCA-peptide fusion protein shows ∼95% of its residual activity with up to 5 cycles of reuse. | [145] | |
| Porous polypropylene and a non-porous polydimethoysilane hollow fiber membrane | Application of CA in situ to the shell side surface of each fiber. | CO2 absorption into K2CO3 increased approximately three-fold when CA is adsorbed onto the Porous polypropylene or non-porous polydimethoxysilane PDMS membrane surface | [146] | |
| Entrapment or encapsulation | Magnetic nanoparticles (sol-gel ferria hydrosol) | 200 mL of ferria hydrosol mixed with CAB solution (10 g/L, 0.05 M Tris, pH 7.4) and dried at 20 °C. | Large thermal stability of CA immobilized, active up to 95 °C | [147] |
| Polyurethane foam (PUF) | 1 mL of CA (1 g/L) or whole-cell solution in 20 mM Tris-sulfate buffer (pH 8.3) was added to a 50 mL containing 1 g of prepolymer. The swelling of PUF continued for 30 min, and then an additional 10 min was allowed for curing. | An immobilization efficiency of 3.4%, 16-fold higher than that for free enzymes. The reusability of the immobilized whole-cell catalyst shows no apparent decrease in activity after 9 reuses. The rate of CO2 capture was accelerated by 80%. | [148] | |
| Ni-based MOFs (Ni-BTC) nanorods, | 100 mL cell lysate of His-HCA II (0.4 mg protein) and 900 mL Milli-Q water were incubated with Ni-based MOFs. | His-HCA II from cell lysate obtained an activity recovery of 99%. After storing for 10 days, the immobilized His-HCA II maintained 40% activity (free enzyme lost 91% activity). Immobilized His-HCA II retained 65% (8 cycles). | [149] | |
| Supported ionic-liquid membranes (SILMs) | 0.25 mg CA/g IL. | SILMs resent permeability (PCO2 = 733.73 barrier) at high temperatures (up to 373 K) and a good transport selectivity towards CO2 against N2. | [150] | |
| Cholinium-based ionic liquids | 0.1 mg CA/g IL | CA samples promote an enhancement of 63% in the carbon dioxide transport rate. | [151] | |
| Covalent binding and cross-linking | Mesoporous SBA-15 surfaces covalently functionalized with amines | HCA immobilization was achieved by mixing 10 mg of amine-functionalized SBA-15 with a 0.1% glutaraldehyde (GA) solution (50 mM sodium phosphate, pH 8.0, 1 h). The product was treated with free HCA (3 mg/mL HCA, pH 7.0), incubated and shaking (1 h, 25 °C). | Immobilized HCA retained activities after long-term storage, exposure to high temperatures, and reuse (40 cycles). CO2 capture efficiency of immobilized HCA was 36 times higher than that of free HCA, 75% of the enzymatic activity was retained after 40 cycles. | [152] |
| Chitosan | CA liquid was slowly added into the pH 5 chitosan solution with continuous stirring. Ratios of 1:0.05–1:2 chitosan:CA (g:mL) | Textile packing with covalently attached enzyme aggregates retained 100% of the initial 66.7% CO2 capture efficiency over 71 days and retained 85% of the initial capture efficiency after 1-year of ambient dry storage. | [153] | |
| Polyethyleneimine and polydopamine in MOF 808 | PBS buffer (pH 8, 10 mM), PEI/PDA-MOF-808 or PDA-MOF-808 was disseminated. 200–400 μL of CA solution (1 mg/mL, Milli-Q) and 10 μL of aqueous solution of GA (0–25 wt%) were added and shaken for a while at 28 C. | CaCO3 produced by CA@PEI/PDA-MOF-808 was 11.0-fold and 2.5-fold higher than free CA and PEI/PDA-MOF-808, respectively. After 8 consecutive rounds, the total production of CaCO3 by CA@PEI/PDA-MOF-808 was 92-fold higher than free CA. | [154] | |
| Alumino-siloxane hybrid aerogel beads | 100 mg of Al/Si-NH2 beads were treated with 0.5% GA for 1 h. 4 mL of free BCA in the buffer (1 mg/mL 100 mM phosphate buffer) was added to GA-treated Al/Si-NH2 beads and stirred (3 h, 25 °C) for BCA immobilization. | Free BCA retained 70% of its maximum activity (immobilized BCA, 88%). Free BCA and BCA-Al/Si-NH2 remained 80% of the activities, after ten days. BCA-Al/Si-NH2 retained 89% of their enzyme activity up to 10 cycles | [155] | |
| Amine-functionalized by co-deposition of polydopamine (PDA) and polyethyleneimine (PEI) | Co-deposition of PDA and PEI and CA covalently anchored on the surface via GA. Surface was treated with a mixture of PDA (2 mg/mL) and PEI, pH 8,5. Amine-functionalized micro-reactor surface was contacted with a mixture of CA and GA (concentration 2.0% (v/v)). | A steady CO2 absorption rate for several hours and good reusability of the immobilized enzyme which maintains its original absorption performance after 10 cycles of operation. | [156] | |
| Polypropylene hollow fiber membranes using GA-activated chitosan | Aminated knitted hollow fiber membranes mats (204 cm2 fiber surface area) were incubated in 5% GA in 100 mM phosphate buffer, pH 8.5 for 1 h under constant rocking at room temperature. | Chitosan/CA coated fibers exhibited accelerated CO2 removal in scaled-down gas exchange devices in buffer and blood (115% enhancement vs. control, 37% enhancement vs. control, respectively). | [157] | |
| Magnetic Cross-Linked Enzyme Aggregates (CLEAs) to bovine carbonic anhydrase (BCA) and magnetic nanoparticle | - Adsorption: the NPs suspension (5 and 2 mg solids) added to 1 mL of 10 mM PBS (pH 7.4), 10 g/L BCA. - Precipitation: NP-enzyme suspension added dropwise to 9 mL containing the precipitating agent (pH 7.4), 1 h or 0.5 h under mixing. - Cross-linking: 25% vol GA added. The system kept under mixing for 3, 16 and 22 h. - CLEAs separated by MF. | BCA-CLEAs can increase the CO2 absorption rate concerning the one observed in the same reactor filled with only alkaline solvent. Biocatalyst reusability analysis showed that BCA-CLEA retained 95% of its initial activity after five CO2 absorption tests at 1000 mg BCA CLEA/L and as many liquid-solid separation steps by membrane filtration. | [158] | |
| Geopolymer micro-spheres (GMS) and covalent attachment by GA | GMS were introduced into a Tris-HCl buffer solution (0.05 M, pH 8.0) with CAs solution (1 mg/mL) in a 50 mL centrifuge tube. After shaking and reacting, the GA was added for the cross-linking between the GMS and CAs. | Kcat/Km values were 61.50 and 12.36 M−1s−1 for the immobilized and free CAs, respectively. At 60 °C, free CAs were inactivated (immobilized CAs kept 34.8% activity). Immobilized CAs maintain 68.73% of activity (8 cycles). | [159] | |
| Microbial transgluta-minase (MTG) acts as a “cross-linking medium” | Iso-peptide bond between glutamine and the primary amine group of a lysine in artificial peptide tags. Equimolar amounts F-CA and M-FDH were added, and a metal constant temperature oscillator was used for the cross-linking reaction (reaction time 12 h, 25 ℃, 400 rpm. The amount of MTG used was 1 U/mL. | The remaining CA activity was more than 93%, and the remaining FDH activity was more than 84%. The efficiency of the cross-linked enzyme is increased by 5.8 times compared with free enzymes. FDH thermal stability at different temperatures is improved the optimal found CA/FDH ratio was 1:2. | [160] |
Table 4.
Relevant studies on CO2 uptake processes performed using CA, and their main features.
| Process | Idea of the Study | CO2 Uptake Conditions | Relevant Conclusions | CA Activities | Ref. |
|---|---|---|---|---|---|
| Chemical absorption | To examine the stability and activity of the enzyme under different pH values and amine solvents. Compare its performance to other common solvents. | CA tested for pH [7,8,9,10,11]. Temperature stability for up to 100 h of incubation. CA stability tested for 7 capture solvents (1 M or 3 M, 150 days, 40 °C). | CA stable at 60 °C, pH range [7,8,9,10,11]: residual activity at pH 5 or 12, ranging from 12 to 91%. The enzyme enhances reaction rates. NaCl, K2CO3, AMP, and MDEA show additive effects. | After 100 h (25 °C), CA activity was kept higher than 75% for 1M NaCl, AMP and MDEA and 125% in 1M K2CO3 | [191] |
| To develop the CA-MOFs composite, with superior catalytic performance and high stability to promote CO2 absorption into a tertiary amine solution. | 0.05 g L−1 and 40 °C, PCO2 15 kPa. | CA loading in ZIF-L-1 increased with the added enzyme amount. CA/ZIF-L-1 composite has higher catalytic activity and stability than the free CA. The immobilization of CA on ZIF-L-1 improves the CA conformational stability | CO2 absorption rate of CA/ZIF-L-1 in 1 M MEA and MDEA at 40 °C: 3.0 × 106 kmol s−1m−2. CA loading reached up to 87 mg g−1. The highest immobilized CA activity was 1.5 times that of the free CA | [192] | |
| Pilot-scale experiments with CA-enhanced MDEA for CC, bench-marking its mass transfer performance against the industrial standard 30 wt% MEA. | Experiments were done using 30 wt% MEA and MDEA solvents varying CA concentrations (0, 0.85, and 3.5 g/L) at different column L/G ratios. | Enzyme-enhanced MDEA solutions exhibit 80% of the mass transfer performance at 30 wt% MEA and have the potential to reduce the size and cost of absorbents in carbon capture. | CO2 capture efficiency higher than 90% for high L/G ratios. The mass transfer increased 20-fold for 25 and 50 wt% MDEA solutions by adding 0.2 g/L CA. | [193] | |
| Use of columns with membranes contractors with polyionic liquids (PIL), amines and CA for CO2 uptake. | PIL blend (F9:1(M10)) with immobilized CA in 30 wt% MDEA at low feed gas (15% CO2 in N2.) pressure (1.3 bar) | Addition of the enzyme to the MDEA solution with PILs significantly improves CO2 uptake rate and reduced the equilibration time. | CA addition to MDEA solution improved the CO2 uptake rate by 1.7 times. CA and membrane improved the uptake rate by twice. | [194] | |
| Carbonation | To present the effects of adding small quantities of immobilized CA on the absorption of CO2 into potassium carbonate. | CO2 absorption (partial pressure 90 kPa) in a 30% K2CO3 solution. A wet-walled column (40–80 °C) with 38 g/L CA. | CA addition improves the CO2 absorption in K2CO3 solvents. The rate of CA-catalyzed CO2 hydration increased with the CA concentration | Increasing CA concentration from 0.4 to 1.8 μM increases the absorption rate by 34% (40 °C) | [195] |
| To explore the potential of using enzymes to catalyze the conversion of CO2 into bicarbonate and on the carbonation rate of brucite, Mg(OH)2. | Gas flow (CO2/N2 10%/90%) at 10 psig. Experimental durations were 11, 7, and 3 days (low, medium, and high flow experiments). | CA accelerates the carbonation of brucite. Higher CO2 gas flow rates results in faster carbonation. Mineralogical compositions depend on the CO2 flow rate. | The carbonation rate of brucite using BCA was accelerated by up to 240% compared to controls. | [196] | |
| To propose a novel method based on microbially induced calcium precipitation to improve the cementitious properties of steel slag. | Bacillus mucilaginosus placed in a sealed container, the air pressure was reduced to −0.05 MPa. CO2 (99.99%) pumped to maintain the gas pressure (0.25 MPa, 30 °C, 32 h). | Ca-silicate promotes the growth of bacteria, enhances the production of polymers and improves bacterial adhesion. Ca-silicate-containing medium (CSCM) can be used as a microbial carrier. | Maximum bacterial growth rate in CSCM was 1.5 times higher than that in the control medium (CM). Bacterial activity in CSCM was 40% of that in CM (30 h). At a dosage of bacterial powders of 1 wt.%, carbonation reached the maximum. | [197] | |
| To develop and characterize a highly efficient and stable biocatalyst for CO2 sequestration using silica nanocomposite with auto-encapsulated CA. (CA)-based biocatalyst encapsulated in a biosilica with a peptide R5. | CaCO3 precipitation was carried out at 30 °C and monitored turbidimetrically at 600 nm. The final pH of the buffer was approximately 9.3 | The encapsulated CA was not leached from the silica matrix. Encapsulation in silica effectively improved the thermostability and activity of the enzyme. | Encapsulation efficiency greater than 95%. Residual activity of the self-encapsulated CA was more than 50% (30 min, 80 °C). Activity of ngCA-R5@silica decreased only after 2.5 days at 60 °C. Encapsulated CA: 98% and 80% of its initial activity (1 day and 5 days of incubation, 50 °C). CaCO3 precipitation is reduced by 5.5-fold when ngCA-R5@silica (30 μg/mL) is present compared to the uncatalyzed reaction. | [198] | |
| Mineralizaton | Mineralization experiments were performed using Curvibacter lanceolatus strain HJ-1, including its secreted extracellular polymeric substances (EPS) and CA (CA). | Three types of mineralization experiments: with CA (duration 96 h), with EPS (96 h), and with bacteria (50 days). | Strain HJ-1, EPS, and CA promote carbonate precipitation. HJ-1 and EPS1 experiments contained calcite and aragonite. CA formed calcite only. HJ-1 and EPS are favorable for aragonite precipitation. | The mass of precipitate (inorganic plus organic substances) increases until ca. 55 mg. The maximum degree of calcification was approximately 6.6%. In the control groups without CA no precipitate was formed. In the absence of HCO3−, the optimized calcification rate followed the order: HJ-1 (49.5%) > CA(6.6%) > EPS2(4.1%). | [199] |
| Production of CA from the bacterium Aeribacillus pallidus TSHB1, a thermostable and alkaline-stable bacterium, highly effective in the formation of CaCO3 from aqueous CO2. | Tris buffer (15 mL, pH 7.4) containing 0.9 g CaCl2-2H2O, CA 0.05 mg (37 °C). | A 3.8-fold higher CA production by A. pallidus than that under unoptimized conditions. Enzyme thermostable that retains activity at alkaline pH: it is useful in carbon sequestration. | The partially purified enzyme produces precipitation of 42.5-mg CO32− mg−1 protein. CO2 sequestration is efficient. | [200] | |
| Study of the biochemical properties, thermostability, and inhibition of CA from Sulfurihydrogenibium azorense, SazCA. | Hepes buffer 10 mM, NaBF4 20 mM, pH 7.5. Phenol red (0.2 mM) as an indicator. | SazCA is highly thermostable and can survive incubation at 90–100 °C. | kcat value 4.40 × 106 s−1, KM value 12.5 mM, kcat/KM 3.5 × 108 M−1 s−1, 5-fold faster than the second CA. SazCA is the second faster enzyme, after superoxide dismutase (SOD). | [201] | |
| To explore wollastonite (calcium silicate) carbonation for the removal of anthropogenic CO2 and evaluate the effectiveness of different natural (CA) and CA biomimetic catalysts (Zr-based MOFs) to enhance CO2 capture. | Water 170 mL, the catalyst (30 ppm), adjusted at pH 4 (25 °C). Wollastonite crystals (100 mg) added to the solution. CA is immobilized on the MOFs UiO-66 and MOF-808@Mg(OH)2 by an impregnation process. | CA accelerates carbonate precipitation but hinders carbonation of wollastonite. Thick carbonate coatings formed on the most reactive surfaces of wollastonite act as passivating layers leading to a reduction in the dissolution and carbonation rates. | The passivating effect explains why the conversion of wollastonite into calcite was so limited (up to 14 mol%). Zr-based MOFs accelerate the dissolution of wollastonite. | [202] |
Table 5.
CO2 reduction to obtain formate using whole-cell approaches.
| Reduction | Organism | Significant Assay Features/Conditions | Formate Production | Ref |
|---|---|---|---|---|
| Hydrogenation | Overexpressed genes of FDH from E. coli, Clostridium carboxidi-vorans, Pyrococcus furiosus and Methanobacterium thermos-formicicum in E. coli JM109(DE3) | H2 atmosphere. Wet cell pellet (0.5 g wet cells/mL) resuspended in 50 mM sodium phosphate buffer, pH 7.0, containing 0.25 M sodium bicarbonate as a source of CO2. Incubation 37 °C. | The highest formic yield (FDH from P. furiosus) was more than 1 g L−1 h−1 | [292] |
| Hydrogenation | Escherichia coli | CO2 and H2 placed under pressure (up to 10 bar). First, pressurized the system to rapidly convert 100% conversion of gaseous CO2 to formic acid. Next, NaOH addition to the E. coli cell suspension (pH > 8). | Formate concentration to 150 (4 bar) and 200 (6 bar) mmol L−1. Increasing the pressure to 10 bar (122.88 mmol L−1 CO2 and 3.61 mmol L−1 H2): > 0.5 mol L−1 formate at 23 h of reaction. | [293] |
| Hydrogenation | Acetobacterium woodii and Thermoanaerobacter kivui | Cells grown with 28 mM glucose or 0.1 M pyruvate (50 mM imidazole, 20 mM KCl, 20 mM MgSO4, 2 mM DTE, 4 μM resazurin, pH 7.0). 1 mg/mL in an anoxic medium. Shaking for 10 min (60 °C), with additional 300 mM KHCO3. The experiment started by replacing the gas phase with H2 + CO2 (80:20%, 2 × 105 Pa). | Optimal formate production rates of 234 mmol g−1protein h−1 | [270] |
| Hydrogenation | H2-dependent CO2 reductase from Acetobacterium woodie expressed in E. coli JM109 | Whole-cell E. coli in presence of formate (10 mM) and methylviologen (10 mM) as electron acceptor. E. coli cells incubated with H2 + CO2 (80:20%, 1.1 × 105 Pa). | 6 mM formic acid in 60 h. Addition of 2.5 mM HCO3− increased 4-fold the formate generation. | [294] |
| Hydrogenation | Acetobacterium woodii and Thermoanaerobacter kivui | 50 mM imidazole, 20 mM MgSO4, 20 mM KCl, 20 mM NaCl, 2 mM DTE, pH 7.0; or 50 mM K-phosphate, 20 mM KCl, 20 mM NaCl, 2 mM DTE, pH 7.0) was maintained at 30 and 60 °C for A. woodii and T. kivui, respectively. Gas flow rate was maintained at a value of 25 mL/min | 60 mM and 80 formic acid generation after 5 h of reaction for A. woodii and T. kivui, respectively. | [295] |
| Electrochemical | Methylobacterium extorquens AM1, | System: 1 mM H2SO4 with a platinum electrode placed in the anode; protons supplied to the cathode through a proton-exchange membrane. 1.9 g wet cells, 10 mM methyl viologen (MV) added to the cathode reactor as an electron mediator | Formate concentrations of up to 60 mM, 80 h (1.9 g wet cells, 10 mM MV, pH 7.0) | [290] |
| Electrochemical | Shewanella oneidensis MR-1 | The electrochemical cell with two compartments divided by a proton-exchange memberane. Copper plates and Ag/AgCl electrodes. Whole-cell S. oneidensis MR-1 (wet-cell, 0.5 g) and MV, 10 mM). RT, anaerobic conditions. | Formic acid at 0.59 mM h−1 for 24 h. Medium supplemented with fumarate and nitrate: 1.9 mM h−1 for 72 h. LB supplemented with 40 mM fumarate, 1mM nitrate and 20 mM DL-lactate: 136.84 mM formic acid at 72 h. (3.8 mM h−1 g−1wet-cell) | [296] |
| Electrochemical | E. coli | NaHCO3 electrolyte saturated with N2 or CO2 media at three different poised potentials, i.e., 0.4, 0.8 and 1.0 V vs. Ag/AgCl. E. coli-immobilized FePc-CDC/ACF and ACF electrodes in the presence of the NR mediator (FePc: iron pfthalocyanine; CDC: carbide-derived carbon; ACF: activated carbon fiber; NR: Neutral Red). | Maximum formate production rate of ~30 mg/L-h under CO2 flow (120 mg/L-h) with NR mediator | [297] |
| Electrochemical | Methylorubrum extorquens AM1 | Nafion 115 (proton permeable) membrane. Cathode: 0.6 g cell pellet (potassium phosphate 200 mM, pH 7.0) as catholyte. Electron mediator: ethyl viologen 10 mM. 0.6 g cell pellet suspended into catholyte (200 mM-potassium phosphate at pH 7) saturated by CO2 purging (30 min). Water splitting reaction in 100 mM-H2SO4. | Formate production: 6 mM h−1 | [298] |
| Electrochemical | Shewanella loihica PV-4 | Cathode: biohydrogel formed by Shewanella loihica PV-4 immobilized in graphene oxide. | High Faradaic efficiency (~99.5%) and 46-fold increase of formate titer without exogenous mediator (4.2 mM formate. 36 h) | [299] |
| Hydrogen/car-bohydrate fermentation | Saccharomyces cerevisiae | Two phases: Glucose fermentation for generating CO2 CO2 Ru catalysis hydrogenation | 26% of the CO2 was hydrogenated. Addition of His 150 mM: 128 mM in formic acid at 48 h. | [300] |
| Photocatalytic hydrogenation | Shewanella oneidensis MR-1 | Anaerobic conditions: N2-filled chamber and samples irradiated from outside the chamber by a KL5125 Cold 150W light source. Irradiation into MV, with triethanolamine (TEOA) as sacrificial agent in 50 mM HEPES, 50 mM NaCl, pH 7, 23 °C. | Formate (∼1500 nmol) was produced when MR-1 was incubated with CO2 (∼5000 nmol) after 48 h incubation | [301] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Villa, R.; Nieto, S.; Donaire, A.; Lozano, P. Direct Biocatalytic Processes for CO2 Capture as a Green Tool to Produce Value-Added Chemicals. Molecules 2023, 28, 5520. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules28145520
AMA Style
Villa R, Nieto S, Donaire A, Lozano P. Direct Biocatalytic Processes for CO2 Capture as a Green Tool to Produce Value-Added Chemicals. Molecules. 2023; 28(14):5520. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules28145520
Chicago/Turabian StyleVilla, Rocio, Susana Nieto, Antonio Donaire, and Pedro Lozano. 2023. "Direct Biocatalytic Processes for CO2 Capture as a Green Tool to Produce Value-Added Chemicals" Molecules 28, no. 14: 5520. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules28145520